
Expression of Many Immunologically Important Genes in ... · Expression of Many Immunologically...
12
of June 16, 2018. This information is current as Receptor and STAT1 β α Dependent on IFN- Independent of Both TLR2 and TLR4 but -Infected Macrophages Is tuberculosis Mycobacterium Important Genes in Expression of Many Immunologically Sheetal Gandotra, David Levy and Sabine Ehrt Shuangping Shi, Antje Blumenthal, Christopher M. Hickey, http://www.jimmunol.org/content/175/5/3318 doi: 10.4049/jimmunol.175.5.3318 2005; 175:3318-3328; ; J Immunol Material Supplementary http://www.jimmunol.org/content/suppl/2005/08/23/175.5.3318.DC1 References http://www.jimmunol.org/content/175/5/3318.full#ref-list-1 , 46 of which you can access for free at: cites 82 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 2005 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on June 16, 2018 http://www.jimmunol.org/ Downloaded from by guest on June 16, 2018 http://www.jimmunol.org/ Downloaded from
Transcript of Expression of Many Immunologically Important Genes in ... · Expression of Many Immunologically...

of June 16, 2018.This information is current as Receptor and STAT1βαDependent on IFN-
Independent of Both TLR2 and TLR4 but -Infected Macrophages Istuberculosis
MycobacteriumImportant Genes in Expression of Many Immunologically
Sheetal Gandotra, David Levy and Sabine EhrtShuangping Shi, Antje Blumenthal, Christopher M. Hickey,
http://www.jimmunol.org/content/175/5/3318doi: 10.4049/jimmunol.175.5.3318
2005; 175:3318-3328; ;J Immunol
MaterialSupplementary http://www.jimmunol.org/content/suppl/2005/08/23/175.5.3318.DC1
Referenceshttp://www.jimmunol.org/content/175/5/3318.full#ref-list-1
, 46 of which you can access for free at: cites 82 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2005 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on June 16, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Expression of Many Immunologically Important Genes inMycobacterium tuberculosis-Infected Macrophages IsIndependent of Both TLR2 and TLR4 but Dependent onIFN-�� Receptor and STAT11
Shuangping Shi,* Antje Blumenthal,† Christopher M. Hickey,† Sheetal Gandotra,* David Levy,‡
and Sabine Ehrt2*†
Macrophages respond to several subcellular products of Mycobacterium tuberculosis (Mtb) through TLR2 or TLR4. However,primary mouse macrophages respond to viable, virulent Mtb by pathways largely independent of MyD88, the common adaptormolecule for TLRs. Using microarrays, quantitative PCR, and ELISA with gene-disrupted macrophages and mice, we now showthat viable Mtb elicits the expression of inducible NO synthase, RANTES, IFN-inducible protein 10, immune-responsive gene 1,and many other key genes in macrophages substantially independently of TLR2, TLR4, their combination, or the TLR adaptorsToll-IL-1R domain-containing adapter protein and Toll-IL-1R domain-containing adapter inducing IFN-�. Mice deficient in bothTLR2 and TLR4 handle aerosol infection with viable Mtb as well as congenic controls. Viable Mtb also up-regulates inducible NOsynthase, RANTES, IFN-inducible protein 10, and IRG1 in macrophages that lack mannose receptor, complement receptors 3 and4, type A scavenger receptor, or CD40. These MyD88, TLR2/4-independent transcriptional responses require IFN-��R andSTAT1, but not IFN-�. Conversely, those genes whose expression is MyD88 dependent do not depend on IFN-��R or STAT1.Transcriptional induction of TNF is TLR2/4, MyD88, STAT1, and IFN-��R independent, but TNF protein release requires theTLR2/4-MyD88 pathway. Thus, macrophages respond transcriptionally to viable Mtb through at least three pathways. TLR2mediates the responses of a numerically minor set of genes that collectively do not appear to affect the course of infection in mice;regulation of TNF requires TLR2/4 for post-transcriptional control, but not for transcriptional induction; and many respondinggenes are regulated through an unknown, TLR2/4-independent pathway that may involve IFN-��R and STAT1. The Journal ofImmunology, 2005, 175: 3318–3328.
M ycobacterium tuberculosis (Mtb)3 is a facultative intra-cellular pathogen that dwells primarily in macro-phages. Macrophages that encounter Mtb can be acti-
vated to produce cytokines critical to controlling infection, such asTNF and IL-12. Activated macrophages also present Mtb-derivedAgs to CD4� T cells, helping to recall adaptive immunity. Withthe help of IFN-� and CD40L from NK and Th1 cells, macro-phages can kill intracellular Mtb, in part through induction of in-
ducible NO synthase (iNOS) (reviewed in Ref. 1). Several differentmacrophage receptors mediate Mtb binding and uptake (2), andmacrophages respond to Mtb by dramatically reprogramming theirtranscriptome (3). However, there is little evidence to establish amechanistic connection between surface receptors and transcrip-tional remodeling when macrophages encounter viable, virulent Mtb.
Macrophage receptors that participate in the uptake of Mtb in-clude complement receptors (CR3, CD11b/CD18; CR4, CD11c/CD18), mannose receptor (MR), and scavenger receptors. How-ever, aside from matrix metalloprotease 9 induction, there is littleevidence that these receptors mediate signaling events during theencounter of macrophage and live Mtb. Conversely, TLRs andCD40 have not been implicated in the uptake of Mtb but do trans-duce signals in macrophages in response to certain Mtb subcellularproducts. The TLR pathway can be implicated by disrupting indi-vidual TLRs or their adaptor proteins, such as MyD88, Toll-IL-1R(TIR) domain-containing adaptor protein (TIRAP), and TIR do-main-containing adaptor inducing IFN-� (TRIF). Engagement ofTLRs can lead to the transcription of proinflammatory cytokinegenes and costimulatory molecules. For example, the mouse mac-rophage-like cell line RAW-TT10 responded to heat-killed MtbH37Rv, H37Ra, and mycobacterial cell wall fractions enriched inlipoarabinomannan, mycolylarabinogalactan-peptidoglycan com-plexes, or other lipids to produce TNF in a TLR2-dependent man-ner (4). Overexpression of TLR2 and TLR4 allowed Chinese ham-ster ovary cells and RAW264.7 macrophage-like cells to respondto live Mtb via both TLR2 and TLR4 (5). Mycobacterial cell wallglycolipid lipoarabinomannan, mannosylated phosphatidylinositol,
*Program in Immunology and Microbial Pathogenesis, Weill Graduate School ofMedical Sciences of Cornell University, New York, NY 10021; †Department of Mi-crobiology and Immunology, Weill Medical College of Cornell University, NewYork, NY 10021; and ‡Department of Pathology and New York University CancerInstitute, New York University School of Medicine, New York, NY 10016
Received for publication January 14, 2005. Accepted for publication June 25, 2005.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by a Cancer Research Institute Predoctoral FellowshipTraining Grant (to S.S.) and National Institutes of Health Grant HL68525 (to S.E.).The Department of Microbiology and Immunology is supported by the William Ran-dolph Hearst Foundation.2 Address correspondence and reprint requests to Dr. Sabine Ehrt, Department ofMicrobiology and Immunology, Weill Cornell Medical College, Box 62, 1300 YorkAvenue, New York, NY 10021. E-mail address: [email protected] Abbreviations used in this paper: Mtb, Mycobacterium tuberculosis; BMM, bonemarrow-derived macrophage; CR, complement receptor; DC, dendritic cell; iNOS,inducible NO synthase; IP10, IFN-inducible protein 10; MOI, multiplicity of infec-tion; MR, mannose receptor; qRT-PCR, quantitative real-time PCR; SR-A, type Ascavenger receptor; TIR, Toll-IL-1R; TIRAP, TIR domain-containing adapter protein;TRIF, TIR domain-containing adapter inducing IFN-�; wt, wild type; IRG1, immune-responsive gene 1.
The Journal of Immunology
Copyright © 2005 by The American Association of Immunologists, Inc. 0022-1767/05/$02.00
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

and a 19-kDa Mtb lipoprotein served as TLR2 agonists on Chinesehamster ovary cells and RAW cells transfected with TLR2,whereas an undefined heat-labile moiety of Mtb acted via TLR4(5). Activation of human macrophages via TLR2 in response to a19-kDa Mtb lipoprotein led to NO-independent inhibition of Mtbreplication (6). Very few such experiments, however, have usedintact, viable, virulent Mtb and primary macrophages expressingonly endogenous receptors at a physiologic level. Our own exper-iments indicated that MyD88 is dispensable for the majority oftranscriptional responses of primary mouse macrophages to live,virulent Mtb (7). This implied either that TLRs are not the majorreceptors used by those cells for recognizing intact, live Mtb, orthat TLR-dependent responses to Mtb mostly involve MyD88-in-dependent signaling pathways. The latter explanation would beunprecedented for TLR2, for which MyD88 serves as an indis-pensable adaptor. Ambiguity concerning the roles of TLR2 andTLR4 in Mtb infection has been compounded by discordant resultsin mice that express a mutant form of TLR4 (the C3H/HeJ allele)or bear disrupted alleles for TLR2 (8–12).
To clarify the roles that TLR2 and TLR4 play in macrophage re-sponses to viable Mtb, we assessed global gene expression in Mtb-infected TLR2-deficient (TLR2�/�) mouse macrophages by microar-ray, susceptibility of TLR2/TLR4-deficient mice to aerosol infectionwith Mtb, and gene expression in Mtb-infected bone marrow-derivedmacrophages (BMM) from mice deficient in TLR2, TLR4, bothTLR2 and TLR4, TIRAP, or TRIF. These studies were also extendedto macrophages deficient in MR, CD18 (and thus both CR3 and CR4),type A scavenger receptor (SR-A), and CD40. Finally, becauseMyD88-independent expression of MCP-5, iNOS, and IFN-inducibleprotein 10 (IP10) in primary mouse macrophages in response to TLR4engagement depended on IFN-� and STAT1 (13), we also examinedthe roles of IFN-�, IFN-��R, and STAT1. Results from our analysessuggest that neither TLR2 nor TLR4 is required for host defenseagainst Mtb and that neither TLR2, TLR4, MyD88, TIRAP, nor TRIFis required for transcriptional induction of iNOS, immune-responsivegene 1 (IRG1), RANTES, and IP10 by live Mtb in primary BMM invitro. In contrast, these TLR2/4, MyD88-independent, Mtb-respon-sive genes require IFN-��R and STAT1 for their expression in Mtb-infected macrophages. The pathway by which macrophages senseMtb and respond via type I IFN signaling does not involve MR,SR-A, CD18 (including CR3/4), or CD40 and remains to be defined.TLR2/4- and MyD88-dependent responses can be subdivided intothose that require TLR2/4 and MyD88 for transcriptional inductionand those that depend on this pathway for post-transcriptional control.
