
Einführung in Simulationen mit Monte Carlo und · PDF fileDie Monte Carlo-Methode 1....
34
Einführung in Simulationen mit Monte Carlo und Brownscher Dynamik Martin Oettel Johannes Bleibel
Transcript of Einführung in Simulationen mit Monte Carlo und · PDF fileDie Monte Carlo-Methode 1....

Einführung in Simulationen mitMonte Carlo und
Brownscher Dynamik
Martin OettelJohannes Bleibel
Die Monte Carlo-Methode
1. Beispiel
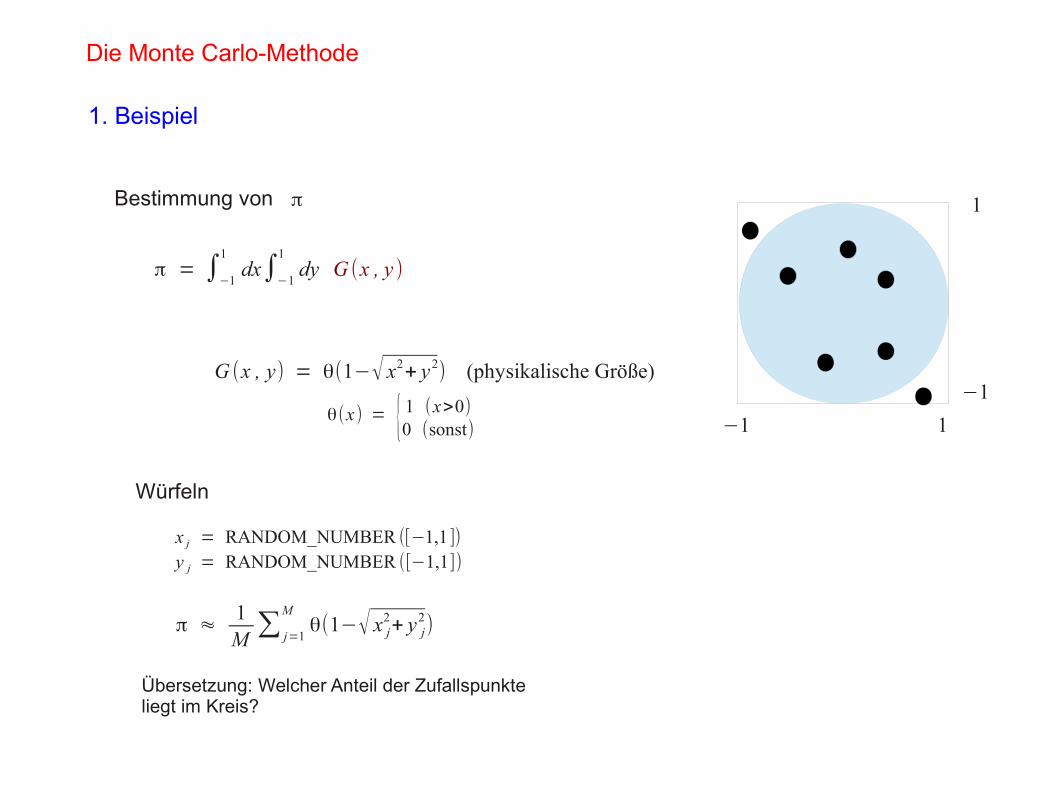
−1 1
−1
1Bestimmung von π
π = ∫−1
1
dx∫−1
1
dy G (x , y )
G (x , y) = θ(1−√ x2+ y2) (physikalische Größe)
θ(x) = {1 (x>0)0 (sonst)
Würfeln
x j = RANDOM_NUMBER ([−1,1 ])y j = RANDOM_NUMBER ([−1,1])
π ≈1M∑ j=1
Mθ(1−√ x j
2+ y j2)
Übersetzung: Welcher Anteil der Zufallspunkteliegt im Kreis?

Die Monte Carlo-Methode
1. Beispiel - Programmieraufgabe im VM Physik für Nanoscience
● Schreiben Sie ein MC-Programm für π
● Unterteilen Sie die Gesamtzahl der “Messungen”:
In jedem “sample” haben Sie Nmeas
Messungen, bilden Sie denMittelwert in jedem “sample”. Berechnen Sie den Mittelwert über alle “samples”.Berechnen Sie die Standardabweichung und die Unsicherheit des Mittelwertes über alle “samples”.
● Veranschaulichen Sie dies entsprechend, vor allem als Funktion von N
samples
N = N samples⋅Nmeas
π
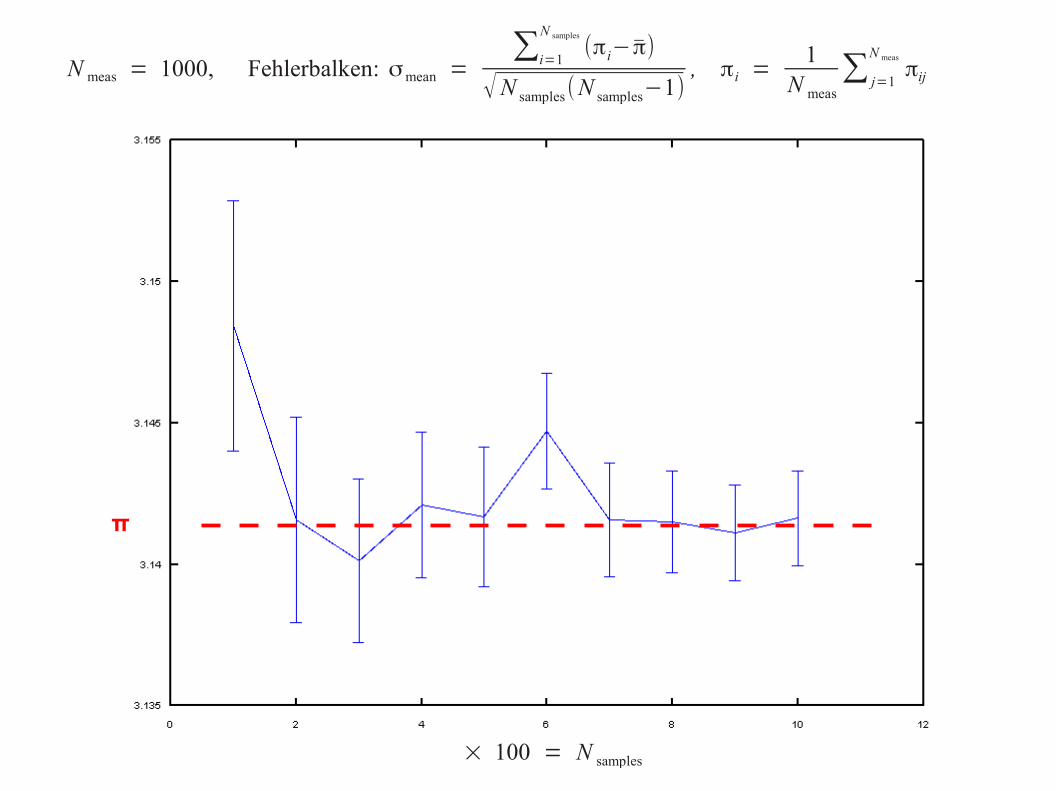
× 100 = N samples
N meas = 1000, Fehlerbalken: σmean =∑i=1
N samples
(πi−π)
√N samples(N samples−1), πi =
1N meas
∑ j=1
N meas
πij

