
Endogenous Cortisol and TGF-β in Human Aqueous Humor ... · iDC and mDC, although inhibition of...
8
of February 2, 2019. This information is current as Cell Function Immune Privilege by Regulating Dendritic Aqueous Humor Contribute to Ocular in Human β Endogenous Cortisol and TGF- Murray and S. John Curnow Graham R. Wallace, Mike Salmon, Saaeha Rauz, Philip I. Kadambari Oswal, Geraint P. Williams, Joseph Abbott, Alastair K. Denniston, Sherine H. Kottoor, Imran Khan, http://www.jimmunol.org/content/186/1/305 doi: 10.4049/jimmunol.1001450 November 2010; 2011; 186:305-311; Prepublished online 24 J Immunol Material Supplementary 0.DC1 http://www.jimmunol.org/content/suppl/2010/11/24/jimmunol.100145 References http://www.jimmunol.org/content/186/1/305.full#ref-list-1 , 13 of which you can access for free at: cites 36 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. All rights reserved. 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on February 2, 2019 http://www.jimmunol.org/ Downloaded from by guest on February 2, 2019 http://www.jimmunol.org/ Downloaded from
Transcript of Endogenous Cortisol and TGF-β in Human Aqueous Humor ... · iDC and mDC, although inhibition of...

of February 2, 2019.This information is current as
Cell FunctionImmune Privilege by Regulating DendriticAqueous Humor Contribute to Ocular
in HumanβEndogenous Cortisol and TGF-
Murray and S. John CurnowGraham R. Wallace, Mike Salmon, Saaeha Rauz, Philip I.Kadambari Oswal, Geraint P. Williams, Joseph Abbott, Alastair K. Denniston, Sherine H. Kottoor, Imran Khan,
http://www.jimmunol.org/content/186/1/305doi: 10.4049/jimmunol.1001450November 2010;
2011; 186:305-311; Prepublished online 24J Immunol
MaterialSupplementary
0.DC1http://www.jimmunol.org/content/suppl/2010/11/24/jimmunol.100145
Referenceshttp://www.jimmunol.org/content/186/1/305.full#ref-list-1
, 13 of which you can access for free at: cites 36 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. All rights reserved.1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on February 2, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Endogenous Cortisol and TGF-b in Human Aqueous HumorContribute to Ocular Immune Privilege by RegulatingDendritic Cell Function
Alastair K. Denniston,*,† Sherine H. Kottoor,*,† Imran Khan,† Kadambari Oswal,†
Geraint P. Williams,*,† Joseph Abbott,† Graham R. Wallace,*,† Mike Salmon,*
Saaeha Rauz,*,† Philip I. Murray,† and S. John Curnow*,†
Aqueous humor (AqH) has been shown to have significant immunosuppressive effects on APCs in animal models. We wanted to
establish whether, in humans, AqH can regulate dendritic cell (DC) function and to identify the dominant mechanism involved. Hu-
man AqH inhibited the capacity of human peripheral blood monocyte-derived DC to induce naive CD4+ T cell proliferation and
cytokine production in vitro, associated with a reduction in DC expression of the costimulatory molecule CD86. This was seen both
for DC cultured under noninflammatory conditions (immature DC) and for DC stimulated by proinflammatory cytokines (mature
DC). DC expression of MHC classes I/II and CD83 was reduced (mature DC only). Myeloid DC from peripheral blood were
similarly sensitive to the effects of human AqH, but only under inflammatory conditions. The addition of a-melanocyte stimulating
hormone and vasoactive intestinal peptide did not cause significant inhibition at physiological levels. However, the addition of
exogenous cortisol at physiological levels recapitulated the AqH-induced reduction in CD86 and inhibition of DC-induced T cell
proliferation, and blockade of cortisol in AqH partially reversed its suppressive effects. TGF-b2 had an additional effect with
cortisol, and although simultaneous blockade of cortisol and TGF-b2 in AqH reduced its effectiveness, there was still a cortisol- and
TGF-b–independent component. In humans, AqH regulates DC maturation and function by the combined actions of cortisol and
TGF-b2, a pathway that is likely to contribute to the maintenance of immune privilege in the eye. The Journal of Immunology,
2011, 186: 305–311.
To maintain a clear visual axis, the eye has a relatively pro-tected relationship with the immune system, known as im-mune privilege (1, 2). Protective immunity is maintained
while a number of different mechanisms restrict any excessive res-ponse that might lead to sight-threatening damage (1, 2).Dendritic cells (DC) are bone marrow-derived leukocytes that
provide a crucial link between the innate and adaptive immune res-ponses. They are highly specialized APCs that respond to the pres-ence of pathogens and inflammation at peripheral tissue sites,migrating to secondary lymphoid organs where they present Agto either naive or memory T cells, leading to T cell proliferationand differentiation toward effector and memory cells (3–6). DC
may be described in terms of their “maturity,” reflecting the tra-ditional paradigm of a linear transition from an immature Ag-capturing DC to a mature, highly immunogenic Ag-presentingDC. Although the linearity and unidirectional nature of the de-velopment of DC has recently been called into question (5), theconcept of DC maturity is still helpful in terms of their capacity toinduce an effective adaptive immune response. The maturity of theDC is reflected in its expression of increased levels of surfacemolecules (such as CD83 and the costimulatory molecules CD80/86) and altered cytokine production, all of which determine boththe magnitude and quality of the T cell response (7).In the ocular microenvironment, and in particular the aqueous hu-
mor (AqH), DC or any other potential APCs are exposed to a numberof molecules that have been reported to have immunomodulatoryeffects. In vivo and in vitro animal studies have suggested thatTGF-b2 (8, 9) and a-melanocyte–stimulating hormone (a-MSH)(10, 11) are the dominant immunosuppressive molecules in AqH,with well-documented inhibitory effects on macrophage inflamma-tory activity and on the generation of Th1 responses. Because al-most all functional studies have used either rabbit AqH (10, 12–18)or murine AqH (18, 19), it is unclear to what extent these moleculesare involved in APC regulation in humans, or whether other mech-anisms such as endogenous glucocorticoids, notably cortisol (20,21), play a role. Additionally, studies of AqH on APC function havetended to focus on the macrophage (9) rather than the DC, eventhough the DC is the most potent APCs in peripheral tissues. Animportant exception is the study by Shen et al. (12), who showedthat murine bone marrow-derived DC cultured in vitro with rabbitAqH are inhibited in their capacity to induce a MLR.Our central hypothesis is that the resting AqHmicroenvironment
maintains a relatively immature DC phenotype. We propose that
*Institute of Biomedical Research and †Academic Unit of Ophthalmology, School ofImmunity and Infection, College of Medical and Dental Sciences, University ofBirmingham, Birmingham and Midland Eye Centre, Birmingham, B15 2TT, UnitedKingdom
Received for publication May 30, 2010. Accepted for publication October 28, 2010.