Materials and MethodsMice
Drs. K. Takeda and S. Akira (Department of Host Defense, Research In-stitute for Microbial Diseases, Osaka University, Osaka, Japan) providedfertilized MyD88�/� ova derived from C57BL/6 mice and MyD88�/�
mice that had been backcrossed to C57BL/6 mice for six generations. Themice to which the zygotes gave rise were intercrossed to generateMyD88�/� and wild-type (wt) control mice. TIRAP�/� mice were pro-vided by Dr. R. Medzhitov (Yale University School of Medicine, NewHaven, CT) (14). TRIF-mutant mice were provided by Dr. B. Beutler (TheScripps Institute, La Jolla CA) (15). TLR2�/� and TLR4�/� mice (16, 17)were also provided by Dr. S. Akira and were crossed to obtain TLR2/TLR4double-deficient mice. Deficiency of both TLRs was confirmed by PCRfrom genomic DNA and lack of response of BMM to TLR2-activatingsynthetic triacylated peptide Pam3CSK4 and TLR4-activating LPS.STAT1�/� and IFN-��R�/� mice were generated as previously described(18, 19). IFN-��/�, CD18�/�, and CD40�/� mice were purchased fromThe Jackson Laboratory. MR knockout mice and SR-A knockout micewere generated as previously described (20, 21). Controls were mice of thesame genetic background. Mice were housed under specific pathogen-freeconditions.
Activation and infection of primary mouse macrophages
For all in vitro studies except microarrays, macrophages were collected intwo or three independent experiments from 8- to 10-wk-old gene-deficientmice and control mice on the same background (three mice per experi-ment). Bone marrow cells were flushed from femurs and differentiated intomacrophages as previously described (3). This resulted in a highly homo-geneous cell population, as judged by the expression of surface markerscharacteristic of macrophages (CD14, Mac-1, CD18, CD16/32, and F4/80)and as assessed by flow cytometry (not shown). Macrophages were in-fected with Mtb from early log phase cultures of a low passage clinicalisolate (strain 1254; American Type Culture Collection 51910) at a mul-tiplicity of infection (MOI) of 5 as described previously (3). Intracellularsurvival of Mtb and measurement of nitrite in the conditioned mediumwere previously reported (3). For microarrays, there were six independentexperiments. The six experiments were independent in the sense that theyinvolved collection of cells from different pools of mice (two mice of eachgenotype per pool) on different days, preparation of cDNA on differentdays, and hybridization to different arrays on different days.
Mouse infections
Mice were infected with logarithmic phase cultures of Mtb by aerosolusing an inhalation exposure system (Glas-Col). Animals were exposed for40 min to an aerosol produced by nebulizing 5 ml of a bacterial suspensionin PBS at a concentration of �2 � 107 bacilli/ml and �2 � 108 bacilli/ml.This resulted in inoculum sizes of 50–70 and 600–700 CFU/lung as de-termined by plating homogenized lungs onto enriched 7H11 plates 24 hafter infection.
Quantification of viable Mtb in mouse organs
Mice were killed by inhalation of CO2 under noncrowded conditions.Lungs were aseptically removed and homogenized in 4 ml of PBS con-taining 0.05% Tween 80. On day 1 after infection, 1.6 ml of lung homog-enate were plated on four 7H11 agar (Difco) plates supplemented with 10%oleic acid/albumin/dextrose catalase and 0.5% glycerol. Plates were incu-bated at 37°C, and CFU were enumerated 14–21 days later. Thereafter, atindicated time points, mice were killed, and their lungs and spleens wereaseptically removed. Spleens and one lobe of the lung were homogenizedin PBS containing 0.05% Tween 80, and serial dilutions were plated onenriched 7H11 plates for CFU. The other lobes were homogenized in 4 mlof TRIzol for RNA preparation.
Array hybridization
Twenty-four hours after infection, macrophage monolayers were lysedwith TRIzol (Invitrogen Life Technologies), and total RNA was isolated.After treatment with DNase I (Ambion) and purification (RNeasy; Qiagen),RNA (2–3 �g) was reverse transcribed (SuperScript II; Invitrogen LifeTechnologies) with a T7-poly(T) primer, and cDNA was transcribed in thepresence of biotinylated UTP and CTP (Enzo). Hybridization to GeneChipoligonucleotide arrays (Mu11KsubA, B) and scanning (Gene-Array scan-ner) followed Affymetrix protocols.
Data processing
Primary image analysis of the arrays was performed using GeneChip Mi-croarray Analysis Suite version 5.0 (Affymetrix), and images were scaledto an average hybridization intensity (average difference) of 250. Dataanalysis was conducted using GeneSpring 6.2 software (Silicon Genetics).Normalization was applied using the distribution of all genes on each chip,i.e., all measurements on each chip were divided by the 50th percentilevalue of that chip. Next, each gene was compared with its control bydividing its intensity by the average intensity of that gene in the six controlsamples (untreated macrophages). Data from six independent replicate ex-periments were used to perform a Wilcoxon two-sample rank test for eachgene. Data from a total of 24 arrays (two genotypes, wt and TLR2�/�; twoconditions, untreated and Mtb infected; six replicates for each conditionand genotype) were included in the analysis. Of the 11,000 genes repre-sented on the Affymetrix arrays, 6640 genes were detected as present (de-tection p � 0.04) by the Affymetrix Microarray Suite software in at leastfour of six samples in one of the two conditions compared. These 6640genes were included in additional analysis; the other genes were excludedbecause of their low, and thus unreliable, signal values. The regulation ofeach of these 6640 genes was tested in a Wilcoxon two-sample rank test inwt macrophages and TLR2�/� macrophages. A gene was considered reg-ulated compared with its control if its regulation changed across six ex-periments with p � 0.05. The Wilcoxon two-sample rank test was chosenbecause it is a nonparametric test that compares two paired groups. Itmakes no rigid assumptions about the distribution of the tested populations
3319The Journal of Immunology
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
(they do not have to follow a Gaussian distribution) and is resilient tooutlier data. To identify TLR2-dependent and TLR2-independent genes,regulation factors (absolute signal intensities in response to a stimulus di-vided by signal intensities of untreated samples) were tested with a Wil-coxon two-sample rank test for each gene in wt vs TLR2�/� macrophages,with p � 0.05 as the cut-off for statistical significance. Data mining wasconducted with FileMakerPro 6. This database program allowed to orga-nize the data and to query for data that meet several criteria. For example,TLR2-dependent genes were regulated in wt macrophages with p � 0.05,but either were not regulated in TLR2�/� macrophages ( p � 0.05) or wereregulated with a fold change that was at least 2-fold reduced in theTLR2�/� macrophages ( p � 0.05 and fold regulation in wt/fold regulationin TLR2�/� � 2; see Results).
Quantitative real-time PCR (qRT-PCR)
RNA was prepared from Mtb-infected BMM, Mtb-infected mouse lungs,as well as uninfected control cells and lungs. Two hundred nanograms ofRNA was transcribed into cDNA with gene-specific primers in 20 �l using50 U of Moloney murine leukemia virus reverse transcriptase(PerkinElmer). cDNA was diluted to 100 �l. PCR was performed in avolume of 15 �l on the ABI PRISM 7900HT sequence detection system(PerkinElmer) as previously described (7). The sequences of primersand probes, except that for IFN-�, were previously described (1). Thesequences of primers and probes for IFN-� were: forward primer, 5�-CTGGAGCAGCTGAATGGAAAG-3�; reverse primer, 5�-TCTCCGTCATCTCATAGGGA-3�; and probe, 5�FAM-TCAACCTCACCTACAGGGCGGACTTC-3� black whole quencher.
Quantification of TNF and IL-10 protein release by ELISA
BMM from wt, MyD88�/�, and TLR2/TLR4�/� mice were seeded in 24-well plates at 5 � 105 cells/well. Cells were infected with live Mtb at aMOI of 3–5 as previously described (3). Supernatants were collected 24 h
after infection. Mouse TNF and IL-10 concentrations were measured byELISA according to the manufacturer’s instructions (R&D Systems).
ResultsNeither TLR2, TLR4, nor their combination is required forMtb-regulated, MyD88-independent gene expression in primarymouse macrophages or for control of Mtb infection in mice
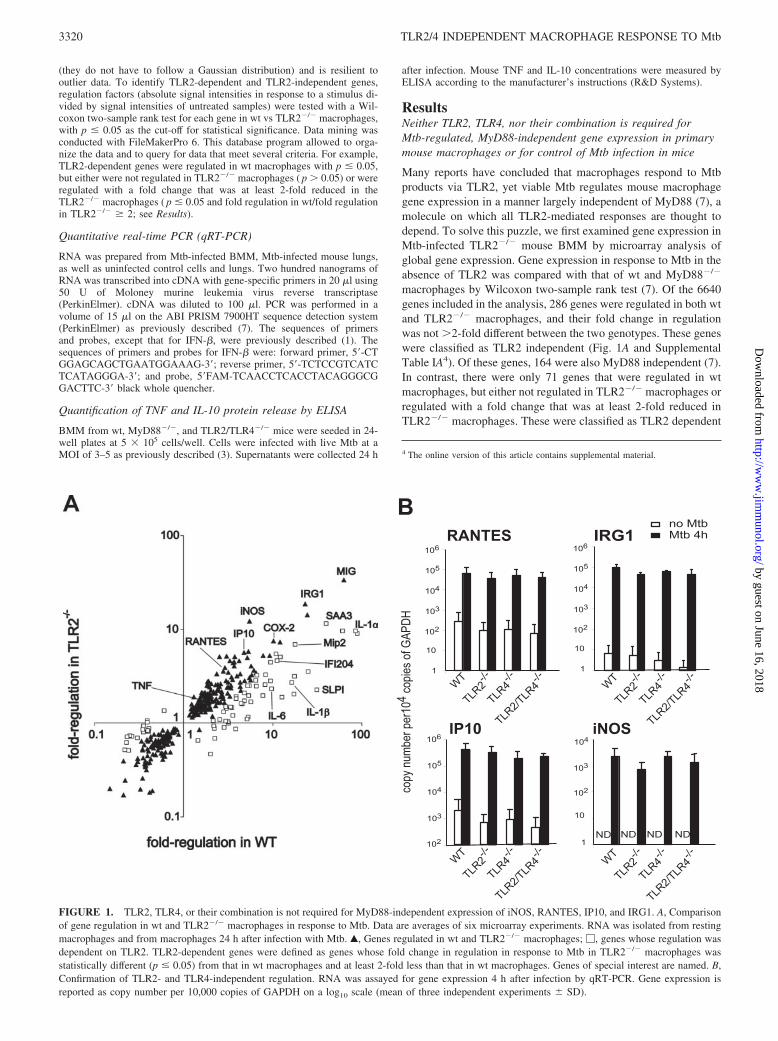
Many reports have concluded that macrophages respond to Mtbproducts via TLR2, yet viable Mtb regulates mouse macrophagegene expression in a manner largely independent of MyD88 (7), amolecule on which all TLR2-mediated responses are thought todepend. To solve this puzzle, we first examined gene expression inMtb-infected TLR2�/� mouse BMM by microarray analysis ofglobal gene expression. Gene expression in response to Mtb in theabsence of TLR2 was compared with that of wt and MyD88�/�
macrophages by Wilcoxon two-sample rank test (7). Of the 6640genes included in the analysis, 286 genes were regulated in both wtand TLR2�/� macrophages, and their fold change in regulationwas not �2-fold different between the two genotypes. These geneswere classified as TLR2 independent (Fig. 1A and SupplementalTable IA4). Of these genes, 164 were also MyD88 independent (7).In contrast, there were only 71 genes that were regulated in wtmacrophages, but either not regulated in TLR2�/� macrophages orregulated with a fold change that was at least 2-fold reduced inTLR2�/� macrophages. These were classified as TLR2 dependent
4 The online version of this article contains supplemental material.
FIGURE 1. TLR2, TLR4, or their combination is not required for MyD88-independent expression of iNOS, RANTES, IP10, and IRG1. A, Comparisonof gene regulation in wt and TLR2�/� macrophages in response to Mtb. Data are averages of six microarray experiments. RNA was isolated from restingmacrophages and from macrophages 24 h after infection with Mtb. Œ, Genes regulated in wt and TLR2�/� macrophages; �, genes whose regulation wasdependent on TLR2. TLR2-dependent genes were defined as genes whose fold change in regulation in response to Mtb in TLR2�/� macrophages wasstatistically different (p � 0.05) from that in wt macrophages and at least 2-fold less than that in wt macrophages. Genes of special interest are named. B,Confirmation of TLR2- and TLR4-independent regulation. RNA was assayed for gene expression 4 h after infection by qRT-PCR. Gene expression isreported as copy number per 10,000 copies of GAPDH on a log10 scale (mean of three independent experiments � SD).