Statistische Theorie der physikalischen Eigenschaften(Statistische Mechanik)
Beispiel: Verteilung der Moleküle in einer Flüssigkeit
● jedes Teilchen kann sich im gesamten Raum aufhalten, ist aber mit allen anderen Teilchen “korreliert”
● Verteilung wird beschrieben durch eine Verteilungsfunktion = Wahrscheinlichkeitsdichte
● Integral über alle Teilchenpositionen ist 1:Verteilungfunktion ist normiert
● Eine physikalische Größe sei Funktion der Teilchenkoordinaten:Der Erwartungswert dieser Größe ist dann:
Einfaches Beispiel: Schwerpunkt
f ( r1 , ... , rN )
N Teilchen
∫d 3 r1 ...∫ d 3 rN f (r1 , ... , r N ) = 1
G = G (r i)
⟨G ⟩ = ∫ d 3 r1 ...∫ d 3 rN G ( ri) f (r1 , ... , r N )
G (r i) →1N∑i=1
Nr i
Selbst wenn f bekannt ist, dann erfordert die Berechnung des Erwartungswert einer physikalischenGröße (wie der Schwerpunkt) im Prinzip eine 3N-dimensionale Integration !

Statistische Theorie der physikalischen Eigenschaften
Strategien zur Lösung des Integrals
⟨G ⟩ = ∫ d 3 r1 ...∫d 3 rN G ( ri) f eq( r1 , ... , rN ) ≈1M∑ j=1
MG ( ri (t j))
N Teilchen zum Zeitpunkt t1. Molekulardynamik
N Teilchen zum Zeitpunkt t+Δt
Man löst die Bewegungsgleichungen von NTeilchen näherungsweise numerisch, erhält also
Dann:
⟨G ⟩ ≈ limT→∞1T∫0
Tdt G ( r i( t))
ri (t)
Was steckt dahinter? Für lange Zeiten strebt ein Systemins Gleichgewicht.
Ergodenhypothese: Die zeitlich gemittelte Verteilung der Teilchen im Raum (aufgrund derLösung ), strebt der Gleichgewichtsverteilung zu.f eq( r1, ... , rN )
ri (t)
M Zeitschritte, t j = j Δ t
⟨G ⟩ = ∫ d 3 r1 ...∫ d 3 rN G ( ri) f (r1 , ... , r N )

Statistische Theorie der physikalischen Eigenschaften
Strategien zur Lösung des Integrals ⟨G ⟩ = ∫ d 3 r1 ...∫ d 3 rN G ( ri) f (r1 , ... , r N )
2. Monte Carlo
“Boltzmann-Faktor”: Die Gleichgewichtsverteilung ist bekannt und lautet
f eq( r1 , ... , rn) =1Z
e−E pot (r1 ,... , rn)
k T k ≈ 1.38⋅10−23 J/K
Boltzmann-Konstante
Z heißt Konfigurationsintegral und normiert feq
Z = ∫d 3 r1 ...∫d 3 rN e−E pot (r1 ,... , rn)
k T
Epot
ist die gesamte potentielle Energie der Moleküle Beispiel: Flüssigkeit an einer Wand
Paar-potentialu
externesPotential(Wand)
E pot = ∑i=1
N
∑ j<iu ( ri−r j) + ∑i=1
N
V ext(r i)

Statistische Theorie der physikalischen Eigenschaften
Strategien zur Lösung des Integrals ⟨G ⟩ = ∫ d 3 r1 ...∫ d 3 rN G ( ri) f (r1 , ... , r N )
2. Monte Carlo (MC)
⟨G ⟩ = ∫ d 3 r1 ...∫ d 3 rN G ( ri) f eq( r1 , ... , rN ) ≈1M∑ j=1
MG ( r i , j)
Anstatt 3N Integrale zu lösen, “würfeln” wir auf eine geschickte Art und WeiseM mal unsere Koordinaten: ri → r i , j ( j=1...M )
Man beachte die formale Ähnlichkeit zu Molekulardynamik:
ri (t j) → r i , j
MD:Lösen der Bewegungs-
gleichung in M Schritten
MC:“Würfeln” der KoordinatenM mal
Die genauen “Würfel”regeln (importance sampling) kommen später.

Die Monte Carlo-Methode
1. Beispiel noch einmal
−1 1
−1
1Bestimmung von π
π = ∫−1
1
dx∫−1
1
dy f (x , y) G (x , y)
f (x , y) = 1 (Gleichverteilung)
G (x , y) = θ(1−√ x2+ y2) (physikalische Größe)
θ(x) = {1 (x>0)0 (sonst)
Würfeln
x j = RANDOM_NUMBER ([−1,1 ])y j = RANDOM_NUMBER ([−1,1])
π ≈1M∑ j=1
Mθ(1−√ x j
2+ y j2)
Übersetzung: Welcher Anteil der Zufallspunkteliegt im Kreis?

Die Monte Carlo-Methode
2. Ein Beispiel von Daan Frenkel: Durchschnittstiefe des Nils
⟨Niltiefe⟩ = ∫dx∫ dy f (x , y) G (x , y )
Nilf (x , y) = {1 (Punkt im Nil)
0 (sonst)
G (x , y) = z (x , y)
Naives Monte Carlo: Zufallspunkte auf quadratischer Karte
⟨Niltiefe⟩ =1M∑ j=1
Mf (x j , y j)G (x j , y j)
ist meistens 0!
Das ist nicht das, was wir wollen!