This work was supported in part by the Medical Research Council (U.K.) ClinicalTraining Fellowship (G0600416 to A.K.D.) and the Marie Curie Early Stage Re-searcher Fellowship (MEST-CT-2005-020996 to S.H.K.). Additionally, the AcademicUnit of Ophthalmology is supported by the Birmingham Eye Foundation (registeredU.K. Charity 257549).
Address correspondence and reprint requests to Dr. S. John Curnow, Academic Unitof Ophthalmology, Institute of Biomedical Research, School of Immunity and In-fection, College of Medical and Dental Sciences, University of Birmingham, Bir-mingham B15 2TT, U.K. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this paper: ALK, activin receptor-like kinase; AqH, aqueoushumor; DC, dendritic cell; iDC, immature dendritic cell; mDC, mature dendritic cell;MFI, median fluorescence intensity; a-MSH, a-melanocyte–stimulating hormone;SS, side light scatter; VIP, vasoactive intestinal peptide.
Copyright� 2010 by The American Association of Immunologists, Inc. 0022-1767/10/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1001450
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
this contributes to immune privilege by reducing the capacity ofthese cells to induce T cell proliferation. In this study, we showthat human AqH inhibits DC induction of naive T cell responsesassociated with reduced expression of MHC and costimulatorymolecules on the DC. We observe that the principal inhibitorsof DC function in human AqH are cortisol and TGF-b2, withcortisol having the dominant effect.
Materials and MethodsSamples
Peripheral blood was taken by venepuncture from normal healthy volunteerdonors into preservative-free heparin, with exclusion of any individuals witha history of inflammatory disease or infection at the time of sampling. AqHwas obtained fromotherwise healthy patients by paracentesis prior to routinecataract surgery, excluding any individuals with a history of inflammatorydisease whether affecting the orbit or not, as well as any taking ocularmedication. AqH was centrifuged at 3003 g for 5 min, and the supernatantremoved and frozen in aliquots at270˚C. Local ethical committee approvalwas granted, and following informed consent all samples were collectedand stored according to the Human Tissue Act (U.K.). These studies con-form to the Declaration of Helsinki.
Generation of monocyte-derived DC and isolation of myeloidDC
PBMC were isolated by density-gradient centrifugation using Ficoll-Paque(GE Healthcare Biosciences, Amersham, U.K.) according to the manu-facturer’s instructions, and cells were washed three times with RPMI 1640to remove platelets. Monocytes were isolated from PBMC using MACSCD14 microbeads (Miltenyi Biotec, Surrey, U.K.), as described by themanufacturer (.98% purity), and cultured for 6 d (37˚C, 5% CO2, hu-midified) in RPMI 1640, 10% pooled human AB+ male serum (Biosera,Ringmer, U.K.), 500 U/ml recombinant human IL-4 (ImmunoTools, Fri-esoythe, Germany), and 1000 U/ml GM-CSF (ImmunoTools) at 2.5 3 106
cells/T25 flask (Starstedt, Leicester, U.K.). At day 3, 2 ml medium wasremoved and 2.5 ml fresh medium was added. At day 6 the nonadherentmonocyte-derived DC were harvested.
Myeloid DC were isolated from PBMC using a two-stage MACSmicrobead kit (Miltenyi Biotec) involving the depletion of CD19+ B cellsprior to the positive selection of BDCA-1+ cells as described by the man-ufacturer (.80% purity).
Culture of DC in the presence of AqH
DC were washed and resuspended in serum-free medium: RPMI 1640medium, 1% ITS+3 liquid media supplement, 1% nonessential amino acidsolution, and 1 mM sodium pyruvate (all Sigma-Aldrich, Dorset, U.K.). DCwere placed in triplicate in a round-bottomed 96-well plate (Greiner,Gloucester, UK) at 20,000 cells/well and cultured in the presence or ab-sence of 50% pooled human AqH with or without the addition of proin-flammatory cytokines in a total volume of 100 ml: 1 mg/ml IL-1b (Pepro-Tech, London, U.K.), 200 ng/ml IL-6 (ImmunoTools), 10 ng/ml TNF-a(PeproTech), and 1 mg/ml PGE2 (Sigma-Aldrich). These populations aresubsequently referred to as immature DC (iDC) and mature DC (mDC),respectively. Pooled AqH was used in all experiments and containedsamples taken from a minimum of six individuals. The role of cortisolwas tested with .98% HPLC cortisol (hydrocortisone; Sigma-Aldrich)at concentrations of 1026–1029 M and inhibited with 1027 M RU486(mifepristone; Sigma-Aldrich). The role of TGF-b2 was tested with re-combinant human TGF-b2 (PeproTech) at 0.1–100 ng/ml and inhibitedwith either the activin receptor-like kinase (ALK) inhibitor SB431542(Sigma-Aldrich) or monoclonal anti–TGF-b1, -2, and -3 (R&D Systems,Abingdon, U.K.). Recombinant a-MSH and vasoactive intestinal peptide(VIP) (Bachem, Weil am Rhein, Germany) were used at 0.1–100 ng/mlafter careful reconstitution and dilution. After 48 h culture, DC super-natants were harvested and the cells were either stained for flow cytometryor washed three times in RPMI 1640, 10% heat-inactivated FCS for use inan allogeneic proliferation assay.
Allogeneic proliferation assay
Naive CD4+ T cells were isolated from PBMC using MACS naive CD4 Tcell isolation kit (Miltenyi Biotec) as described by the manufacturer andlabeled with 1 mM CFSE (Invitrogen, Paisley, U.K.) for 10 min at 37˚C.They were then washed three times in RPMI 1640, 10% heat-inactivatedFCS, rested overnight, and washed once further before being placed in
triplicate in the washed DC-culture plates at 100,000 cells per well inRPMI, 10% heat-inactivated FCS. After a 4-d culture, supernatants wereharvested, cells were stained with the dead cell exclusion dye propidiumiodide, and proliferation of live cells was analyzed with a flow cytometer.The number of cells that had proliferated was calculated by gating on cellswith a lower level of positive CFSE staining than unstimulated cells andassessing total numbers using counting beads (Invitrogen).