3320 TLR2/4 INDEPENDENT MACROPHAGE RESPONSE TO Mtb
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
(Fig. 1A and Supplemental Table IB). Most of these were alsoMyD88 dependent. Independent analyses of the microarray datausing Welch’s approximate t test for two groups and Student’stwo-sample t test identified similar numbers of TLR2-dependentand TLR2-independent genes. Thus, more than twice as manygenes required neither TLR2 nor MyD88 for their regulation byMtb than those that were MyD88 and TLR2 dependent.
Unexpectedly, 20 genes appeared TLR2 dependent, but MyD88independent (Supplemental Table IC). Although unprecedented,dependency on TLR2, but not MyD88, was confirmed by inde-pendent methods for one of these genes: secretory leukocyte pro-tease inhibitor. However, when we tested two other potentiallyTLR2-dependent, MyD88-independent genes by qRT-PCR, MIP-2and IFI-204 homologue, their expression in response to Mtbproved to be independent of both TLR2 and MyD88, a rare in-stance of dissociation between microarray and qRT-PCR results inour experience (data not shown). We conclude that a MyD88-independent route to TLR2-mediated signaling exists, but it is usedinfrequently by macrophages responding to viable Mtb.
Leaving aside the role of MyD88, we focused on specific ex-amples of immunologically important genes to confirm TLR2-in-dependent signaling in response to viable Mtb. For this, we appliedqRT-PCR to mRNA from wt and TLR2�/� macrophages. This setof experiments also included TLR4�/� and TLR2/TLR4�/� mac-rophages to evaluate possible redundancy between TLR2 andTLR4 (Fig. 1B). Indeed, the expression of iNOS, IP10, RANTES,and IRG1 was regulated by Mtb in the absence of either or bothTLR2 and TLR4.
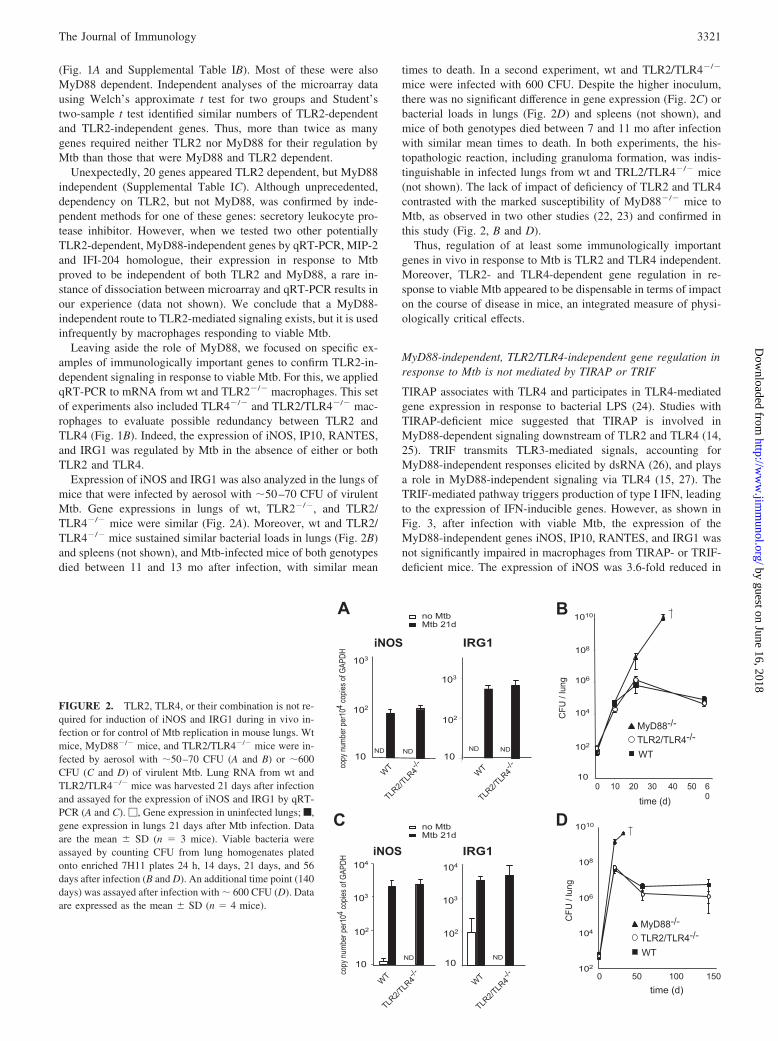
Expression of iNOS and IRG1 was also analyzed in the lungs ofmice that were infected by aerosol with �50–70 CFU of virulentMtb. Gene expressions in lungs of wt, TLR2�/�, and TLR2/TLR4�/� mice were similar (Fig. 2A). Moreover, wt and TLR2/TLR4�/� mice sustained similar bacterial loads in lungs (Fig. 2B)and spleens (not shown), and Mtb-infected mice of both genotypesdied between 11 and 13 mo after infection, with similar mean
times to death. In a second experiment, wt and TLR2/TLR4�/�
mice were infected with 600 CFU. Despite the higher inoculum,there was no significant difference in gene expression (Fig. 2C) orbacterial loads in lungs (Fig. 2D) and spleens (not shown), andmice of both genotypes died between 7 and 11 mo after infectionwith similar mean times to death. In both experiments, the his-topathologic reaction, including granuloma formation, was indis-tinguishable in infected lungs from wt and TRL2/TLR4�/� mice(not shown). The lack of impact of deficiency of TLR2 and TLR4contrasted with the marked susceptibility of MyD88�/� mice toMtb, as observed in two other studies (22, 23) and confirmed inthis study (Fig. 2, B and D).
Thus, regulation of at least some immunologically importantgenes in vivo in response to Mtb is TLR2 and TLR4 independent.Moreover, TLR2- and TLR4-dependent gene regulation in re-sponse to viable Mtb appeared to be dispensable in terms of impacton the course of disease in mice, an integrated measure of physi-ologically critical effects.
MyD88-independent, TLR2/TLR4-independent gene regulation inresponse to Mtb is not mediated by TIRAP or TRIF
TIRAP associates with TLR4 and participates in TLR4-mediatedgene expression in response to bacterial LPS (24). Studies withTIRAP-deficient mice suggested that TIRAP is involved inMyD88-dependent signaling downstream of TLR2 and TLR4 (14,25). TRIF transmits TLR3-mediated signals, accounting forMyD88-independent responses elicited by dsRNA (26), and playsa role in MyD88-independent signaling via TLR4 (15, 27). TheTRIF-mediated pathway triggers production of type I IFN, leadingto the expression of IFN-inducible genes. However, as shown inFig. 3, after infection with viable Mtb, the expression of theMyD88-independent genes iNOS, IP10, RANTES, and IRG1 wasnot significantly impaired in macrophages from TIRAP- or TRIF-deficient mice. The expression of iNOS was 3.6-fold reduced in
FIGURE 2. TLR2, TLR4, or their combination is not re-quired for induction of iNOS and IRG1 during in vivo in-fection or for control of Mtb replication in mouse lungs. Wtmice, MyD88�/� mice, and TLR2/TLR4�/� mice were in-fected by aerosol with �50–70 CFU (A and B) or �600CFU (C and D) of virulent Mtb. Lung RNA from wt andTLR2/TLR4�/� mice was harvested 21 days after infectionand assayed for the expression of iNOS and IRG1 by qRT-PCR (A and C). �, Gene expression in uninfected lungs; f,gene expression in lungs 21 days after Mtb infection. Dataare the mean � SD (n 3 mice). Viable bacteria wereassayed by counting CFU from lung homogenates platedonto enriched 7H11 plates 24 h, 14 days, 21 days, and 56days after infection (B and D). An additional time point (140days) was assayed after infection with � 600 CFU (D). Dataare expressed as the mean � SD (n 4 mice).
3321The Journal of Immunology
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
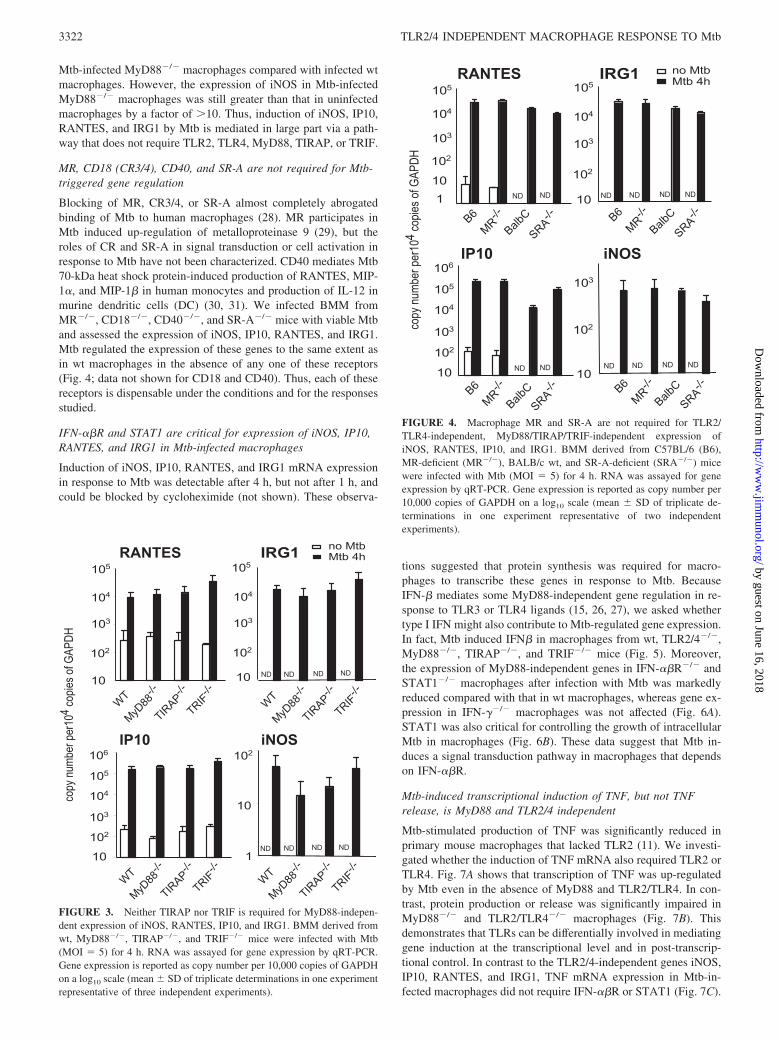
Mtb-infected MyD88�/� macrophages compared with infected wtmacrophages. However, the expression of iNOS in Mtb-infectedMyD88�/� macrophages was still greater than that in uninfectedmacrophages by a factor of �10. Thus, induction of iNOS, IP10,RANTES, and IRG1 by Mtb is mediated in large part via a path-way that does not require TLR2, TLR4, MyD88, TIRAP, or TRIF.