Die Monte Carlo-Methode
3. “Importance sampling” und Metropolis-Algorithmus
⟨G ⟩ = ∫ d 3 r1 ...∫ d 3 rN G ( ri) f eq( r1 , ... , rN ) ≈1M∑ j=1
MG ( r i , j)Wir wollen:
und nicht: ⟨G ⟩ ≈1M∑ j=1
MG ( r i , j) f eq( r1, j , ... , r N , j)
( f eq( r1 ,... , r n) =1Z
e−E pot( r1 , ... , rn)
k T )
Wir wollen also Zufallszahlen erzeugen, deren Verteilung mit dem
Boltzmann-Faktor gewichtet sind.
r i , j
f eq

Die Monte Carlo-Methode
3. “Importance sampling” und Metropolis-Algorithmus
Der Algorithmus: 1. Starte mit einer beliebigen Anfangskonfiguration: 2. Für jedes Teilchen: Berechne potentielle Energie vor Verrückung
Verrücke Position zufällig:
Berechne Energieänderung:
Akzeptiere oder verwerfe die neuen Koordinaten:
Wiederhole die Schritte unter 2. für jedes Teilchen. Benutze immer die aktuellen Koordinaten der Teilchen.
3. Berechne die physikalische Größe
4. Wiederhole Schritte 2 und 3 M-1 mal
5. Endresultat:
ri = r i ,0
E pot,vor = E pot( r1 , ... , r N )
(x ' iy ' iz ' i
) = (xiy iz i) + Δ x (
r1
r2
r3)
r1, r2, r3 ...Zufallszahlen zwischen -1 und 1
Δ E pot = E pot( r 1 , ... , r ' i , ... , r N )−E pot,vor
Δ E pot < 0 : r i = r ' i
Δ E pot > 0 : r i = r ' i nur mit Wahrscheinlichkeit e−ΔE pot
k T
G1 = G ( r1 , ... , r N )
⟨G ⟩ =1M∑ j=1
MG j

Die Monte Carlo-Methode
3. “Importance sampling” und Metropolis-Algorithmus
Warum funktioniert dieser Algorithmus?
Der Monte Carlo-Schritt (2.) würfelt uns eine neue Konfiguration.Minimale Bedingung: Haben wir schon Konfigurationen erreicht, die mit verteilt sind, so bleiben die neuen Konfigurationen auch mit verteilt.
Gedankenexperiment: - Konfigurationen seien numerierbar: (z.B. durch Diskretisierung des Raumes) - sehr viele Monte Carlo-Schritte, angewandt auf sehr viele Konfigurationen - Monte Carlo-Schritt definiert uns eine Übergangsrate = Übergangswahrscheinlichkeit pro Schritt zwischen den Konfigurationen
- Gleichgewicht: Es gilt für alle Konfigurationen
f eq
f eq
pk→l = p(ck → cl )
ck = {r1 , ... , r N }k
∑k≠lf eq(cl ) p l→ k = ∑k≠l
f eq(ck ) pk→l
c l
Rate, mit der cl
verlassen wirdRate, mit der c
l
erreicht wird
“balance”

Die Monte Carlo-Methode
3. “Importance sampling” und Metropolis-Algorithmus
Warum funktioniert dieser Algorithmus?
Gedankenexperiment: - Gleichgewicht, stärkere Bedingung: Es gilt für alle Paare von Konfigurationen
f eq(c l) p l→k = f eq(ck ) pk→l
ck , cl
“detailed balance”
Wenn “detailed balance”, dann auch “balance”!
- Also:
- Check Metropolis-Algorithmus: dies wird genau erfüllt!
p l→kpk→l
=f eq(ck )
f eq(c l)= e
Epot,l−Epot , k
k T
Entwicklung zum Gleichgewicht: Wir haben gesehen, dass Metropolis uns im Gleichgewicht lässt. Aber kommen wir auch dahin, wenn wir mit einer beliebigen Konfiguration beginnen? Das ist nicht einfach zu beantworten, muss in jedem Fall praktisch getestet werden.

Die Monte Carlo-Methode
4. Praktische Hinweise zur Implementierung von Metropolis
● Harte Potentiale und “MC move” (Verrückung) :kein Überlapp mit anderen Teilchen → Akzeptanz
Überlapp → Ablehnung
● periodische Randbedingungen: bei “move” und Überlappsbestimmungbeachten!
● “acceptance ratio”: 0.1 ... 0.9 (Bedingung an !)
● Observable (Messgrößen) als unabhängige Funktionen definieren.Maximal einmal pro “sweep” (“attempted move for all particles”) messen!
Δ x
Die Monte Carlo-Methode
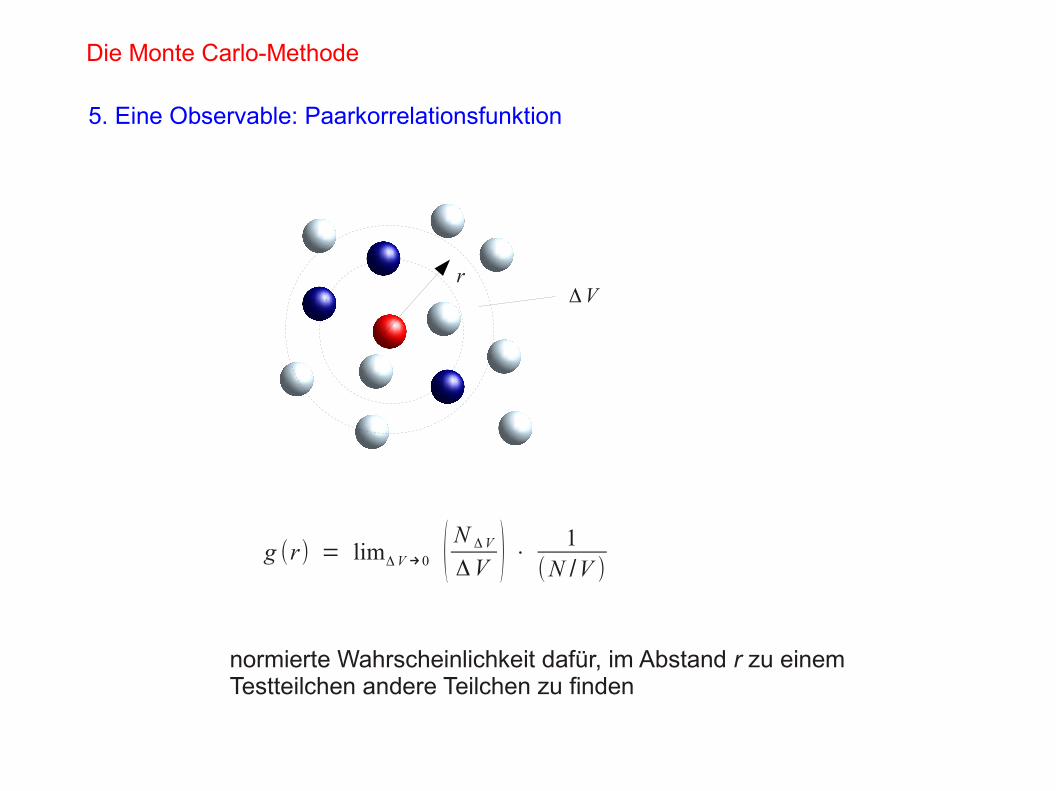
5. Eine Observable: Paarkorrelationsfunktion
g (r) = limΔV→0 ( N ΔV
ΔV ) ⋅ 1(N /V )
rΔV
normierte Wahrscheinlichkeit dafür, im Abstand r zu einemTestteilchen andere Teilchen zu finden