Multiplex bead immunoassay
Culture supernatants were analyzed using a Human Cytokine Twenty-Five-Plex multiplex bead immunoassay (BioSource, Nivelles, Belgium) detect-ing (range, pg/ml) IL-1a (1.6–6,380), IL-1b (9.9–27,900), IL-2 (0.8–2,500),IL-2R (9.6–38,800), IL-4 (7.9–10,000), IL-5 (0.5–650), IL-6 (1.8–7,410),IL-7 (60–4,500) , IL-10 (4.5–15,000), IL-12p40/p70 (3.5–3,460), IL-13
C
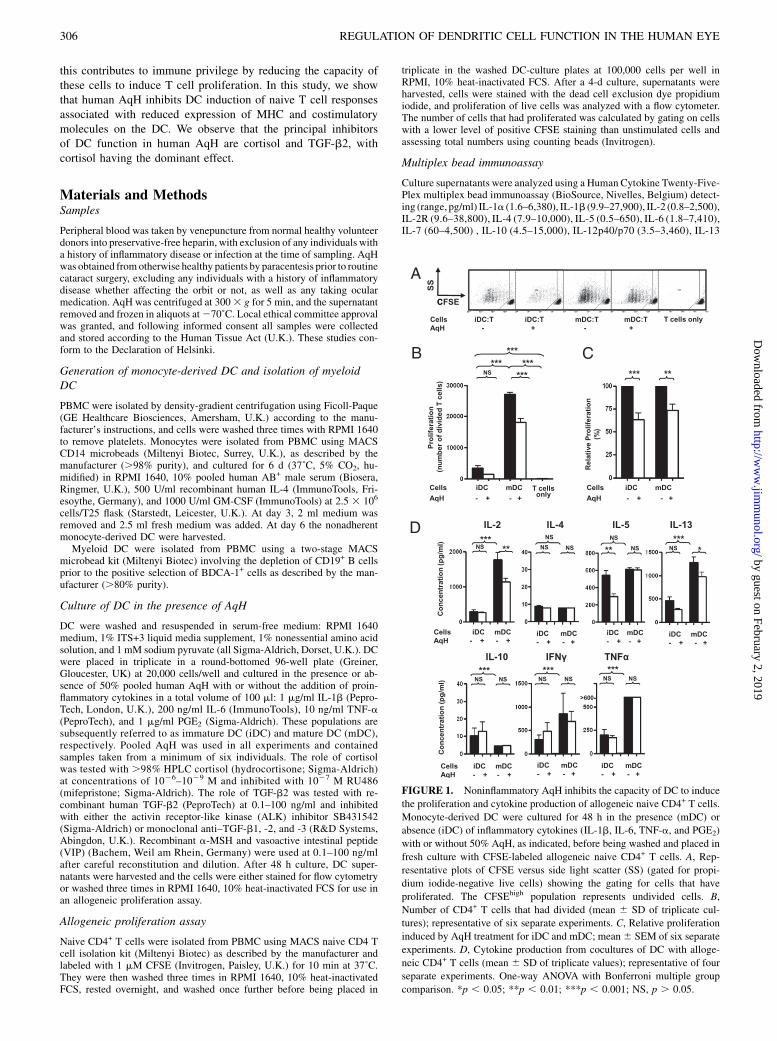
FIGURE 1. Noninflammatory AqH inhibits the capacity of DC to induce
the proliferation and cytokine production of allogeneic naive CD4+ T cells.
Monocyte-derived DC were cultured for 48 h in the presence (mDC) or
absence (iDC) of inflammatory cytokines (IL-1b, IL-6, TNF-a, and PGE2)
with or without 50% AqH, as indicated, before being washed and placed in
fresh culture with CFSE-labeled allogeneic naive CD4+ T cells. A, Rep-
resentative plots of CFSE versus side light scatter (SS) (gated for propi-
dium iodide-negative live cells) showing the gating for cells that have
proliferated. The CFSEhigh population represents undivided cells. B,
Number of CD4+ T cells that had divided (mean 6 SD of triplicate cul-
tures); representative of six separate experiments. C, Relative proliferation
induced by AqH treatment for iDC and mDC; mean6 SEM of six separate
experiments. D, Cytokine production from cocultures of DC with alloge-
neic CD4+ T cells (mean 6 SD of triplicate values); representative of four
separate experiments. One-way ANOVA with Bonferroni multiple group
comparison. *p , 0.05; **p , 0.01; ***p , 0.001; NS, p . 0.05.
306 REGULATION OF DENDRITIC CELL FUNCTION IN THE HUMAN EYE
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
(0.8–3,500), IL-15 (1.4–17,500), IL-17 (1.4–921), TNF-a (2.4–600), IFN-a(10.4–3,050), IFN-g (2.5–3,500), GM-CSF (3.2–5,500), CCL3 (MIP-1a;5.9–7,500), CCL4 (MIP-1b; 2.9–5,500), CXCL10 (IP-10; 34–4,500),CXCL9 (MIG; 58–1,500), CCL11 (eotaxin; 1–1,500),CCL5 (RANTES; 20–2,000), and CCL2 (MCP-1; 21–5,500). The procedure was carried out ac-cording to the manufacturer’s instructions. Samples were analyzed usinga microbead analyzer (Luminex 100; Luminex, Austin, TX).
Flow cytometry
DC were labeled with a combination of FITC-anti-CD80 (2D10; Bio-Legend, San Diego, CA), PE-anti-CD83 (HB15e; AbD Serotec, Oxford,U.K.), PE-Cy5-anti-CD86 (2331; BD Biosciences, Oxford, U.K.), PacificBlue-anti–HLA-A,B,C (W6/32; BioLegend), PE-Texas Red-anti–HLA-DR(Immu-357; Beckman Coulter, High Wycombe, U.K.), PE-Cy7-CCR5(2D7; BD Pharmingen, San Diego, CA), and FITC-CCR7 (150503; R&D
Systems) for 20 min at 4˚C in PBS/2% BSA (Sigma-Aldrich). The extent ofpositive staining was determined using an isotype-matched negative controlAb (data not shown). All Abs were used at predetermined optimal dilutions.Dead cells were stained with propidium iodide (Sigma-Aldrich), accordingto the manufacturer’s instructions, and excluded from further analysis. Cellswere analyzed using a CyAn three-laser, nine-color flow cytometer withSummit software (Beckman Coulter, High Wycombe, U.K.).
Cortisol ELISA
Human AqH was diluted 25-fold prior to testing in a cortisol acetylcho-linesterase competitive enzyme immunoassay (Cayman Chemical, AnnArbor, MI) according to the manufacturer’s instruction. Absorbance wasmeasured with a multiwell plate reader (Anthos HT 111; Anthos LabtecInstruments, Salzburg, Austria). Standard curves were plotted using a four-parameter logistic equation fitted to the logarithmic transformation of thestandard concentrations versus the percentage cortisol bound.
Statistical analysis
All data were analyzed using GraphPad Prism 4 (GraphPad Software, SanDiego, CA). The figure legends provide details of the statistical tests used.
ResultsHuman monocyte-derived DC were treated with noninflammatoryAqH and assayed for their ability to induce proliferation of alloge-neic CFSE-labeled CD4+ naive T cells (Fig. 1A). AqH treatment ofDC led to a significant reduction in the number of proliferatedcells in these DC/T cell cocultures both for nonstimulated DC(iDC) and for DC stimulated by a mixture of inflammatory cyto-kines (mDC) (Fig. 1B, 1C). Mean reduction by AqH was 37% foriDC (p , 0.001) and 26% for mDC (p , 0.01).