MR, CD18 (CR3/4), CD40, and SR-A are not required for Mtb-triggered gene regulation
Blocking of MR, CR3/4, or SR-A almost completely abrogatedbinding of Mtb to human macrophages (28). MR participates inMtb induced up-regulation of metalloproteinase 9 (29), but theroles of CR and SR-A in signal transduction or cell activation inresponse to Mtb have not been characterized. CD40 mediates Mtb70-kDa heat shock protein-induced production of RANTES, MIP-1�, and MIP-1� in human monocytes and production of IL-12 inmurine dendritic cells (DC) (30, 31). We infected BMM fromMR�/�, CD18�/�, CD40�/�, and SR-A�/� mice with viable Mtband assessed the expression of iNOS, IP10, RANTES, and IRG1.Mtb regulated the expression of these genes to the same extent asin wt macrophages in the absence of any one of these receptors(Fig. 4; data not shown for CD18 and CD40). Thus, each of thesereceptors is dispensable under the conditions and for the responsesstudied.
IFN-��R and STAT1 are critical for expression of iNOS, IP10,RANTES, and IRG1 in Mtb-infected macrophages
Induction of iNOS, IP10, RANTES, and IRG1 mRNA expressionin response to Mtb was detectable after 4 h, but not after 1 h, andcould be blocked by cycloheximide (not shown). These observa-
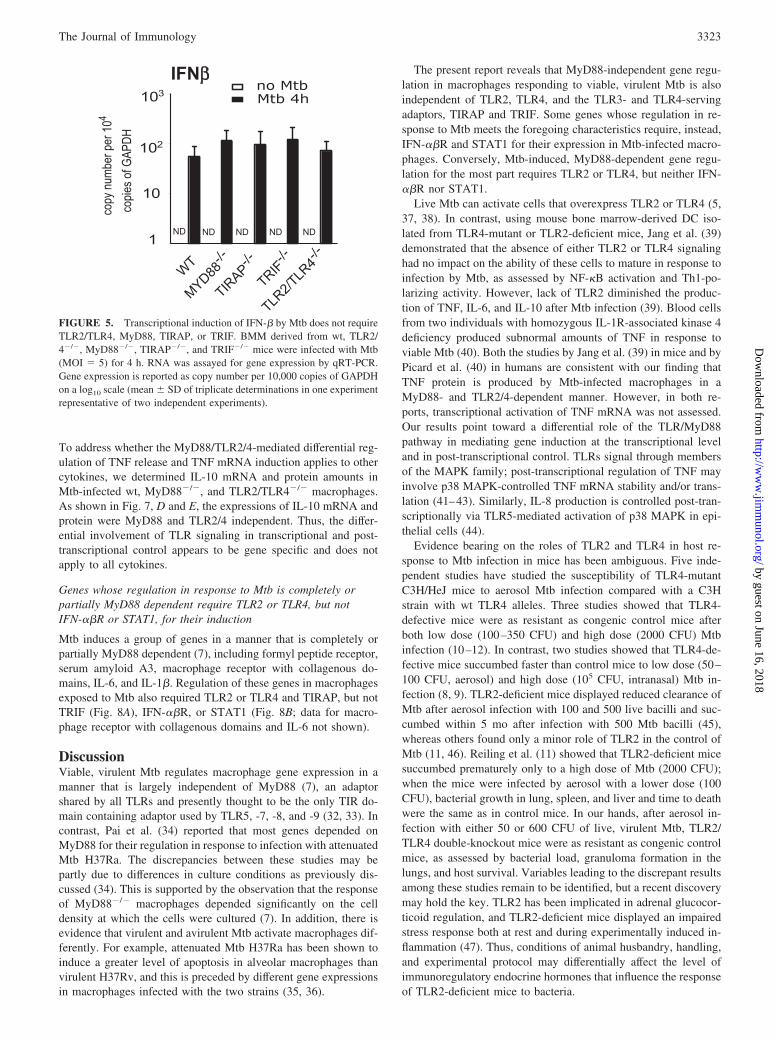
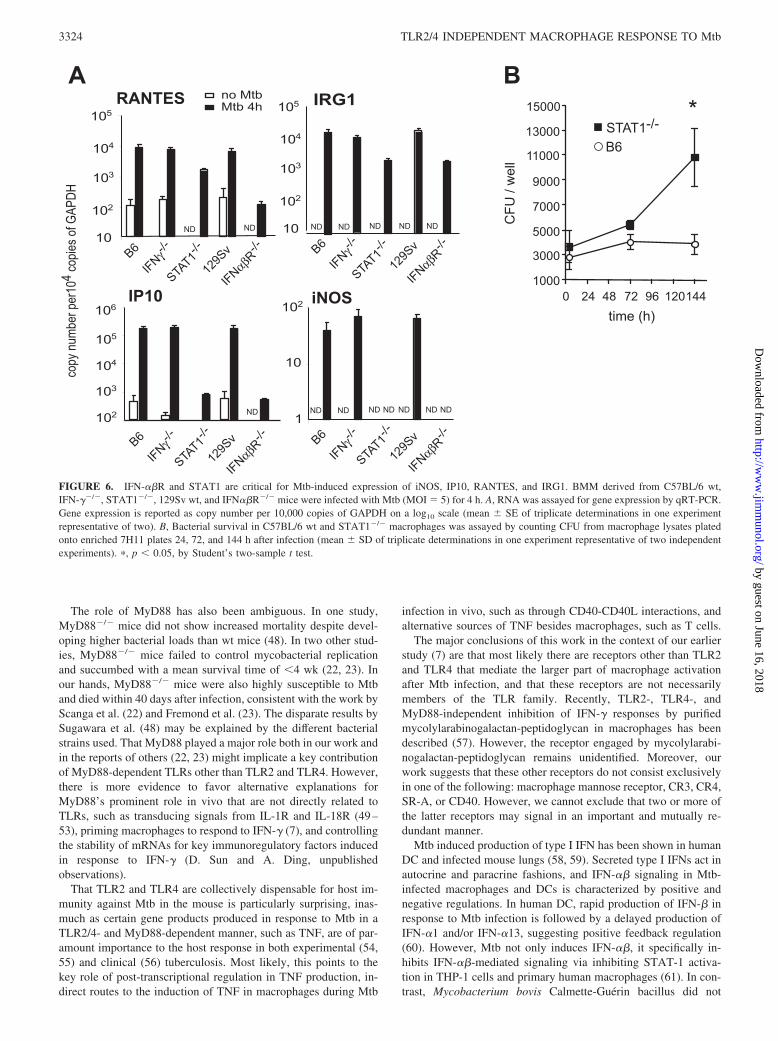
tions suggested that protein synthesis was required for macro-phages to transcribe these genes in response to Mtb. BecauseIFN-� mediates some MyD88-independent gene regulation in re-sponse to TLR3 or TLR4 ligands (15, 26, 27), we asked whethertype I IFN might also contribute to Mtb-regulated gene expression.In fact, Mtb induced IFN� in macrophages from wt, TLR2/4�/�,MyD88�/�, TIRAP�/�, and TRIF�/� mice (Fig. 5). Moreover,the expression of MyD88-independent genes in IFN-��R�/� andSTAT1�/� macrophages after infection with Mtb was markedlyreduced compared with that in wt macrophages, whereas gene ex-pression in IFN-��/� macrophages was not affected (Fig. 6A).STAT1 was also critical for controlling the growth of intracellularMtb in macrophages (Fig. 6B). These data suggest that Mtb in-duces a signal transduction pathway in macrophages that dependson IFN-��R.
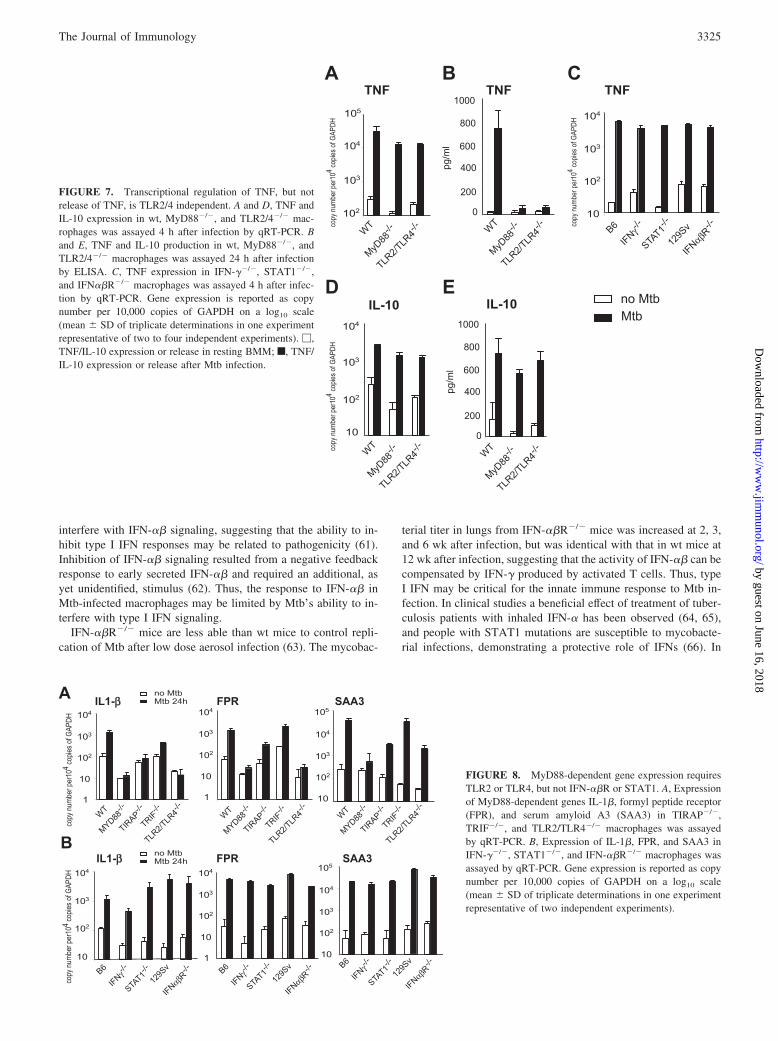
Mtb-induced transcriptional induction of TNF, but not TNFrelease, is MyD88 and TLR2/4 independent
Mtb-stimulated production of TNF was significantly reduced inprimary mouse macrophages that lacked TLR2 (11). We investi-gated whether the induction of TNF mRNA also required TLR2 orTLR4. Fig. 7A shows that transcription of TNF was up-regulatedby Mtb even in the absence of MyD88 and TLR2/TLR4. In con-trast, protein production or release was significantly impaired inMyD88�/� and TLR2/TLR4�/� macrophages (Fig. 7B). Thisdemonstrates that TLRs can be differentially involved in mediatinggene induction at the transcriptional level and in post-transcrip-tional control. In contrast to the TLR2/4-independent genes iNOS,IP10, RANTES, and IRG1, TNF mRNA expression in Mtb-in-fected macrophages did not require IFN-��R or STAT1 (Fig. 7C).