Monte-Carlo Mehode
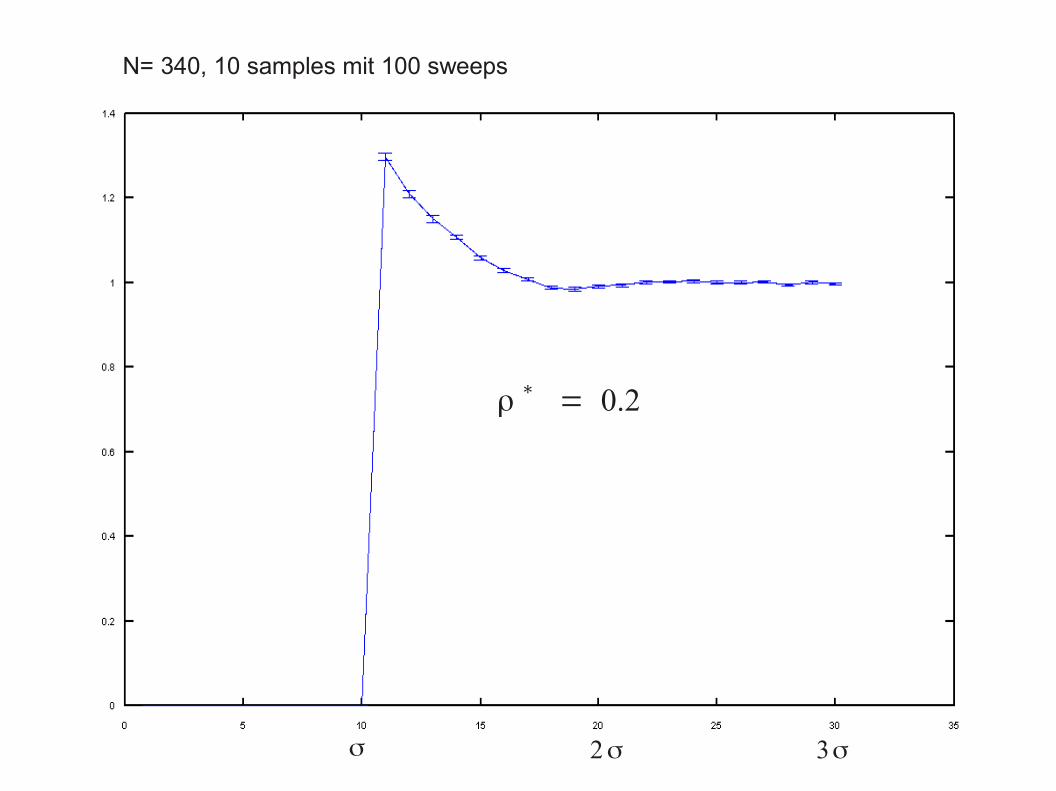
Beispiel: Paarkorrelationsfunktionen für harte Kugeln Programmieraufgabe im VM Physik für Nanoscience
Bestimmen Sie die Paarkorrelationsfunktion mit Fehlerbalkenfür harte Kugeln:
● Teilchenzahlen N = O(100)
● Dichten ρ* = 0.2 , 0.5 und 0.94( ρ* = ρσ3, σ = 2 R: Hartkugeldurchmesser )
σ 2σ 3σ
N= 340, 10 samples mit 100 sweeps
ρ∗= 0.2
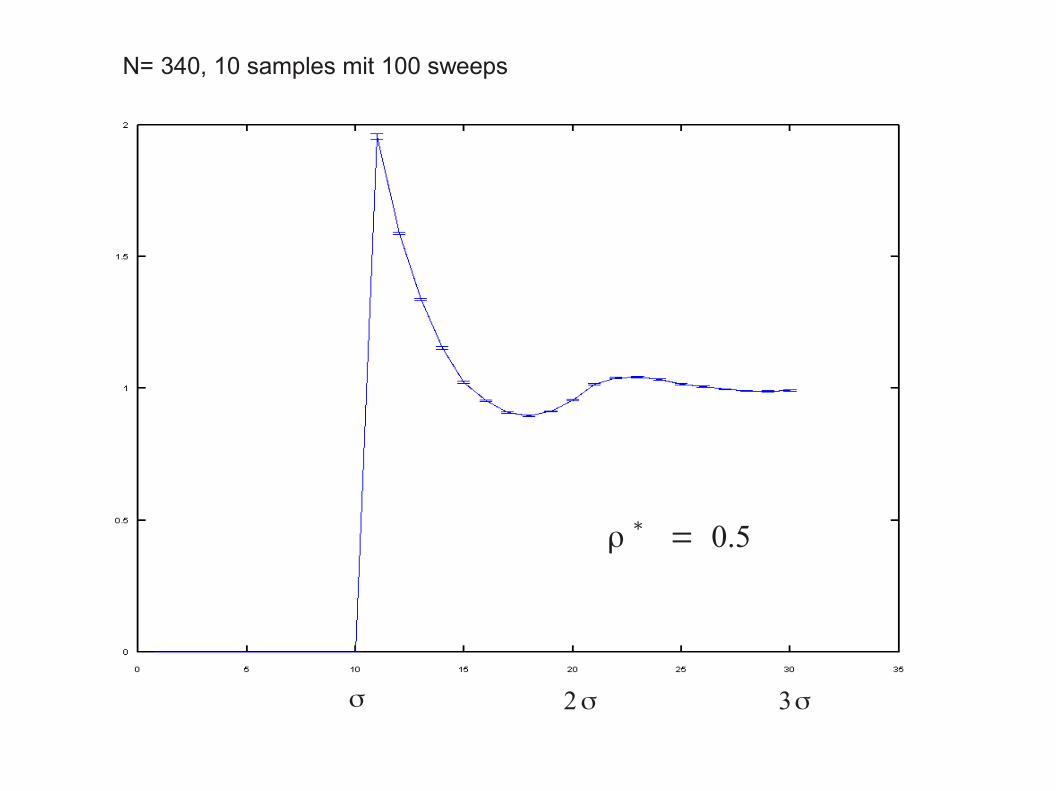
N= 340, 10 samples mit 100 sweeps
ρ∗= 0.5
σ 2σ 3σ

Part II
Brownian Dynamics

Reminder: Statistical Mechanics
Beispiel: Verteilung der Moleküle in einer Flüssigkeit
● jedes Teilchen kann sich im gesamten Raum aufhalten, ist aber mit allen anderen Teilchen “korreliert”
● Verteilung wird beschrieben durch eine Verteilungsfunktion = Wahrscheinlichkeitsdichte
● Integral über alle Teilchenpositionen ist 1:Verteilungfunktion ist normiert
● Eine physikalische Größe sei Funktion der Teilchenkoordinaten:Der Erwartungswert dieser Größe ist dann:
Einfaches Beispiel: Schwerpunkt
f ( r1 , ... , rN )
N Teilchen
∫d 3 r1 ...∫ d 3 rN f (r1 , ... , r N ) = 1
G = G (r i)
⟨G ⟩ = ∫ d 3 r1 ...∫ d 3 rN G ( ri) f (r1 , ... , r N )
G (r i) →1N∑i=1
Nr i
Selbst wenn f bekannt ist, dann erfordert die Berechnung des Erwartungswert einer physikalischenGröße (wie der Schwerpunkt) im Prinzip eine 3N-dimensionale Integration !