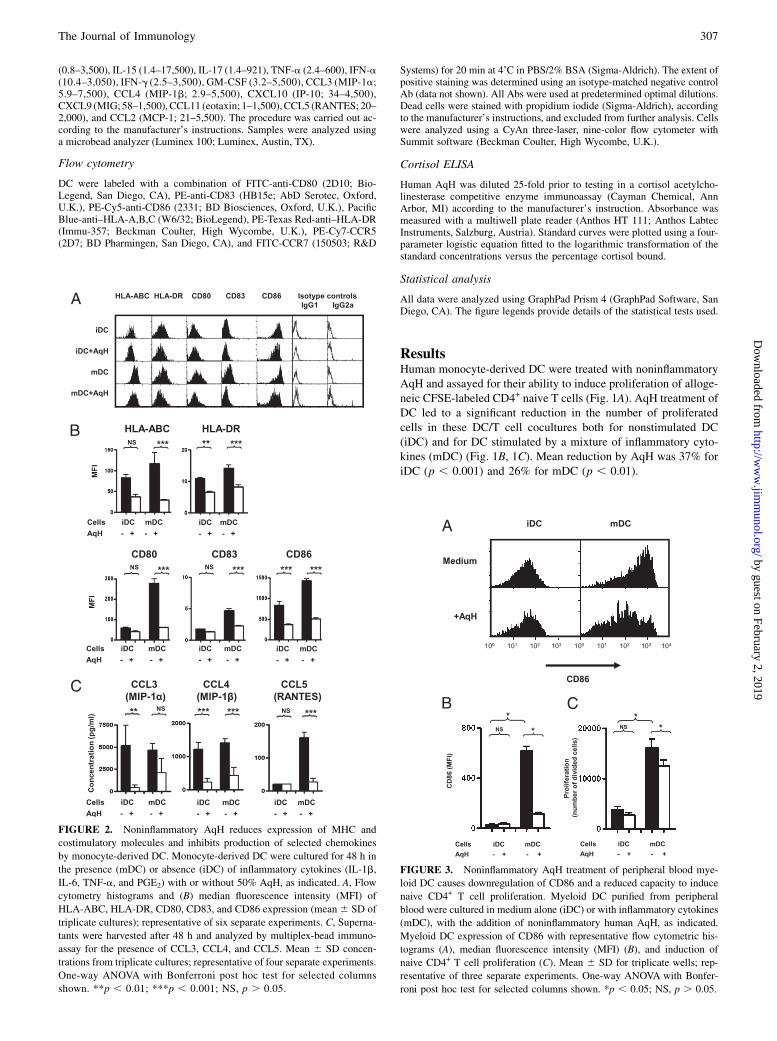
FIGURE 2. Noninflammatory AqH reduces expression of MHC and
costimulatory molecules and inhibits production of selected chemokines
by monocyte-derived DC. Monocyte-derived DC were cultured for 48 h in
the presence (mDC) or absence (iDC) of inflammatory cytokines (IL-1b,
IL-6, TNF-a, and PGE2) with or without 50% AqH, as indicated. A, Flow
cytometry histograms and (B) median fluorescence intensity (MFI) of
HLA-ABC, HLA-DR, CD80, CD83, and CD86 expression (mean6 SD of
triplicate cultures); representative of six separate experiments. C, Superna-
tants were harvested after 48 h and analyzed by multiplex-bead immuno-
assay for the presence of CCL3, CCL4, and CCL5. Mean 6 SD concen-
trations from triplicate cultures; representative of four separate experiments.
One-way ANOVA with Bonferroni post hoc test for selected columns
shown. **p , 0.01; ***p , 0.001; NS, p . 0.05.
FIGURE 3. Noninflammatory AqH treatment of peripheral blood mye-
loid DC causes downregulation of CD86 and a reduced capacity to induce
naive CD4+ T cell proliferation. Myeloid DC purified from peripheral
blood were cultured in medium alone (iDC) or with inflammatory cytokines
(mDC), with the addition of noninflammatory human AqH, as indicated.
Myeloid DC expression of CD86 with representative flow cytometric his-
tograms (A), median fluorescence intensity (MFI) (B), and induction of
naive CD4+ T cell proliferation (C). Mean 6 SD for triplicate wells; rep-
resentative of three separate experiments. One-way ANOVA with Bonfer-
roni post hoc test for selected columns shown. *p , 0.05; NS, p . 0.05.
The Journal of Immunology 307
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
Multiplex-bead immunoassay analysis of DC/T cell coculturesdemonstrated a reduction in coculture levels of IL-2 (mDC), IL-5(iDC), and IL-13 (mDC) following AqH treatment of DC (Fig.1D). Treatment with AqH did not cause significant skewing of theT cell cytokine profile toward either a Th1 or Th2 response; IL-17levels were undetectable in all cases. IL-10 was found at lowlevels (10 pg/ml) and, unlike all other cytokines studied, washigher in iDC rather than mDC/T cell cocultures (Fig. 1D). Allother T cell cytokines measured were either unchanged in re-sponse to AqH or were undetectable.Treatment of iDC with AqH caused significant reduction in class
I and II MHC and CD86 and prevented inflammatory cytokine-induced upregulation of the costimulatory molecule CD80 and ofthe marker of DC maturation, CD83 (Fig. 2A, 2B). In contrast,levels of the chemokine receptors CCR5 and CCR7 were notaffected by the addition of AqH (Supplemental Fig. 1).Multiplex-bead immunoassay analysis showed a significant re-
duction in DC-produced chemokines that bind to CCR5 (Fig. 2C),of which CCL5 (RANTES) showed a weak correlation with DCinduction of T cell proliferation (r = 0.65; p , 0.05). All othermolecules measured were either unchanged in response to AqH orwere undetectable.The effect of AqH was also assessed on myeloid DC purified
directly from peripheral blood. In myeloid DC, noninflammatoryAqH was found to cause similar changes to those seen withmonocyte-derived DC, with reduction in CD86 expression of ma-ture myeloid DC (Fig. 3A, 3B) and a significant fall in maturemyeloid DC capacity to induce naive CD4+ T cell proliferation(Fig. 3C). Because the myeloid DC are only .80% pure, it waspossible that the effects of AqH could be only on the contaminat-ing cells. However, the expression of CD86 suggests that mostcells have a lower level of CD86, rather than a change in anyminor population (Fig. 3A).