FIGURE 3. Neither TIRAP nor TRIF is required for MyD88-indepen-dent expression of iNOS, RANTES, IP10, and IRG1. BMM derived fromwt, MyD88�/�, TIRAP�/�, and TRIF�/� mice were infected with Mtb(MOI 5) for 4 h. RNA was assayed for gene expression by qRT-PCR.Gene expression is reported as copy number per 10,000 copies of GAPDHon a log10 scale (mean � SD of triplicate determinations in one experimentrepresentative of three independent experiments).
FIGURE 4. Macrophage MR and SR-A are not required for TLR2/TLR4-independent, MyD88/TIRAP/TRIF-independent expression ofiNOS, RANTES, IP10, and IRG1. BMM derived from C57BL/6 (B6),MR-deficient (MR�/�), BALB/c wt, and SR-A-deficient (SRA�/�) micewere infected with Mtb (MOI 5) for 4 h. RNA was assayed for geneexpression by qRT-PCR. Gene expression is reported as copy number per10,000 copies of GAPDH on a log10 scale (mean � SD of triplicate de-terminations in one experiment representative of two independentexperiments).
3322 TLR2/4 INDEPENDENT MACROPHAGE RESPONSE TO Mtb
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
To address whether the MyD88/TLR2/4-mediated differential reg-ulation of TNF release and TNF mRNA induction applies to othercytokines, we determined IL-10 mRNA and protein amounts inMtb-infected wt, MyD88�/�, and TLR2/TLR4�/� macrophages.As shown in Fig. 7, D and E, the expressions of IL-10 mRNA andprotein were MyD88 and TLR2/4 independent. Thus, the differ-ential involvement of TLR signaling in transcriptional and post-transcriptional control appears to be gene specific and does notapply to all cytokines.
Genes whose regulation in response to Mtb is completely orpartially MyD88 dependent require TLR2 or TLR4, but notIFN-��R or STAT1, for their induction
Mtb induces a group of genes in a manner that is completely orpartially MyD88 dependent (7), including formyl peptide receptor,serum amyloid A3, macrophage receptor with collagenous do-mains, IL-6, and IL-1�. Regulation of these genes in macrophagesexposed to Mtb also required TLR2 or TLR4 and TIRAP, but notTRIF (Fig. 8A), IFN-��R, or STAT1 (Fig. 8B; data for macro-phage receptor with collagenous domains and IL-6 not shown).
DiscussionViable, virulent Mtb regulates macrophage gene expression in amanner that is largely independent of MyD88 (7), an adaptorshared by all TLRs and presently thought to be the only TIR do-main containing adaptor used by TLR5, -7, -8, and -9 (32, 33). Incontrast, Pai et al. (34) reported that most genes depended onMyD88 for their regulation in response to infection with attenuatedMtb H37Ra. The discrepancies between these studies may bepartly due to differences in culture conditions as previously dis-cussed (34). This is supported by the observation that the responseof MyD88�/� macrophages depended significantly on the celldensity at which the cells were cultured (7). In addition, there isevidence that virulent and avirulent Mtb activate macrophages dif-ferently. For example, attenuated Mtb H37Ra has been shown toinduce a greater level of apoptosis in alveolar macrophages thanvirulent H37Rv, and this is preceded by different gene expressionsin macrophages infected with the two strains (35, 36).
The present report reveals that MyD88-independent gene regu-lation in macrophages responding to viable, virulent Mtb is alsoindependent of TLR2, TLR4, and the TLR3- and TLR4-servingadaptors, TIRAP and TRIF. Some genes whose regulation in re-sponse to Mtb meets the foregoing characteristics require, instead,IFN-��R and STAT1 for their expression in Mtb-infected macro-phages. Conversely, Mtb-induced, MyD88-dependent gene regu-lation for the most part requires TLR2 or TLR4, but neither IFN-��R nor STAT1.
Live Mtb can activate cells that overexpress TLR2 or TLR4 (5,37, 38). In contrast, using mouse bone marrow-derived DC iso-lated from TLR4-mutant or TLR2-deficient mice, Jang et al. (39)demonstrated that the absence of either TLR2 or TLR4 signalinghad no impact on the ability of these cells to mature in response toinfection by Mtb, as assessed by NF-�B activation and Th1-po-larizing activity. However, lack of TLR2 diminished the produc-tion of TNF, IL-6, and IL-10 after Mtb infection (39). Blood cellsfrom two individuals with homozygous IL-1R-associated kinase 4deficiency produced subnormal amounts of TNF in response toviable Mtb (40). Both the studies by Jang et al. (39) in mice and byPicard et al. (40) in humans are consistent with our finding thatTNF protein is produced by Mtb-infected macrophages in aMyD88- and TLR2/4-dependent manner. However, in both re-ports, transcriptional activation of TNF mRNA was not assessed.Our results point toward a differential role of the TLR/MyD88pathway in mediating gene induction at the transcriptional leveland in post-transcriptional control. TLRs signal through membersof the MAPK family; post-transcriptional regulation of TNF mayinvolve p38 MAPK-controlled TNF mRNA stability and/or trans-lation (41–43). Similarly, IL-8 production is controlled post-tran-scriptionally via TLR5-mediated activation of p38 MAPK in epi-thelial cells (44).
Evidence bearing on the roles of TLR2 and TLR4 in host re-sponse to Mtb infection in mice has been ambiguous. Five inde-pendent studies have studied the susceptibility of TLR4-mutantC3H/HeJ mice to aerosol Mtb infection compared with a C3Hstrain with wt TLR4 alleles. Three studies showed that TLR4-defective mice were as resistant as congenic control mice afterboth low dose (100–350 CFU) and high dose (2000 CFU) Mtbinfection (10–12). In contrast, two studies showed that TLR4-de-fective mice succumbed faster than control mice to low dose (50–100 CFU, aerosol) and high dose (105 CFU, intranasal) Mtb in-fection (8, 9). TLR2-deficient mice displayed reduced clearance ofMtb after aerosol infection with 100 and 500 live bacilli and suc-cumbed within 5 mo after infection with 500 Mtb bacilli (45),whereas others found only a minor role of TLR2 in the control ofMtb (11, 46). Reiling et al. (11) showed that TLR2-deficient micesuccumbed prematurely only to a high dose of Mtb (2000 CFU);when the mice were infected by aerosol with a lower dose (100CFU), bacterial growth in lung, spleen, and liver and time to deathwere the same as in control mice. In our hands, after aerosol in-fection with either 50 or 600 CFU of live, virulent Mtb, TLR2/TLR4 double-knockout mice were as resistant as congenic controlmice, as assessed by bacterial load, granuloma formation in thelungs, and host survival. Variables leading to the discrepant resultsamong these studies remain to be identified, but a recent discoverymay hold the key. TLR2 has been implicated in adrenal glucocor-ticoid regulation, and TLR2-deficient mice displayed an impairedstress response both at rest and during experimentally induced in-flammation (47). Thus, conditions of animal husbandry, handling,and experimental protocol may differentially affect the level ofimmunoregulatory endocrine hormones that influence the responseof TLR2-deficient mice to bacteria.
FIGURE 5. Transcriptional induction of IFN-� by Mtb does not requireTLR2/TLR4, MyD88, TIRAP, or TRIF. BMM derived from wt, TLR2/4�/�, MyD88�/�, TIRAP�/�, and TRIF�/� mice were infected with Mtb(MOI 5) for 4 h. RNA was assayed for gene expression by qRT-PCR.Gene expression is reported as copy number per 10,000 copies of GAPDHon a log10 scale (mean � SD of triplicate determinations in one experimentrepresentative of two independent experiments).
3323The Journal of Immunology
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
The role of MyD88 has also been ambiguous. In one study,MyD88�/� mice did not show increased mortality despite devel-oping higher bacterial loads than wt mice (48). In two other stud-ies, MyD88�/� mice failed to control mycobacterial replicationand succumbed with a mean survival time of �4 wk (22, 23). Inour hands, MyD88�/� mice were also highly susceptible to Mtband died within 40 days after infection, consistent with the work byScanga et al. (22) and Fremond et al. (23). The disparate results bySugawara et al. (48) may be explained by the different bacterialstrains used. That MyD88 played a major role both in our work andin the reports of others (22, 23) might implicate a key contributionof MyD88-dependent TLRs other than TLR2 and TLR4. However,there is more evidence to favor alternative explanations forMyD88’s prominent role in vivo that are not directly related toTLRs, such as transducing signals from IL-1R and IL-18R (49–53), priming macrophages to respond to IFN-� (7), and controllingthe stability of mRNAs for key immunoregulatory factors inducedin response to IFN-� (D. Sun and A. Ding, unpublishedobservations).
That TLR2 and TLR4 are collectively dispensable for host im-munity against Mtb in the mouse is particularly surprising, inas-much as certain gene products produced in response to Mtb in aTLR2/4- and MyD88-dependent manner, such as TNF, are of par-amount importance to the host response in both experimental (54,55) and clinical (56) tuberculosis. Most likely, this points to thekey role of post-transcriptional regulation in TNF production, in-direct routes to the induction of TNF in macrophages during Mtb
infection in vivo, such as through CD40-CD40L interactions, andalternative sources of TNF besides macrophages, such as T cells.