Die Molekulardynamik-Methode
1. Übersicht
⟨G ⟩ ≈ limT→∞1T∫0
Tdt G ( r i( t))Ziel: Berechnung von gemittelten physikalischen Größen:
→ man benötigt ri (t)
d.h. ausgehend von einer Anfangsverteilung der Teilchen im Raum, werden die Bewegungsgleichungen integriert:
m ¨r i=F i
gesamte Kraft auf Teilchen i2. zeitliche Ableitung → 2 mal integrieren!
F i : im allgemeinen schwierig zu bestimmen (WW zwischen N Teilchen!)
→ Ansatz: berücksichtige nur paarweise Wechselwirkungen
F i=∑ j=1, j≠i
NF ij
mit F ij=∇ V ( r i , r j)
Schwierigkeiten:
- zweifache numerische Integration
-

Die Molekulardynamik-Methode
2. Integration mittels Euler-Verfahren
x=F x /mdv xdt=F x /m
vx (t )=v x ,0+dv xdt
|t=t0∗(t−t 0)
vx (t )−v x ,0=dv xdt
|t=t0Δ t
Δ v x=FmΔ t
Betrachte x-Komponente eines Teilchens: bzw.
Taylor-Entwicklung der Geschwindigkeit:
Änderung der Geschwindigkeit des Teilchens
diskreter Zeitschritt
(zweite Integration für die Position analog)
Vorteile: - einfaches Verfahren
- unaufwendige Implementierung
Nachteile: - benötigt kleine Zeitschritte
- Stabilität
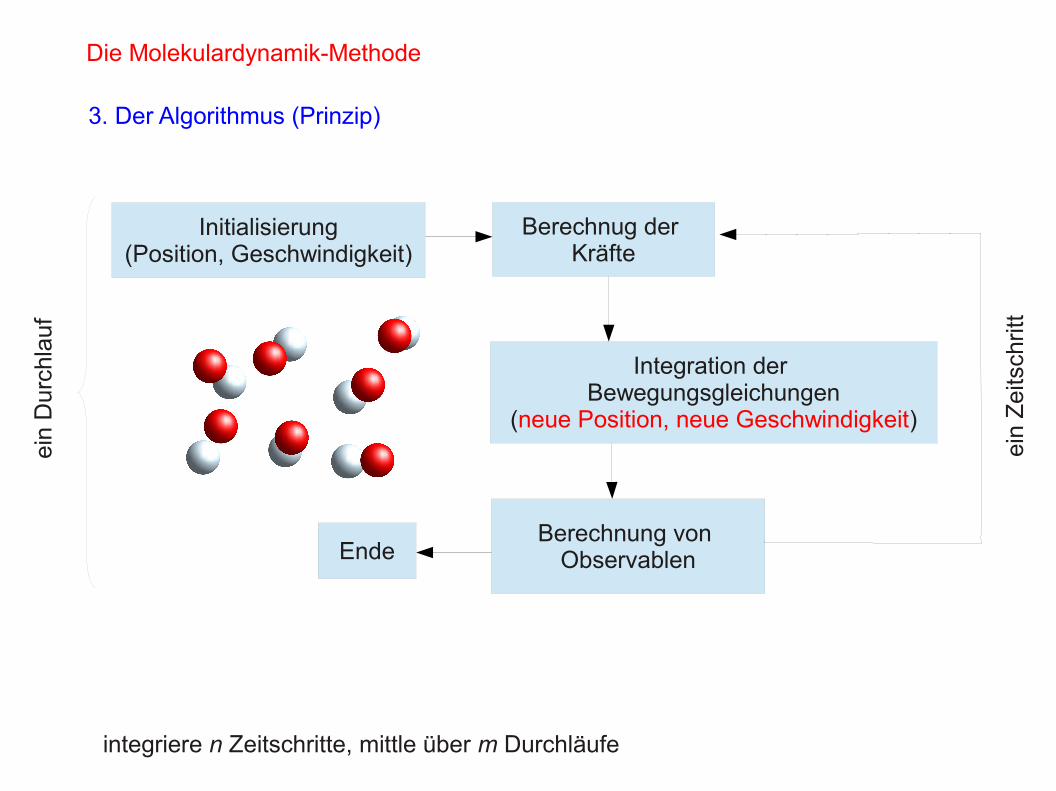
Die Molekulardynamik-Methode
3. Der Algorithmus (Prinzip)
Initialisierung(Position, Geschwindigkeit)
Berechnug der Kräfte
Integration der Bewegungsgleichungen
(neue Position, neue Geschwindigkeit)
Berechnung von Observablen
ein
Zei
tsch
ritt
integriere n Zeitschritte, mittle über m Durchläufe