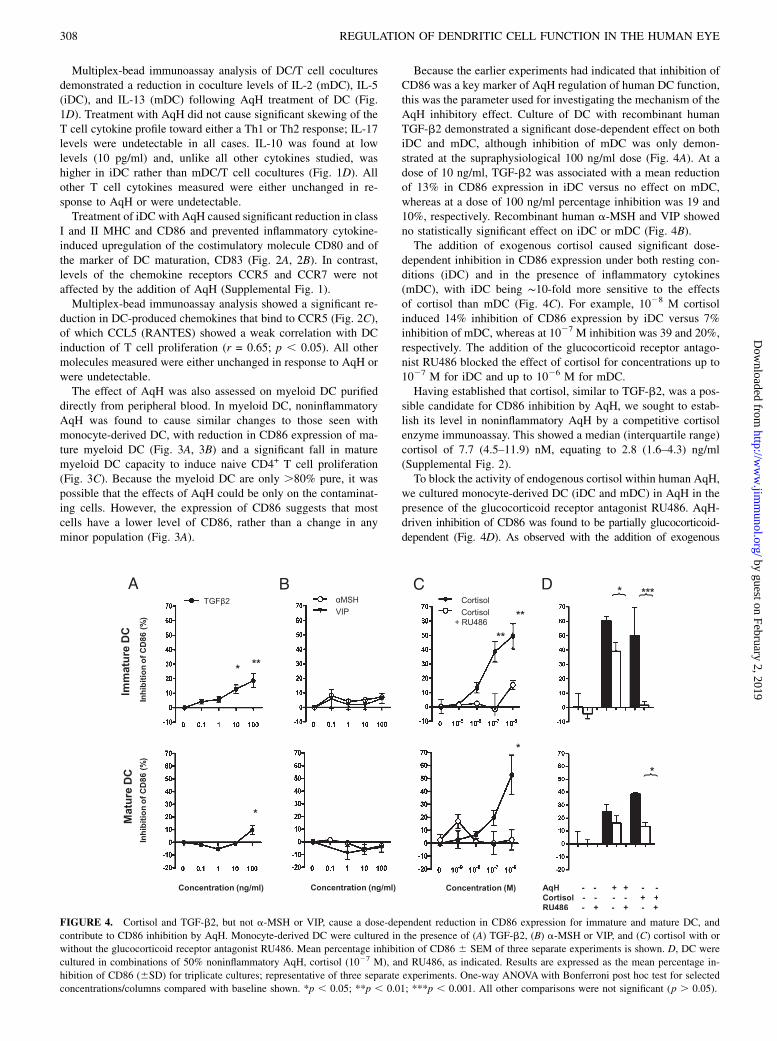
Because the earlier experiments had indicated that inhibition ofCD86 was a key marker of AqH regulation of human DC function,this was the parameter used for investigating the mechanism of theAqH inhibitory effect. Culture of DC with recombinant humanTGF-b2 demonstrated a significant dose-dependent effect on bothiDC and mDC, although inhibition of mDC was only demon-strated at the supraphysiological 100 ng/ml dose (Fig. 4A). At adose of 10 ng/ml, TGF-b2 was associated with a mean reductionof 13% in CD86 expression in iDC versus no effect on mDC,whereas at a dose of 100 ng/ml percentage inhibition was 19 and10%, respectively. Recombinant human a-MSH and VIP showedno statistically significant effect on iDC or mDC (Fig. 4B).The addition of exogenous cortisol caused significant dose-
dependent inhibition in CD86 expression under both resting con-ditions (iDC) and in the presence of inflammatory cytokines(mDC), with iDC being ∼10-fold more sensitive to the effectsof cortisol than mDC (Fig. 4C). For example, 1028 M cortisolinduced 14% inhibition of CD86 expression by iDC versus 7%inhibition of mDC, whereas at 1027 M inhibition was 39 and 20%,respectively. The addition of the glucocorticoid receptor antago-nist RU486 blocked the effect of cortisol for concentrations up to1027 M for iDC and up to 1026 M for mDC.Having established that cortisol, similar to TGF-b2, was a pos-
sible candidate for CD86 inhibition by AqH, we sought to estab-lish its level in noninflammatory AqH by a competitive cortisolenzyme immunoassay. This showed a median (interquartile range)cortisol of 7.7 (4.5–11.9) nM, equating to 2.8 (1.6–4.3) ng/ml(Supplemental Fig. 2).To block the activity of endogenous cortisol within human AqH,
we cultured monocyte-derived DC (iDC and mDC) in AqH in thepresence of the glucocorticoid receptor antagonist RU486. AqH-driven inhibition of CD86 was found to be partially glucocorticoid-dependent (Fig. 4D). As observed with the addition of exogenous
FIGURE 4. Cortisol and TGF-b2, but not a-MSH or VIP, cause a dose-dependent reduction in CD86 expression for immature and mature DC, and
contribute to CD86 inhibition by AqH. Monocyte-derived DC were cultured in the presence of (A) TGF-b2, (B) a-MSH or VIP, and (C) cortisol with or
without the glucocorticoid receptor antagonist RU486. Mean percentage inhibition of CD86 6 SEM of three separate experiments is shown. D, DC were
cultured in combinations of 50% noninflammatory AqH, cortisol (1027 M), and RU486, as indicated. Results are expressed as the mean percentage in-
hibition of CD86 (6SD) for triplicate cultures; representative of three separate experiments. One-way ANOVA with Bonferroni post hoc test for selected
concentrations/columns compared with baseline shown. *p , 0.05; **p , 0.01; ***p , 0.001. All other comparisons were not significant (p . 0.05).
308 REGULATION OF DENDRITIC CELL FUNCTION IN THE HUMAN EYE
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
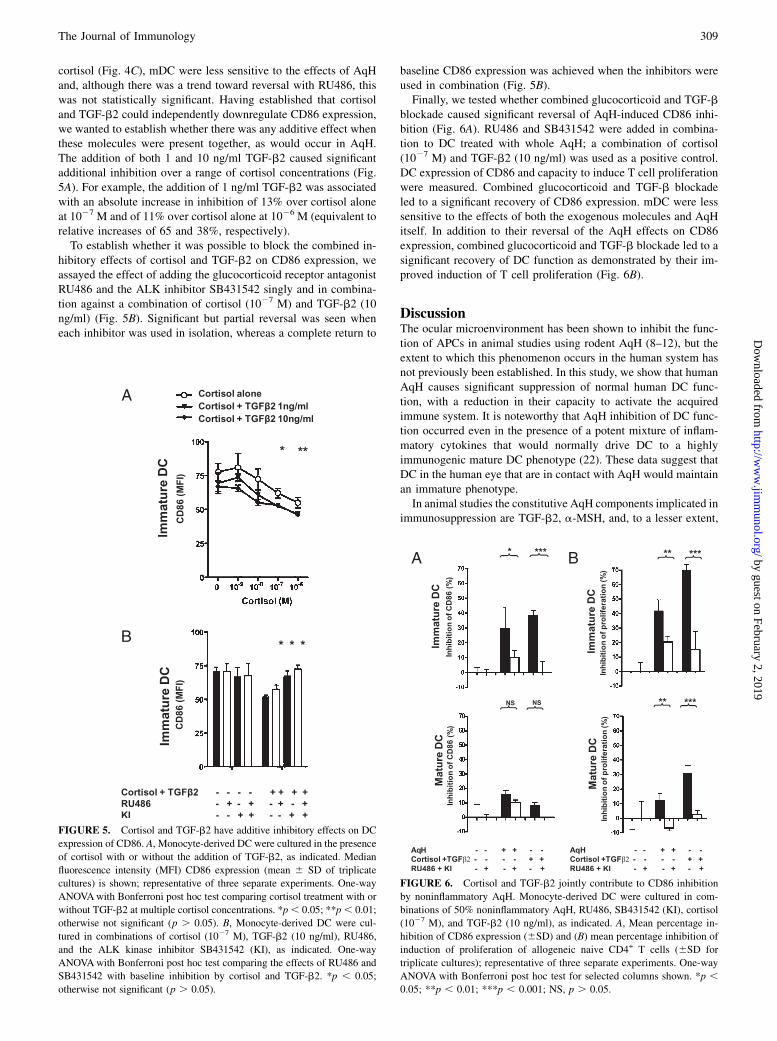
cortisol (Fig. 4C), mDC were less sensitive to the effects of AqHand, although there was a trend toward reversal with RU486, thiswas not statistically significant. Having established that cortisoland TGF-b2 could independently downregulate CD86 expression,we wanted to establish whether there was any additive effect whenthese molecules were present together, as would occur in AqH.The addition of both 1 and 10 ng/ml TGF-b2 caused significantadditional inhibition over a range of cortisol concentrations (Fig.5A). For example, the addition of 1 ng/ml TGF-b2 was associatedwith an absolute increase in inhibition of 13% over cortisol aloneat 1027 M and of 11% over cortisol alone at 1026 M (equivalent torelative increases of 65 and 38%, respectively).To establish whether it was possible to block the combined in-
hibitory effects of cortisol and TGF-b2 on CD86 expression, weassayed the effect of adding the glucocorticoid receptor antagonistRU486 and the ALK inhibitor SB431542 singly and in combina-tion against a combination of cortisol (1027 M) and TGF-b2 (10ng/ml) (Fig. 5B). Significant but partial reversal was seen wheneach inhibitor was used in isolation, whereas a complete return to
baseline CD86 expression was achieved when the inhibitors wereused in combination (Fig. 5B).Finally, we tested whether combined glucocorticoid and TGF-b
blockade caused significant reversal of AqH-induced CD86 inhi-bition (Fig. 6A). RU486 and SB431542 were added in combina-tion to DC treated with whole AqH; a combination of cortisol(1027 M) and TGF-b2 (10 ng/ml) was used as a positive control.DC expression of CD86 and capacity to induce T cell proliferationwere measured. Combined glucocorticoid and TGF-b blockadeled to a significant recovery of CD86 expression. mDC were lesssensitive to the effects of both the exogenous molecules and AqHitself. In addition to their reversal of the AqH effects on CD86expression, combined glucocorticoid and TGF-b blockade led to asignificant recovery of DC function as demonstrated by their im-proved induction of T cell proliferation (Fig. 6B).
DiscussionThe ocular microenvironment has been shown to inhibit the func-tion of APCs in animal studies using rodent AqH (8–12), but theextent to which this phenomenon occurs in the human system hasnot previously been established. In this study, we show that humanAqH causes significant suppression of normal human DC func-tion, with a reduction in their capacity to activate the acquiredimmune system. It is noteworthy that AqH inhibition of DC func-tion occurred even in the presence of a potent mixture of inflam-matory cytokines that would normally drive DC to a highlyimmunogenic mature DC phenotype (22). These data suggest thatDC in the human eye that are in contact with AqH would maintainan immature phenotype.In animal studies the constitutive AqH components implicated in