The major conclusions of this work in the context of our earlierstudy (7) are that most likely there are receptors other than TLR2and TLR4 that mediate the larger part of macrophage activationafter Mtb infection, and that these receptors are not necessarilymembers of the TLR family. Recently, TLR2-, TLR4-, andMyD88-independent inhibition of IFN-� responses by purifiedmycolylarabinogalactan-peptidoglycan in macrophages has beendescribed (57). However, the receptor engaged by mycolylarabi-nogalactan-peptidoglycan remains unidentified. Moreover, ourwork suggests that these other receptors do not consist exclusivelyin one of the following: macrophage mannose receptor, CR3, CR4,SR-A, or CD40. However, we cannot exclude that two or more ofthe latter receptors may signal in an important and mutually re-dundant manner.
Mtb induced production of type I IFN has been shown in humanDC and infected mouse lungs (58, 59). Secreted type I IFNs act inautocrine and paracrine fashions, and IFN-�� signaling in Mtb-infected macrophages and DCs is characterized by positive andnegative regulations. In human DC, rapid production of IFN-� inresponse to Mtb infection is followed by a delayed production ofIFN-�1 and/or IFN-�13, suggesting positive feedback regulation(60). However, Mtb not only induces IFN-��, it specifically in-hibits IFN-��-mediated signaling via inhibiting STAT-1 activa-tion in THP-1 cells and primary human macrophages (61). In con-trast, Mycobacterium bovis Calmette-Guerin bacillus did not
FIGURE 6. IFN-��R and STAT1 are critical for Mtb-induced expression of iNOS, IP10, RANTES, and IRG1. BMM derived from C57BL/6 wt,IFN-��/�, STAT1�/�, 129Sv wt, and IFN��R�/� mice were infected with Mtb (MOI 5) for 4 h. A, RNA was assayed for gene expression by qRT-PCR.Gene expression is reported as copy number per 10,000 copies of GAPDH on a log10 scale (mean � SE of triplicate determinations in one experimentrepresentative of two). B, Bacterial survival in C57BL/6 wt and STAT1�/� macrophages was assayed by counting CFU from macrophage lysates platedonto enriched 7H11 plates 24, 72, and 144 h after infection (mean � SD of triplicate determinations in one experiment representative of two independentexperiments). �, p � 0.05, by Student’s two-sample t test.
3324 TLR2/4 INDEPENDENT MACROPHAGE RESPONSE TO Mtb
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
interfere with IFN-�� signaling, suggesting that the ability to in-hibit type I IFN responses may be related to pathogenicity (61).Inhibition of IFN-�� signaling resulted from a negative feedbackresponse to early secreted IFN-�� and required an additional, asyet unidentified, stimulus (62). Thus, the response to IFN-�� inMtb-infected macrophages may be limited by Mtb’s ability to in-terfere with type I IFN signaling.
IFN-��R�/� mice are less able than wt mice to control repli-cation of Mtb after low dose aerosol infection (63). The mycobac-
terial titer in lungs from IFN-��R�/� mice was increased at 2, 3,and 6 wk after infection, but was identical with that in wt mice at12 wk after infection, suggesting that the activity of IFN-�� can becompensated by IFN-� produced by activated T cells. Thus, typeI IFN may be critical for the innate immune response to Mtb in-fection. In clinical studies a beneficial effect of treatment of tuber-culosis patients with inhaled IFN-� has been observed (64, 65),and people with STAT1 mutations are susceptible to mycobacte-rial infections, demonstrating a protective role of IFNs (66). In
FIGURE 7. Transcriptional regulation of TNF, but notrelease of TNF, is TLR2/4 independent. A and D, TNF andIL-10 expression in wt, MyD88�/�, and TLR2/4�/� mac-rophages was assayed 4 h after infection by qRT-PCR. Band E, TNF and IL-10 production in wt, MyD88�/�, andTLR2/4�/� macrophages was assayed 24 h after infectionby ELISA. C, TNF expression in IFN-��/�, STAT1�/�,and IFN��R�/� macrophages was assayed 4 h after infec-tion by qRT-PCR. Gene expression is reported as copynumber per 10,000 copies of GAPDH on a log10 scale(mean � SD of triplicate determinations in one experimentrepresentative of two to four independent experiments). �,TNF/IL-10 expression or release in resting BMM; f, TNF/IL-10 expression or release after Mtb infection.
FIGURE 8. MyD88-dependent gene expression requiresTLR2 or TLR4, but not IFN-��R or STAT1. A, Expressionof MyD88-dependent genes IL-1�, formyl peptide receptor(FPR), and serum amyloid A3 (SAA3) in TIRAP�/�,TRIF�/�, and TLR2/TLR4�/� macrophages was assayedby qRT-PCR. B, Expression of IL-1�, FPR, and SAA3 inIFN-��/�, STAT1�/�, and IFN-��R�/� macrophages wasassayed by qRT-PCR. Gene expression is reported as copynumber per 10,000 copies of GAPDH on a log10 scale(mean � SD of triplicate determinations in one experimentrepresentative of two independent experiments).
3325The Journal of Immunology
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

contrast, treatment of human monocytes with IFN-�� resulted inincreased growth of M. bovis Calmette-Guerin bacillus, and intra-nasal instillation of IFN-�� in Mtb-infected mice increased my-cobacterial growth and impaired mouse survival (59, 67). Thus, theroles of type I IFN for the pathogenesis of and protection againstMtb are complex and are likely to be beneficial as well as detri-mental depending on the context. Similarly, type I IFNs playedvariable roles in innate immunity against other intracellular patho-gens. IFN-��R-deficient mice showed increased resistance to in-fection with Listeria monocytogenes, which appeared to be due toa release of IFN-�-mediated TNF-� suppression and a block ofIFN-��-induced apoptosis of splenic lymphocytes (68–70). Incontrast, IFN-�� inhibited growth of intracellular Legionellapneumophilia in primary mouse macrophages via an IFN-�-inde-pendent, as yet unidentified, mechanism (71). IFN-�� was alsofound to increase resistance against Toxoplasma gondii, Leishma-nia major, and Clamydia psittaci (72–74). Taken together, the im-pact of type I IFN may vary and depend on several aspects of thehost-pathogen interaction (75).
We demonstrated that Mtb induces IFN-� mRNA expression inmouse macrophages and that the expression of immunologicallyrelevant genes requires IFN-��R and STAT1, but not TLR2/4,MyD88, TIRAP, or TRIF. Similar to our observation, expressionof IP-10 in Mtb-infected human DCs was also dependent on IFN-��, suggesting that chemokine responses in DCs are modulated byIFN-�� in an autocrine and paracrine manner (76). Induction oftype I IFN via TLR7, TLR8, and TLR9 in DCs requires MyD88(77–80), in contrast to the pathway activated by Mtb. IFN-� canalso be induced in a TRIF-dependent manner by TLR4 and TLR3ligands (27, 81). However, neither TLR4 nor TRIF was requiredfor induction of IFN-� mRNA by Mtb. Thus, none of the forego-ing pathways appears to be essential for type I IFN induction byMtb. Without testing TRIF/MyD88 double-deficient mice, we can-not rule out that the TLR4/TRIF pathway might be redundant withthe TLR7/TLR8/TLR9/MyD88 pathway. However, this seems un-likely, because one would expect at least a partial loss of responsein the single adaptor mutants, which we did not observe. Similar toour observations, Stockinger et al. (82) showed that macrophagesinfected with L. monocytogenes produced type I IFN via a pathwayindependent of TLR4, TLR9, MyD88, TRIF, TRIF-related adaptormolecule, and nucleotide-binding oligomerization domain 2.
In summary, we have provided evidence for four pathways lead-ing to gene regulation in response to intact, viable Mtb in primarymouse macrophages. In pathway 1, Mtb signals through TLRs,chiefly TLR2, in a manner dependent on MyD88. In pathway 2,Mtb signals through TLR2, but not through MyD88; this pathway,although novel and mechanistically intriguing, is used sparingly.In pathway 3, Mtb signals through an unknown receptor(s), ap-parently leading as a primary response to production of type I IFN,because the transcriptional responses to Mtb appear to be second-ary and depend, at least for the genes tested in this study, on IFN-��R and STAT1. In pathway 4, Mtb transcriptionally activatesgene expression in a TLR2/4-, MyD88-, IFN-��R-independentmanner, but protein production requires MyD88 and TLR2/4. It isthe third pathway that dominates the response to viable Mtb quan-titatively and physiologically in primary mouse macrophages, asjudged by global gene expression analysis. Thus, efforts to seek themajor signaling receptors of macrophages for intact, viable Mtbshould continue.
AcknowledgmentsWe are grateful to C. Nathan for helpful discussions and for critical readingof the manuscript. We thank B. Beutler, M. C. Nussenzweig, K. Takeda,S. Akira, E. Pamer, R. Medzhitov, J. Husemann, and S. C. Silverstein for
help in obtaining mutant mice. We thank Lindsay McGann for excellenttechnical assistance.
DisclosuresThe authors have no financial conflict of interest.
References1. Flynn, J. L., and J. Chan. 2001. Immunology of tuberculosis. Annu. Rev. Immu-
nol. 19: 93–129.2. Ernst, J. D. 1998. Macrophage receptors for Mycobacterium tuberculosis. Infect.
Immun. 66: 1277–1281.3. Ehrt, S., D. Schnappinger, S. Bekiranov, J. Drenkow, S. Shi, T. R. Gingeras,
T. Gaasterland, G. Schoolnik, and C. Nathan. 2001. Reprogramming of the mac-rophage transcriptome in response to interferon-� and Mycobacterium tubercu-losis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J. Exp.Med. 194: 1123–1140.
4. Underhill, D. M., A. Ozinsky, K. D. Smith, and A. Aderem. 1999. Toll-likereceptor-2 mediates mycobacteria-induced proinflammatory signaling in macro-phages. Proc. Natl. Acad. Sci. USA 96: 14459–14463.
5. Means, T. K., S. Wang, E. Lien, A. Yoshimura, D. T. Golenbock, andM. J. Fenton. 1999. Human Toll-like receptors mediate cellular activation byMycobacterium tuberculosis. J. Immunol. 163: 3920–3927.
6. Thoma-Uszynski, S., S. Stenger, O. Takeuchi, M. T. Ochoa, M. Engele,P. A. Sieling, P. F. Barnes, M. Rollinghoff, P. L. Bolcskei, M. Wagner, et al.2001. Induction of direct antimicrobial activity through mammalian Toll-likereceptors. Science 291: 1544–1547.
7. Shi, S., C. Nathan, D. Schnappinger, J. Drenkow, M. Fuortes, E. Block, A. Ding,T. R. Gingeras, G. Schoolnik, S. Akira, et al. 2003. MyD88 primes macrophagesfor full-scale activation by interferon-� yet mediates few responses to Mycobac-terium tuberculosis. J. Exp. Med. 198: 987–997.
8. Abel, B., N. Thieblemont, V. J. Quesniaux, N. Brown, J. Mpagi, K. Miyake,F. Bihl, and B. Ryffel. 2002. Toll-like receptor 4 expression is required to controlchronic Mycobacterium tuberculosis infection in mice. J. Immunol. 169:3155–3162.
9. Branger, J., J. C. Leemans, S. Florquin, S. Weijer, P. Speelman, andT. Van Der Poll. 2004. Toll-like receptor 4 plays a protective role in pulmonarytuberculosis in mice. Int. Immunol. 16: 509–516.