Ende
ein
Dur
chla
uf
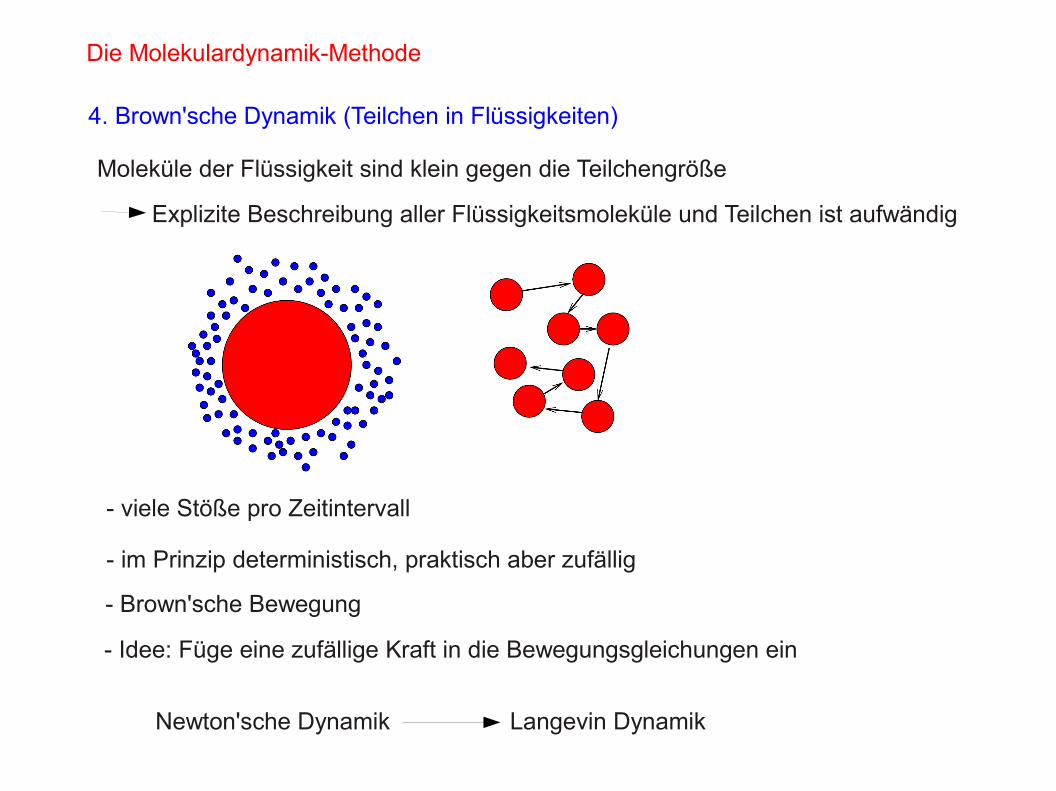
Die Molekulardynamik-Methode
4. Brown'sche Dynamik (Teilchen in Flüssigkeiten)
Moleküle der Flüssigkeit sind klein gegen die Teilchengröße
Explizite Beschreibung aller Flüssigkeitsmoleküle und Teilchen ist aufwändig
- viele Stöße pro Zeitintervall
- im Prinzip deterministisch, praktisch aber zufällig
- Brown'sche Bewegung
- Idee: Füge eine zufällige Kraft in die Bewegungsgleichungen ein
Newton'sche Dynamik Langevin Dynamik
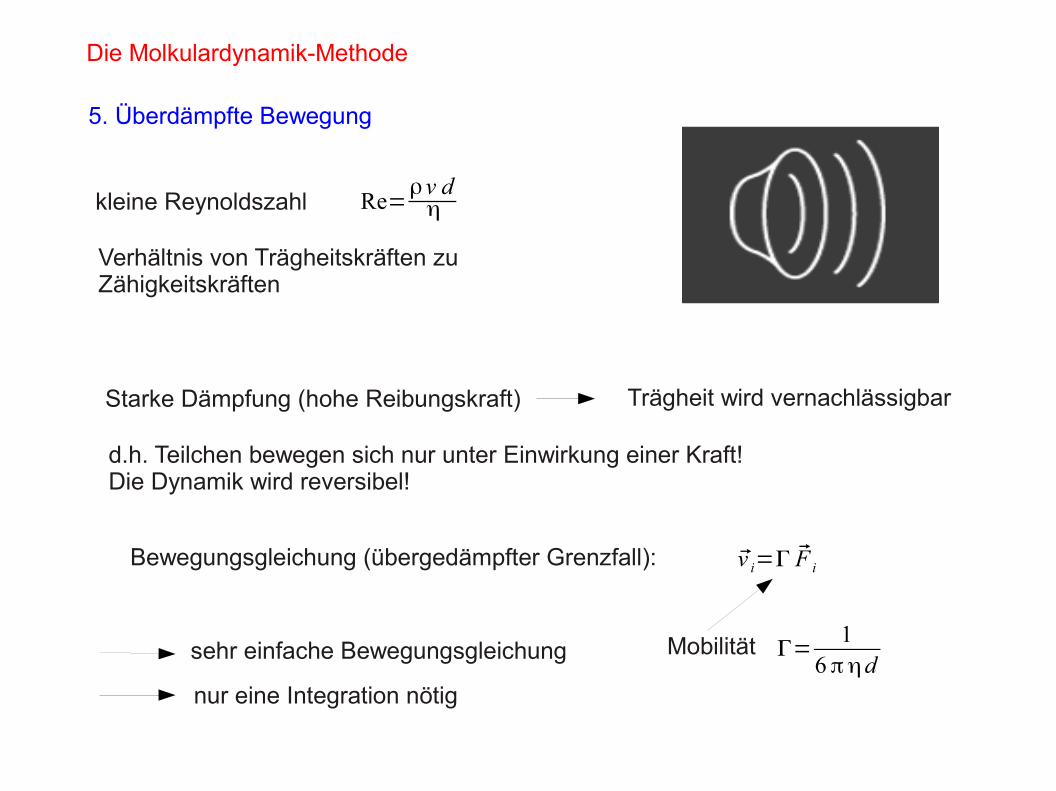
Die Molkulardynamik-Methode
5. Überdämpfte Bewegung
kleine Reynoldszahl Re=ρv dη
Verhältnis von Trägheitskräften zu Zähigkeitskräften
Starke Dämpfung (hohe Reibungskraft) Trägheit wird vernachlässigbar
d.h. Teilchen bewegen sich nur unter Einwirkung einer Kraft!Die Dynamik wird reversibel!
Bewegungsgleichung (übergedämpfter Grenzfall): v i=Γ F i
Mobilität Γ=1
6 πηdsehr einfache Bewegungsgleichung
nur eine Integration nötig

Die Molekulardynamik-Methode
5. Überdämpfte Bewegung
Bewegungsgleichung (übergedämpfter Grenzfall): v i=Γ F i
F i=−∇ V + F randGesamtkraft enthält Zufallskraft:
v i=Γ(−∇ V )+Γ F rand
zufällige Geschwindigkeit
Mit Euler Verfahren: Δ r i=(Γ(−∇ V )+Γ F rand )Δ t
konservative Kräfte
Δ r i=Γ(−∇ V )Δ t+ r rand
konservative Kräfte (Drift)Änderung der Positionzufällige Verschiebung
Position-Langevin Gleichung: ˙r=Γ F + r rand