immunosuppression are TGF-b2, a-MSH, and, to a lesser extent,
FIGURE 6. Cortisol and TGF-b2 jointly contribute to CD86 inhibition
by noninflammatory AqH. Monocyte-derived DC were cultured in com-
binations of 50% noninflammatory AqH, RU486, SB431542 (KI), cortisol
(1027 M), and TGF-b2 (10 ng/ml), as indicated. A, Mean percentage in-
hibition of CD86 expression (6SD) and (B) mean percentage inhibition of
induction of proliferation of allogeneic naive CD4+ T cells (6SD for
triplicate cultures); representative of three separate experiments. One-way
ANOVA with Bonferroni post hoc test for selected columns shown. *p ,0.05; **p , 0.01; ***p , 0.001; NS, p . 0.05.
FIGURE 5. Cortisol and TGF-b2 have additive inhibitory effects on DC
expression of CD86. A, Monocyte-derived DC were cultured in the presence
of cortisol with or without the addition of TGF-b2, as indicated. Median
fluorescence intensity (MFI) CD86 expression (mean 6 SD of triplicate
cultures) is shown; representative of three separate experiments. One-way
ANOVAwith Bonferroni post hoc test comparing cortisol treatment with or
without TGF-b2 at multiple cortisol concentrations. *p, 0.05; **p, 0.01;
otherwise not significant (p . 0.05). B, Monocyte-derived DC were cul-
tured in combinations of cortisol (1027 M), TGF-b2 (10 ng/ml), RU486,
and the ALK kinase inhibitor SB431542 (KI), as indicated. One-way
ANOVAwith Bonferroni post hoc test comparing the effects of RU486 and
SB431542 with baseline inhibition by cortisol and TGF-b2. *p , 0.05;
otherwise not significant (p . 0.05).
The Journal of Immunology 309
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

VIP, calcitonin gene-related peptide, somatostatin, and cortisol. InAqH the concentration of TGF-b2 is 1–10 ng/ml, with .90%being in latent form, bound to the latency-associated peptide(23, 24). In the mouse, TGF-b2 treatment of APCs inhibits theirproduction of IL-12 (i.e., deviates them away from a Th1 res-ponse) and reduces CD40 activation accessory signals (8, 9). Addi-tionally, TGF-b2 prevents macrophage proinflammatory functions,such as secretion of inflammatory cytokines or generation of re-active oxygen species. In contrast, Tsukahara et al. (25) observedthat TGF-b treatment of peritoneal exudate cells with 5 ng/mlporcine TGF-b2 did not alter expression of CD80, CD86, or 4-1BBL (the latter was undetectable in either case), and yet TGF-b2diverted the peritoneal exudate cells to produce IL-4 and IL-10 butnot IFN-g. Recently, Shen et al. (12) demonstrated that murinebone marrow-derived DC are maintained in an immature MHClo
CD80/86lo state by the addition of rabbit AqH, and that this effectcan be recapitulated by recombinant TGF-b2 or blocked by AqHpretreatment with anti–TGF-b2. In our studies we found TGF-b2to have a weak inhibitory effect on DC maturation at levels $10ng/ml for iDC but only at the supraphysiological dose of 100 ng/ml for mDC. This suggests that under conditions of intraocularinflammation, TGF-b2 would no longer be a significant immu-noregulatory of DC function, although it should be recognizedthat TGF-b2 is a key regulator for many other elements of theimmune response.We observed a reduction in concentrations of CCL5, as well as
other CXCR3 ligands, secreted by DC cultured in AqH, and thatthese levels correlated with the induction of T cell proliferation.This suggests that the recruitment and/or retention of lymphocytesto DC by controlling their chemokine secretion may be an impor-tant factor influencing the degree of T cell proliferation. This pos-sibility remains to be formally tested.Although DC may mediate many of their actions within the af-
fected tissue, some of these DCmay also migrate through the drain-ing lymphatics. Despite the obvious inhibition of DCmaturation bynoninflammatory AqH, we did not detect a decrease in CCR7 ex-pression. This suggests that the DC, remaining in a relatively“immature” state, would still be able to migrate through any lym-phatics that might be present and therefore control T cell res-ponses outside of the affected eye.In our experimental model we chose to add the proinflammatory
cytokines simultaneously with the AqH to mimic the DC popula-tion that would be recruited to the inflamed eye. However, note thatthe resident DC population would have been preconditioned by theAqH, which may only serve to increase the suppressive effects ofAqH. We were not able to incorporate this aspect into our model,and therefore this issue may warrant further investigation.AqH contains a range of neuromediators including a-MSH
(∼32 pg/ml in human AqH) and VIP (∼40 ng/ml in rabbitAqH) (10, 15). It is interesting that in our study a-MSH did notsignificantly affect human DC maturation. This finding is sup-ported by the recent study by Shen et al. (12), who found, despitethe previously well-documented inhibitory effects on murine mac-rophage function (26, 27), that a-MSH and calcitonin gene-relatedpeptide were not significantly inhibitory for murine DC. The find-ing that VIP similarly had no effect at physiological levels is per-haps to be expected since its main immunomodulatory effects arethought to be directed against T cells (28).In contrast, the endogenous glucocorticoid cortisol did have a sig-