10. Kamath, A. B., J. Alt, H. Debbabi, and S. M. Behar. 2003. Toll-like receptor4-defective C3H/HeJ mice are not more susceptible than other C3H substrains toinfection with Mycobacterium tuberculosis. Infect. Immun. 71: 4112–4118.
11. Reiling, N., C. Holscher, A. Fehrenbach, S. Kroger, C. J. Kirschning, S. Goyert,and S. Ehlers. 2002. Cutting edge: Toll-like receptor (TLR)2- and TLR4-medi-ated pathogen recognition in resistance to airborne infection with Mycobacteriumtuberculosis. J. Immunol. 169: 3480–3484.
12. Shim, T. S., O. C. Turner, and I. M. Orme. 2003. Toll-like receptor 4 plays no rolein susceptibility of mice to Mycobacterium tuberculosis infection. Tuberculosis(Edinb.) 83: 367–371.
13. Toshchakov, V., B. W. Jones, P. Y. Perera, K. Thomas, M. J. Cody, S. Zhang,B. R. Williams, J. Major, T. A. Hamilton, M. J. Fenton, et al. 2002. TLR4, butnot TLR2, mediates IFN-�-induced STAT1�/�-dependent gene expression inmacrophages. Nat. Immunol. 3: 392–398.
14. Horng, T., G. M. Barton, R. A. Flavell, and R. Medzhitov. 2002. The adaptormolecule TIRAP provides signalling specificity for Toll-like receptors. Nature420: 329–333.
15. Hoebe, K., X. Du, P. Georgel, E. Janssen, K. Tabeta, S. O. Kim, J. Goode, P. Lin,N. Mann, S. Mudd, et al. 2003. Identification of Lps2 as a key transducer ofMyD88-independent TIR signalling. Nature 424: 743–748.
16. Hoshino, K., O. Takeuchi, T. Kawai, H. Sanjo, T. Ogawa, Y. Takeda, K. Takeda,and S. Akira. 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice arehyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene prod-uct. J. Immunol. 162: 3749–3752.
17. Takeuchi, O., K. Hoshino, T. Kawai, H. Sanjo, H. Takada, T. Ogawa, K. Takeda,and S. Akira. 1999. Differential roles of TLR2 and TLR4 in recognition of Gram-negative and Gram-positive bacterial cell wall components. Immunity 11: 443.
18. Durbin, J. E., R. Hackenmiller, M. C. Simon, and D. E. Levy. 1996. Targeteddisruption of the mouse Stat1 gene results in compromised innate immunity toviral disease. Cell 84: 443–451.
19. Muller, U., U. Steinhoff, L. F. Reis, S. Hemmi, J. Pavlovic, R. M. Zinkernagel,and M. Aguet. 1994. Functional role of type I and type II interferons in antiviraldefense. Science 264: 1918–1921.
20. Lee, S. J., S. Evers, D. Roeder, A. F. Parlow, J. Risteli, L. Risteli, Y. C. Lee,T. Feizi, H. Langen, and M. C. Nussenzweig. 2002. Mannose receptor-mediatedregulation of serum glycoprotein homeostasis. Science 295: 1898–1901.
21. Suzuki, H., Y. Kurihara, M. Takeya, N. Kamada, M. Kataoka, K. Jishage,O. Ueda, H. Sakaguchi, T. Higashi, T. Suzuki, et al. 1997. A role for macrophagescavenger receptors in atherosclerosis and susceptibility to infection. Nature 386:292–296.
22. Scanga, C. A., A. Bafica, C. G. Feng, A. W. Cheever, S. Hieny, and A. Sher.2004. MyD88-deficient mice display a profound loss in resistance to Mycobac-terium tuberculosis associated with partially impaired Th1 cytokine and nitricoxide synthase 2 expression. Infect. Immun. 72: 2400–2404.
23. Fremond, C. M., V. Yeremeev, D. M. Nicolle, M. Jacobs, V. F. Quesniaux, andB. Ryffel. 2004. Fatal Mycobacterium tuberculosis infection despite adaptiveimmune response in the absence of MyD88. J. Clin. Invest. 114: 1790–1799.
24. Horng, T., G. M. Barton, and R. Medzhitov. 2001. TIRAP: an adapter moleculein the Toll signaling pathway. Nat. Immunol. 2: 835–841.
3326 TLR2/4 INDEPENDENT MACROPHAGE RESPONSE TO Mtb
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

25. Yamamoto, M., S. Sato, H. Hemmi, H. Sanjo, S. Uematsu, T. Kaisho,K. Hoshino, O. Takeuchi, M. Kobayashi, T. Fujita, et al. 2002. Essential role forTIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature420: 324–329.
26. Yamamoto, M., S. Sato, K. Mori, K. Hoshino, O. Takeuchi, K. Takeda, andS. Akira. 2002. Cutting edge: a novel Toll/IL-1 receptor domain-containingadapter that preferentially activates the IFN-� promoter in the Toll-like receptorsignaling. J. Immunol. 169: 6668–6672.
27. Yamamoto, M., S. Sato, H. Hemmi, K. Hoshino, T. Kaisho, H. Sanjo,O. Takeuchi, M. Sugiyama, M. Okabe, K. Takeda, et al. 2003. Role of adaptorTRIF in the MyD88-independent toll-like receptor signaling pathway. Science301: 640–693.
28. Zimmerli, S., S. Edwards, and J. D. Ernst. 1996. Selective receptor blockadeduring phagocytosis does not alter the survival and growth of Mycobacteriumtuberculosis in human macrophages. Am. J. Respir. Cell Mol. Biol. 15: 760–770.
29. Rivera-Marrero, C. A., W. Schuyler, S. Roser, J. D. Ritzenthaler, S. A. Newburn,and J. Roman. 2000. Tuberculosis induction of matrix metalloproteinase-9: therole of mannose and receptor-mediated mechanisms. Am. J. Physiol. 282:L546–L555.
30. Lazarevic, V., A. J. Myers, C. A. Scanga, and J. L. Flynn. 2003. CD40, but notCD40L, is required for the optimal priming of T cells and control of aerosol M.tuberculosis infection. Immunity 19: 823–835.
31. Wang, Y., C. G. Kelly, J. T. Karttunen, T. Whittall, P. J. Lehner, L. Duncan,P. MacAry, J. S. Younson, M. Singh, W. Oehlmann, et al. 2001. CD40 is acellular receptor mediating mycobacterial heat shock protein 70 stimulation ofCC-chemokines. Immunity 15: 971–983.
32. Akira, S., and K. Takeda. 2004. Toll-like receptor signalling. Nat. Rev. Immunol.4: 499–511.
33. Yamamoto, M., K. Takeda, and S. Akira. 2004. TIR domain-containing adaptorsdefine the specificity of TLR signaling. Mol. Immunol. 40: 861–868.
34. Pai, R. K., M. E. Pennini, A. A. Tobian, D. H. Canaday, W. H. Boom, andC. V. Harding. 2004. Prolonged toll-like receptor signaling by Mycobacteriumtuberculosis and its 19-kilodalton lipoprotein inhibits � interferon-induced reg-ulation of selected genes in macrophages. Infect. Immun. 72: 6603–6614.
35. Keane, J., M. K. Balcewicz-Sablinska, H. G. Remold, G. L. Chupp, B. B. Meek,M. J. Fenton, and H. Kornfeld. 1997. Infection by Mycobacterium tuberculosispromotes human alveolar macrophage apoptosis. Infect. Immun. 65: 298–304.
36. Spira, A., J. D. Carroll, G. Liu, Z. Aziz, V. Shah, H. Kornfeld, and J. Keane.2003. Apoptosis genes in human alveolar macrophages infected with virulent orattenuated Mycobacterium tuberculosis: a pivotal role for tumor necrosis factor.Am. J. Respir. Cell Mol. Biol. 29: 545–551.
37. Lien, E., T. J. Sellati, A. Yoshimura, T. H. Flo, G. Rawadi, R. W. Finberg,J. D. Carroll, T. Espevik, R. R. Ingalls, J. D. Radolf, et al. 1999. Toll-like receptor2 functions as a pattern recognition receptor for diverse bacterial products.J. Biol. Chem. 274: 33419–33425.
38. Means, T. K., B. W. Jones, A. B. Schromm, B. A. Shurtleff, J. A. Smith, J. Keane,D. T. Golenbock, S. N. Vogel, and M. J. Fenton. 2001. Differential effects of aToll-like receptor antagonist on Mycobacterium tuberculosis-induced macro-phage responses. J. Immunol. 166: 4074–4082.
39. Jang, S., S. Uematsu, S. Akira, and P. Salgame. 2004. IL-6 and IL-10 inductionfrom dendritic cells in response to Mycobacterium tuberculosis is predominantlydependent on TLR2-mediated recognition. J. Immunol. 173: 3392–3397.
40. Picard, C., A. Puel, M. Bonnet, C. L. Ku, J. Bustamante, K. Yang, C. Soudais,S. Dupuis, J. Feinberg, C. Fieschi, et al. 2003. Pyogenic bacterial infections inhumans with IRAK-4 deficiency. Science 299: 2076–2079.
41. Brook, M., G. Sully, A. R. Clark, and J. Saklatvala. 2000. Regulation of tumournecrosis factor � mRNA stability by the mitogen-activated protein kinase p38signalling cascade. FEBS Lett. 483: 57–61.
42. Kontoyiannis, D., M. Pasparakis, T. T. Pizarro, F. Cominelli, and G. Kollias.1999. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Im-munity 10: 387–398.
43. Lee, J. C., J. T. Laydon, P. C. McDonnell, T. F. Gallagher, S. Kumar, D. Green,D. McNulty, M. J. Blumenthal, J. R. Heys, S. W. Landvatter, et al. 1994. Aprotein kinase involved in the regulation of inflammatory cytokine biosynthesis.Nature 372: 739–746.
44. Yu, Y., H. Zeng, S. Lyons, A. Carlson, D. Merlin, A. S. Neish, and A. T. Gewirtz.2003. TLR5-mediated activation of p38 MAPK regulates epithelial IL-8 expres-sion via posttranscriptional mechanism. Am. J. Physiol. 285: G282–G290.
45. Drennan, M. B., D. Nicolle, V. J. Quesniaux, M. Jacobs, N. Allie, J. Mpagi,C. Fremond, H. Wagner, C. Kirschning, and B. Ryffel. 2004. Toll-like receptor2-deficient mice succumb to Mycobacterium tuberculosis infection.Am. J. Pathol. 164: 49–57.
46. Sugawara, I., H. Yamada, C. Li, S. Mizuno, O. Takeuchi, and S. Akira. 2003.Mycobacterial infection in TLR2 and TLR6 knockout mice. Microbiol. Immunol.47: 327–336.