Die Molekulardynamik-Methode
6. Mittleres Verschiebungsquadrat - Diffusion
Einstein (1905) und Smoluchowski (1906): D=Γk BT
Diffusionskonstante Boltzmann Konstante
Messung der Diffusion: Mittleres Verschiebungsquadrat
⟨r 2(t )⟩=⟨ r (t )− r (t=0)⟩
2 (gemittelt über alle Teilchen)
D=14t⟨ r2(t )⟩damit:
1. Ziel der Brown'schen Dynamik: korrekte Beschreibung der Diffusion!
Steuerung des MSD über die zufälligen Verschiebungen
Breite der Zufalssverteilung soll von Temperatur und Mobilität abhängen
⟨ r rand ⟩=0 σ=√2Γ k BT Δ t=√2DΔ t
bzw. ⟨r 2(t )⟩=4Dt
⟨r 2⟩
(2D)
Die Molekulardynamik-Methode
7. Details zur Implementierung – Einheiten, Potentiale
Abstände als Vielfache des Teilchendurchmessers:
Energie als Vielfaches von
damit:
r=rσ
k BT V=Vk BT
F=−∇ V=−σ ∇Vk BT
üblich: k BT=1
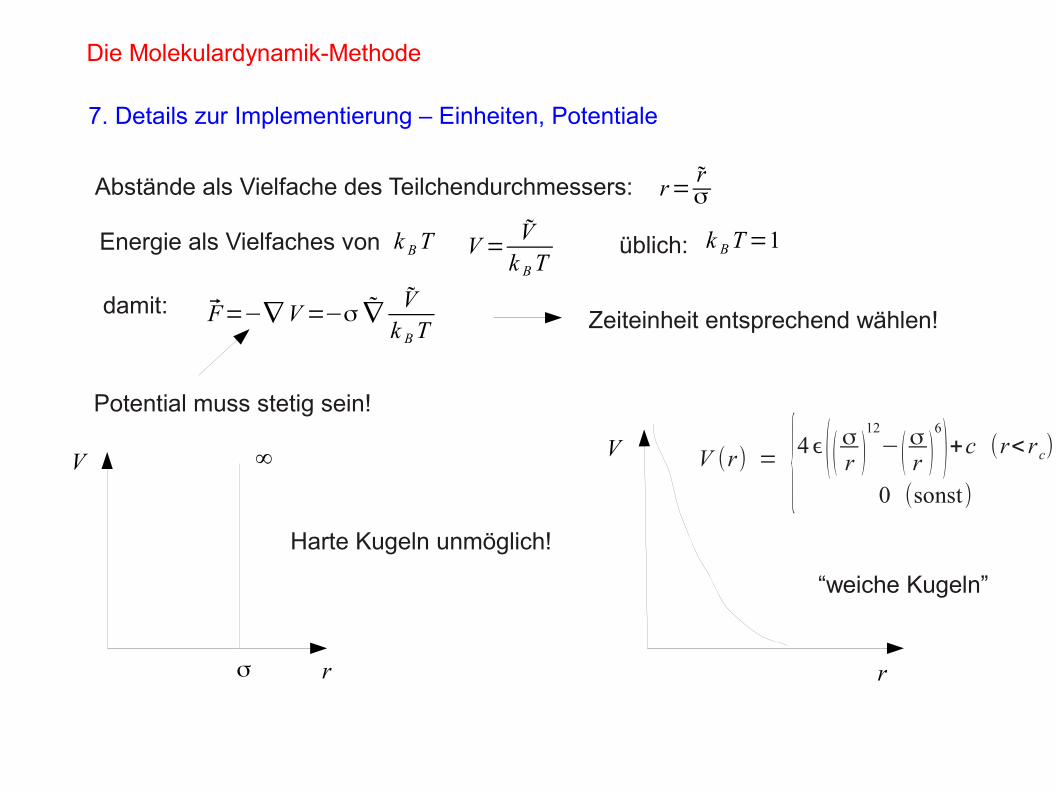
Potential muss stetig sein!
σ
∞
Harte Kugeln unmöglich!
“weiche Kugeln”
VV V (r) = {4ϵ (( σr )
12
−(σr )
6
)+c (r< rc)0 (sonst)
r r
Zeiteinheit entsprechend wählen!
Die Molekulardynamik-Methode
7. Details zur Implementierung – Zufallskräfte, zufällige Verschiebung
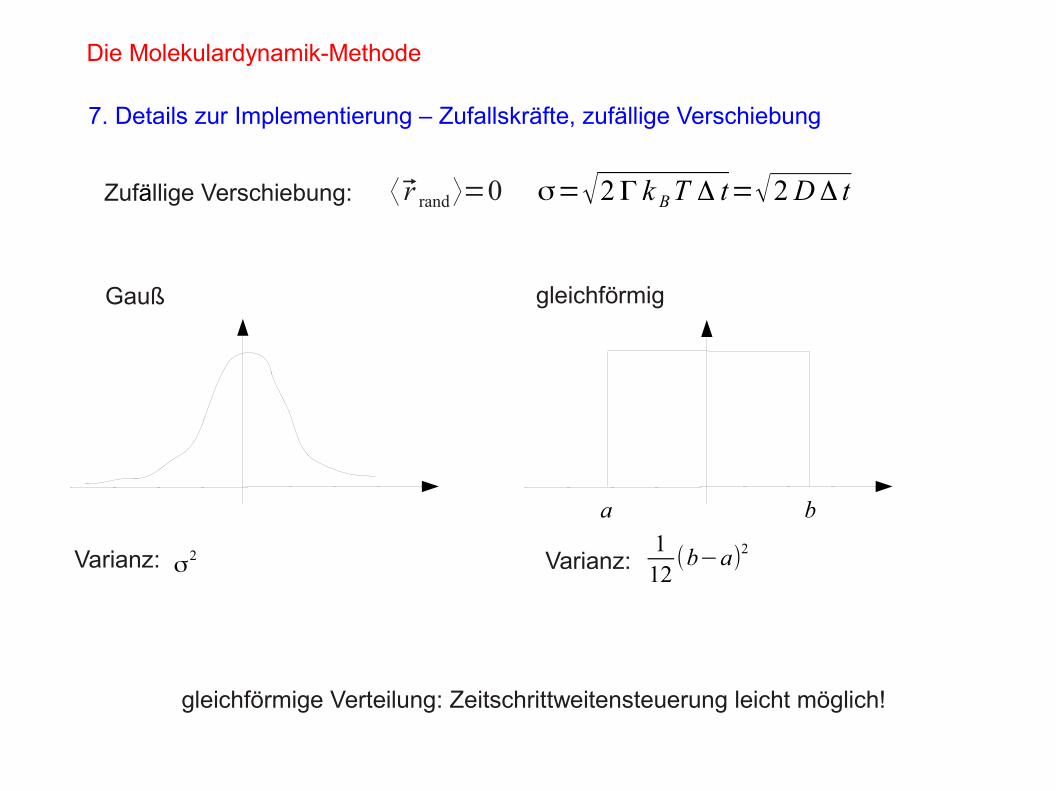
⟨ r rand ⟩=0 σ=√2Γ k BT Δ t=√2DΔ tZufällige Verschiebung:
Gauß gleichförmig
Varianz: Varianz:σ2
112(b−a)2
a b
gleichförmige Verteilung: Zeitschrittweitensteuerung leicht möglich!