nificant suppressive effect on DCmaturation, inhibiting both CD86expression and induction of naive CD4+ T cell proliferation. Cor-tisol has been observed at biologically significant levels in humanand rodent AqH. In our study we observed median (interquartilerange) levels of cortisol in human AqH to be 7.7 (4.5–11.9) nM,
equating to 2.8 (1.6–4.3) ng/ml (10, 15). Despite this variation inconcentrations of cortisol, there was a very consistent inhibition ofDC function, consistent with the dose-response curve to exoge-nous cortisol over this range (Fig. 4C). Other studies detectingcortisol by RIA have reported values ranging from 4.8 to 49.7nM (equivalent to 1.7–18.0 ng/ml) (20, 21, 29, 30). Interestingly,the binding protein cortisol-binding globulin was not present atdetectable levels (by RIA) (20). Thus, it would appear that the vastmajority of cortisol in AqH exists in free form, in contrast to serumwhere 80–90% is bound to cortisol-binding globulin, 4–10% isbound to albumin, and the remaining 6–10% is free cortisol.The potential immunoregulatory function of cortisol in AqH hasreceived little attention previously, and there are no previous stud-ies on its potential role on APC function in the eye. Knisely et al.(20) noted that cortisol at the levels present in AqH could inhibitPHA/IL-1–stimulated thymocyte proliferation, and interestinglythat there was an additive effect with physiological levels ofTGF-b. In vitro studies of human DC have shown that cortisoland other glucocorticoids may interfere with DC differentiationand maturation. The addition of dexamethasone reduces class IIMHC, CD80/86, and CD83 expression with consequent reductionin T cell proliferation in an MLR (31). Similarly, cortisol has beenshown to inhibit DC maturation with failure of upregulation ofCD83 in response to TNF-a, reduced IL-12p70 production, andreduced allostimulatory activity (32, 33). Our study demonstratedthat cortisol at the levels present in AqH can inhibit DC matura-tion, and that blockade of endogenous glucocorticoids within theAqH leads to a significant (albeit partial) reduction in the inhib-itory effect of AqH. Our study also demonstrated that TGF-b2may cause additional inhibition of DC maturation, and it suggeststhat the levels of cortisol and TGF-b2 present within human AqHwould be sufficient to significantly inhibit DC maturation in thehuman eye. Although our data clearly show a role for humanAqH-derived cortisol and TGF-b in the regulation of DC functionin the eye, it is less clear to what degree this pathway is respon-sible for maintaining immune privilege in the eye and for whichanatomical parts of the eye (e.g., cornea and iris). These issues canonly be truly addressed in vivo in animal models.Note that there is also evidence that AqH has a number of im-
munosuppressive properties directly on T cells (i.e., not via itseffects on APCs). These effects include inhibition of Th1 function(10, 34) and the induction of regulatory T cell function (35, 36).However, in this study we wanted to specifically investigate theeffects of AqH on the DC, and so we carefully removed AqH fromthe DC cultures before commencing DC/T cell coculture.These data support the model that the normal resting ocular mi-
croenvironment maintains a relatively immature DC phenotype.We propose that this contributes to immune privilege by reducingtheir capacity to induce an adaptive immune response. Our studyprovides evidence that human AqH inhibits DC induction of naiveT cell responses associated with reduced expression of MHC andcostimulatory molecules on the DC, and that in human AqH the keyimmunoregulators are cortisol and TGF-b2, with cortisol havingthe dominant effect.
DisclosuresThe authors have no financial conflicts of interest.
References1. Streilein, J. W. 2003. Ocular immune privilege: therapeutic opportunities from
an experiment of nature. Nat. Rev. Immunol. 3: 879–889.2. Streilein, J. W. 2003. Ocular immune privilege: the eye takes a dim but practical
view of immunity and inflammation. J. Leukoc. Biol. 74: 179–185.3. Steinman, R. M. 1991. The dendritic cell system and its role in immunogenicity.
Annu. Rev. Immunol. 9: 271–296.
310 REGULATION OF DENDRITIC CELL FUNCTION IN THE HUMAN EYE
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

4. Reis e Sousa, C. 2004. Activation of dendritic cells: translating innate into adap-tive immunity. Curr. Opin. Immunol. 16: 21–25.
5. Reis e Sousa, C. 2006. Dendritic cells in a mature age. Nat. Rev. Immunol. 6:476–483.
6. Rossi, M., and J. W. Young. 2005. Human dendritic cells: potent antigen-presenting cells at the crossroads of innate and adaptive immunity. J. Immu-nol. 175: 1373–1381.
7. Banchereau, J., F. Briere, C. Caux, J. Davoust, S. Lebecque, Y. J. Liu,B. Pulendran, and K. Palucka. 2000. Immunobiology of dendritic cells. Annu. Rev.Immunol. 18: 767–811.
8. Takeuchi, M., M. M. Kosiewicz, P. Alard, and J. W. Streilein. 1997. On themechanisms by which transforming growth factor-b2 alters antigen-presentingabilities of macrophages on T cell activation. Eur. J. Immunol. 27: 1648–1656.
9. Takeuchi, M., P. Alard, and J. W. Streilein. 1998. TGF-b promotes immunedeviation by altering accessory signals of antigen-presenting cells. J. Immunol.160: 1589–1597.
10. Taylor, A. W., J. W. Streilein, and S. W. Cousins. 1992. Identification ofa-melanocyte stimulating hormone as a potential immunosuppressive factor inaqueous humor. Curr. Eye Res. 11: 1199–1206.
11. Taylor, A. W., D. G. Yee, T. Nishida, and K. Namba. 2000. Neuropeptide reg-ulation of immunity: the immunosuppressive activity of a-melanocyte-stimulating hormone (a-MSH). Ann. N. Y. Acad. Sci. 917: 239–247.