47. Bornstein, S. R., P. Zacharowski, R. R. Schumann, A. Barthel, N. Tran,C. Papewalis, V. Rettori, S. M. McCann, K. Schulze-Osthoff, W. A. Scherbaum,et al. 2004. Impaired adrenal stress response in Toll-like receptor 2-deficientmice. Proc. Natl. Acad. Sci. USA 101: 16695–16700.
48. Sugawara, I., H. Yamada, S. Mizuno, K. Takeda, and S. Akira. 2003. Mycobac-terial infection in MyD88-deficient mice. Microbiol. Immunol. 47: 841–847.
49. Juffermans, N. P., S. Florquin, L. Camoglio, A. Verbon, A. H. Kolk, P. Speelman,S. J. van Deventer, and T. van Der Poll. 2000. Interleukin-1 signaling is essentialfor host defense during murine pulmonary tuberculosis. J. Infect. Dis. 182:902–908.
50. Kinjo, Y., K. Kawakami, K. Uezu, S. Yara, K. Miyagi, Y. Koguchi, T. Hoshino,M. Okamoto, Y. Kawase, K. Yokota, et al. 2002. Contribution of IL-18 to Th1response and host defense against infection by Mycobacterium tuberculosis: acomparative study with IL-12p40. J. Immunol. 169: 323–329.
51. Sugawara, I., H. Yamada, S. Hua, and S. Mizuno. 2001. Role of interleukin (IL)-1type 1 receptor in mycobacterial infection. Microbiol. Immunol. 45: 743–750.
52. Sugawara, I., H. Yamada, H. Kaneko, S. Mizuno, K. Takeda, and S. Akira. 1999.Role of interleukin-18 (IL-18) in mycobacterial infection in IL-18-gene-disruptedmice. Infect. Immun. 67: 2585–2589.
53. Yamada, H., S. Mizumo, R. Horai, Y. Iwakura, and I. Sugawara. 2000. Protectiverole of interleukin-1 in mycobacterial infection in IL-1 �/� double-knockoutmice. Lab. Invest. 80: 759–767.
54. Bean, A. G., D. R. Roach, H. Briscoe, M. P. France, H. Korner, J. D. Sedgwick,and W. J. Britton. 1999. Structural deficiencies in granuloma formation in TNFgene-targeted mice underlie the heightened susceptibility to aerosol Mycobacte-rium tuberculosis infection, which is not compensated for by lymphotoxin. J. Im-munol. 162: 3504–3511.
55. Flynn, J. L., M. M. Goldstein, J. Chan, K. J. Triebold, K. Pfeffer,C. J. Lowenstein, R. Schreiber, T. W. Mak, and B. R. Bloom. 1995. Tumornecrosis factor-� is required in the protective immune response against Myco-bacterium tuberculosis in mice. Immunity 2: 561–572.
56. Keane, J., S. Gershon, R. P. Wise, E. Mirabile-Levens, J. Kasznica,W. D. Schwieterman, J. N. Siegel, and M. M. Braun. 2001. Tuberculosis asso-ciated with infliximab, a tumor necrosis factor �-neutralizing agent. N. Engl.J. Med. 345: 1098–1104.
57. Fortune, S. M., A. Solache, A. Jaeger, P. J. Hill, J. T. Belisle, B. R. Bloom,E. J. Rubin, and J. D. Ernst. 2004. Mycobacterium tuberculosis inhibits macro-phage responses to IFN-� through myeloid differentiation factor 88-dependentand -independent mechanisms. J. Immunol. 172: 6272–6280.
58. Giacomini, E., E. Iona, L. Ferroni, M. Miettinen, L. Fattorini, G. Orefici,I. Julkunen, and E. M. Coccia. 2001. Infection of human macrophages and den-dritic cells with Mycobacterium tuberculosis induces a differential cytokine geneexpression that modulates T cell response. J. Immunol. 166: 7033–7041.
59. Manca, C., L. Tsenova, A. Bergtold, S. Freeman, M. Tovey, J. M. Musser,C. E. Barry III, V. H. Freedman, and G. Kaplan. 2001. Virulence of a Mycobac-terium tuberculosis clinical isolate in mice is determined by failure to induce Th1type immunity and is associated with induction of IFN-�/�. Proc. Natl. Acad. Sci.USA 98: 5752–5757.
60. Remoli, M. E., E. Giacomini, G. Lutfalla, E. Dondi, G. Orefici, A. Battistini,G. Uze, S. Pellegrini, and E. M. Coccia. 2002. Selective expression of type I IFNgenes in human dendritic cells infected with Mycobacterium tuberculosis. J. Im-munol. 169: 366–374.
61. Prabhakar, S., Y. Qiao, Y. Hoshino, M. Weiden, A. Canova, E. Giacomini,E. Coccia, and R. Pine. 2003. Inhibition of response to � interferon by Myco-bacterium tuberculosis. Infect. Immun. 71: 2487–2497.
62. Prabhakar, S., Y. Qiao, A. Canova, D. B. Tse, and R. Pine. 2005. IFN-�� secretedduring infection is necessary but not sufficient for negative feedback regulation ofIFN-�� signaling by Mycobacterium tuberculosis. J. Immunol. 174: 1003–1012.
63. Cooper, A. M., J. E. Pearl, J. V. Brooks, S. Ehlers, and I. M. Orme. 2000.Expression of the nitric oxide synthase 2 gene is not essential for early control ofMycobacterium tuberculosis in the murine lung. Infect. Immun. 68: 6879–6882.
64. Giosue, S., M. Casarini, L. Alemanno, G. Galluccio, P. Mattia, G. Pedicelli,L. Rebek, A. Bisetti, and F. Ameglio. 1998. Effects of aerosolized interferon-� inpatients with pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 158:1156–1162.
65. Giosue, S., M. Casarini, F. Ameglio, L. Alemanno, C. Saltini, and A. Bisetti.1996. Minimal dose of aerosolized interferon-� in human subjects: biologicalconsequences and side-effects. Eur. Respir. J. 9: 42–46.
66. Dupuis, S., C. Dargemont, C. Fieschi, N. Thomassin, S. Rosenzweig, J. Harris,S. M. Holland, R. D. Schreiber, and J. L. Casanova. 2001. Impairment of my-cobacterial but not viral immunity by a germline human STAT1 mutation. Sci-ence 293: 300–303.
67. Bouchonnet, F., N. Boechat, M. Bonay, and A. J. Hance. 2002. �/� interferonimpairs the ability of human macrophages to control growth of Mycobacteriumbovis BCG. Infect. Immun. 70: 3020–3025.
68. Auerbuch, V., D. G. Brockstedt, N. Meyer-Morse, M. O’Riordan, andD. A. Portnoy. 2004. Mice lacking the type I interferon receptor are resistant toListeria monocytogenes. J. Exp. Med. 200: 527–533.
69. Carrero, J. A., B. Calderon, and E. R. Unanue. 2004. Type I interferon sensitizeslymphocytes to apoptosis and reduces resistance to Listeria infection. J. Exp.Med. 200: 535–540.
70. O’Connell, R. M., S. K. Saha, S. A. Vaidya, K. W. Bruhn, G. A. Miranda,B. Zarnegar, A. K. Perry, B. O. Nguyen, T. F. Lane, T. Taniguchi, et al. 2004.Type I interferon production enhances susceptibility to Listeria monocytogenesinfection. J. Exp. Med. 200: 437–445.
71. Schiavoni, G., C. Mauri, D. Carlei, F. Belardelli, M. C. Pastoris, and E. Proietti.2004. Type I IFN protects permissive macrophages from Legionella pneumophilainfection through an IFN-�-independent pathway. J. Immunol. 173: 1266–1275.
72. Carlin, J. M., E. C. Borden, and G. I. Byrne. 1989. Interferon-induced indoleam-ine 2,3-dioxygenase activity inhibits Chlamydia psittaci replication in humanmacrophages. J. Interferon Res. 9: 329–337.
73. Orellana, M. A., Y. Suzuki, F. Araujo, and J. S. Remington. 1991. Role of �interferon in resistance to Toxoplasma gondii infection. Infect. Immun. 59:3287–3290.
74. Shankar, A. H., P. Morin, and R. G. Titus. 1996. Leishmania major: differentialresistance to infection in C57BL/6 (high interferon-�/�) and congenic B6.C-H-28c (low interferon-�/�) mice. Exp. Parasitol. 84: 136–143.
3327The Journal of Immunology
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

75. Decker, T., S. Stockinger, M. Karaghiosoff, M. Muller, and P. Kovarik. 2002.IFNs and STATs in innate immunity to microorganisms. J. Clin. Invest. 109:1271–1277.
76. Lande, R., E. Giacomini, T. Grassi, M. E. Remoli, E. Iona, M. Miettinen,I. Julkunen, and E. M. Coccia. 2003. IFN-�� released by Mycobacterium tuber-culosis-infected human dendritic cells induces the expression of CXCL10: selec-tive recruitment of NK and activated T cells. J. Immunol. 170: 1174–1182.
77. Diebold, S. S., T. Kaisho, H. Hemmi, S. Akira, and C. Reis e Sousa. 2004. Innateantiviral responses by means of TLR7-mediated recognition of single-strandedRNA. Science 303: 1529–1531.
78. Heil, F., H. Hemmi, H. Hochrein, F. Ampenberger, C. Kirschning, S. Akira,G. Lipford, H. Wagner, and S. Bauer. 2004. Species-specific recognition of sin-gle-stranded RNA via toll-like receptor 7 and 8. Science 303: 1526–1529.
79. Hemmi, H., T. Kaisho, K. Takeda, and S. Akira. 2003. The roles of Toll-likereceptor 9, MyD88, and DNA-dependent protein kinase catalytic subunit in theeffects of two distinct CpG DNAs on dendritic cell subsets. J. Immunol. 170:3059–3064.
80. Kawai, T., S. Sato, K. J. Ishii, C. Coban, H. Hemmi, M. Yamamoto, K. Terai,M. Matsuda, J. Inoue, S. Uematsu, et al. 2004. Interferon-� induction throughToll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6.Nat. Immunol. 5: 1061–1068.
81. Kawai, T., O. Takeuchi, T. Fujita, J. Inoue, P. F. Muhlradt, S. Sato,K. Hoshino, and S. Akira. 2001. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 andthe expression of a subset of lipopolysaccharide-inducible genes. J. Immunol.167: 5887–5894.
82. Stockinger, S., B. Reutterer, B. Schaljo, C. Schellack, S. Brunner, T. Materna,M. Yamamoto, S. Akira, T. Taniguchi, P. J. Murray, et al. 2004. IFN regu-latory factor 3-dependent induction of type I IFNs by intracellular bacteria ismediated by a TLR- and Nod2-independent mechanism. J. Immunol. 173:7416 –7425.
83. Ding, A., H. Yu, J. Yang, S. Shin, and S. Ehrt. Induction of macrophage-derivedsecretory leukocyte protease inhibitor by Mycobacterium tuberculosis depends onTLR2 but not MyD88. Immunology. In press.
3328 TLR2/4 INDEPENDENT MACROPHAGE RESPONSE TO Mtb
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from


