Beispiel: Programmieraufgabe aus dem VM Nanoscience
Die Molekulardynamik-Methode
Schreiben Sie ein Programm zur Simulation einer Brown'schen Dynamik
- Berechnen sie das mittlere Verschiebungsquadrata) ohne Wechselwirkungb) mit einer rein repulsiven Wechselwirkung*(c) mit einer zusätzlich attraktiven Wechselwirkung)
- Berechnen Sie die Paarkorrelationsfunktion g(r) für den Fall b) und vergleichen Sie das Ergebnis mit Ihren MC-Resultaten (gleiche Dichte, gleiche Teilchenanzahl)
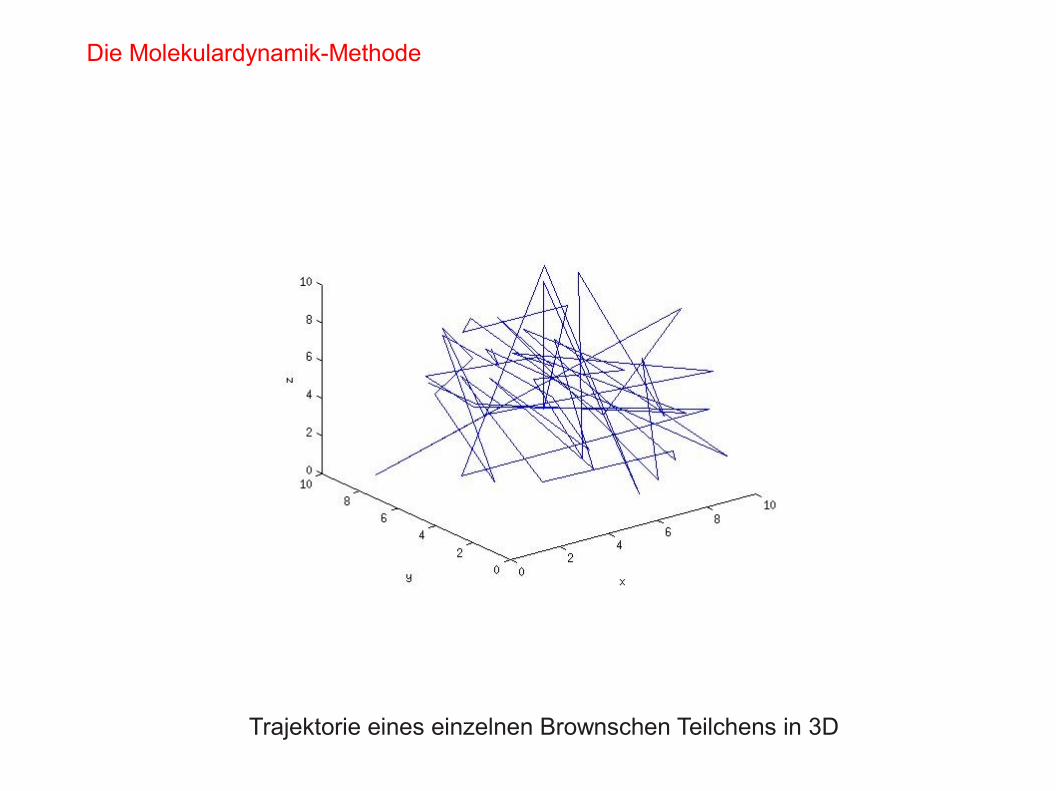
- Erstellen Sie “Snapshots” des Systems zur Visualisierung der Brown'sche Bewegung
Die Molekulardynamik-Methode
Trajektorie eines einzelnen Brownschen Teilchens in 3D
Die Molekulardynamik-Methode
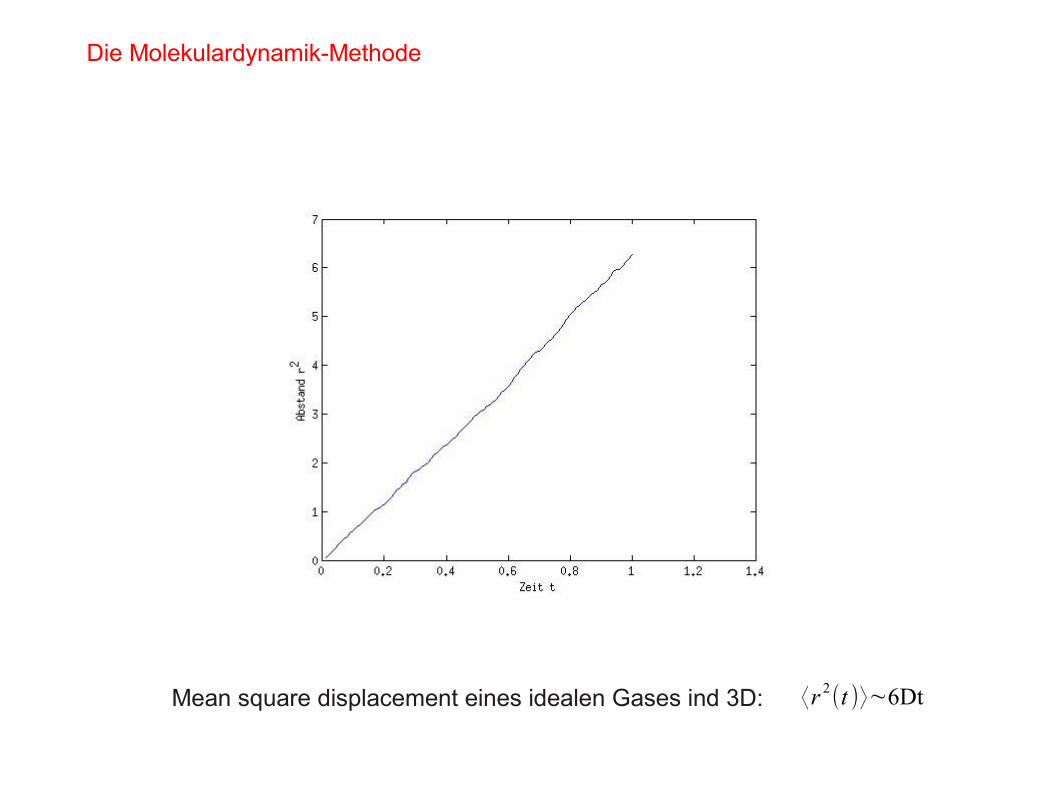
Mean square displacement eines idealen Gases ind 3D: ⟨r 2(t )⟩∼6Dt

Die Molekulardynamik-Methode
Vergleich MC (Dieter, 500 Teilchen) – BD (Malte, 800 Teilchen), weiches repulsives Potential


