12. Shen, L., S. Barabino, A. W. Taylor, and M. R. Dana. 2007. Effect of the ocularmicroenvironment in regulating corneal dendritic cell maturation. Arch. Oph-thalmol. 125: 908–915.
13. Taylor, A. W., P. Alard, D. G. Yee, and J. W. Streilein. 1997. Aqueous humorinduces transforming growth factor-b (TGF-b)-producing regulatory T-cells.Curr. Eye Res. 16: 900–908.
14. Taylor, A. W., and D. G. Yee. 2003. Somatostatin is an immunosuppressivefactor in aqueous humor. Invest. Ophthalmol. Vis. Sci. 44: 2644–2649.
15. Taylor, A. W., J. W. Streilein, and S. W. Cousins. 1994. Immunoreactive vaso-active intestinal peptide contributes to the immunosuppressive activity of normalaqueous humor. J. Immunol. 153: 1080–1086.
16. Cousins, S. W., M. M. McCabe, D. Danielpour, and J. W. Streilein. 1991.Identification of transforming growth factor-b as an immunosuppressive factor inaqueous humor. Invest. Ophthalmol. Vis. Sci. 32: 2201–2211.
17. Nishida, T., and A. W. Taylor. 1999. Specific aqueous humor factors induceactivation of regulatory T cells. Invest. Ophthalmol. Vis. Sci. 40: 2268–2274.
18. Kaiser, C. J., B. R. Ksander, and J. W. Streilein. 1989. Inhibition of lymphocyteproliferation by aqueous humor. Reg. Immunol. 2: 42–49.
19. Granstein, R. D., R. Staszewski, T. L. Knisely, E. Zeira, R. Nazareno, M. Latina,and D. M. Albert. 1990. Aqueous humor contains transforming growth factor-band a small (less than 3500 daltons) inhibitor of thymocyte proliferation.J. Immunol. 144: 3021–3027.
20. Knisely, T. L., J. Hosoi, R. Nazareno, and R. D. Granstein. 1994. The presence ofbiologically significant concentrations of glucocorticoids but little or no cortisolbinding globulin within aqueous humor: relevance to immune privilege in theanterior chamber of the eye. Invest. Ophthalmol. Vis. Sci. 35: 3711–3723.
21. Rauz, S., C. M. Cheung, P. J. Wood, M. Coca-Prados, E. A. Walker, P. I. Murray,and P. M. Stewart. 2003. Inhibition of 11b-hydroxysteroid dehydrogenase type 1lowers intraocular pressure in patients with ocular hypertension. QJM 96: 481–490.
22. Jonuleit, H., U. Kuhn, G. Muller, K. Steinbrink, L. Paragnik, E. Schmitt, J. Knop,and A. H. Enk. 1997. Pro-inflammatory cytokines and prostaglandins inducematuration of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur. J. Immunol. 27: 3135–3142.
23. Taylor, A. W. 2007. Ocular immunosuppressive microenvironment. Chem.Immunol. Allergy 92: 71–85.
24. Curnow, S. J., F. Falciani, O. M. Durrani, C. M. Cheung, E. J. Ross, K. Wloka,S. Rauz, G. R. Wallace, M. Salmon, and P. I. Murray. 2005. Multiplex beadimmunoassay analysis of aqueous humor reveals distinct cytokine profiles inuveitis. Invest. Ophthalmol. Vis. Sci. 46: 4251–4259.
25. Tsukahara, R., M. Takeuchi, H. Akiba, T. Kezuka, K. Takeda, Y. Usui, M. Usui,H. Yagita, and K. Okumura. 2005. Critical contribution of CD80 and CD86 toinduction of anterior chamber-associated immune deviation. Int. Immunol. 17:523–530.
26. Taylor, A. W. 2005. The immunomodulating neuropeptide a-melanocyte-stimulating hormone (a-MSH) suppresses LPS-stimulated TLR4 with IRAK-Min macrophages. J. Neuroimmunol. 162: 43–50.
27. Taylor, A. W., D. G. Yee, and J. W. Streilein. 1998. Suppression of nitric oxidegenerated by inflammatory macrophages by calcitonin gene-related peptide inaqueous humor. Invest. Ophthalmol. Vis. Sci. 39: 1372–1378.
28. Taylor, A. W., J. W. Streilein, and S. W. Cousins. 1994. a-Melanocyte-stimulating hormone suppresses antigen-stimulated T cell production ofg-interferon. Neuroimmunomodulation 1: 188–194.
29. Weinstein, B. I., N. Kandalaft, R. Ritch, C. B. Camras, D. J. Morris, S. A. Latif,P. Vecsei, J. Vittek, G. G. Gordon, and A. L. Southren. 1991. 5a-Dihydrocortisolin human aqueous humor and metabolism of cortisol by human lenses in vitro.Invest. Ophthalmol. Vis. Sci. 32: 2130–2135.
30. Rozsıval, P., R. Hampl, J. Obenberger, L. Starka, and S. Rehak. 1981. Aqueoushumour and plasma cortisol levels in glaucoma and cataract patients. Curr. EyeRes. 1: 391–396.
31. Piemonti, L., P. Monti, P. Allavena, M. Sironi, L. Soldini, B. E. Leone, C. Socci,and V. Di Carlo. 1999. Glucocorticoids affect human dendritic cell differentia-tion and maturation. J. Immunol. 162: 6473–6481.
32. Freeman, L., M. Hewison, S. V. Hughes, K. N. Evans, D. Hardie, T. K. Means,and R. Chakraverty. 2005. Expression of 11b-hydroxysteroid dehydrogenasetype 1 permits regulation of glucocorticoid bioavailability by human dendriticcells. Blood 106: 2042–2049.
33. de Jong, E. C., P. L. Vieira, P. Kalinski, and M. L. Kapsenberg. 1999. Cortico-steroids inhibit the production of inflammatory mediators in immaturemonocyte-derived DC and induce the development of tolerogenic DC3.J. Leukoc. Biol. 66: 201–204.
34. Cousins, S. W., W. B. Trattler, and J. W. Streilein. 1991. Immune privilege andsuppression of immunogenic inflammation in the anterior chamber of the eye.Curr. Eye Res. 10: 287–297.
35. Taylor, A., and K. Namba. 2001. In vitro induction of CD25+ CD4+ regulatoryT cells by the neuropeptide a-melanocyte stimulating hormone (a-MSH).Immunol. Cell Biol. 79: 358–367.
36. Namba, K., N. Kitaichi, T. Nishida, and A. W. Taylor. 2002. Induction of reg-ulatory T cells by the immunomodulating cytokines a-melanocyte-stimulating hormone and transforming growth factor-beta2. J. Leukoc. Biol.72: 946–952.
The Journal of Immunology 311
by guest on February 2, 2019http://w
ww
.jimm
unol.org/D
ownloaded from


















