

Ehlers-Danlos Syndrome Rare Types - Cloud Object … · Ehlers-Danlos Syndrome Rare Types ......
91
Transcript of Ehlers-Danlos Syndrome Rare Types - Cloud Object … · Ehlers-Danlos Syndrome Rare Types ......


Ehlers-Danlos Syndrome Rare Types
Clair A. Francomano, MD On behalf of Fransiska Malfait, MD and the
Rare Disease Committee
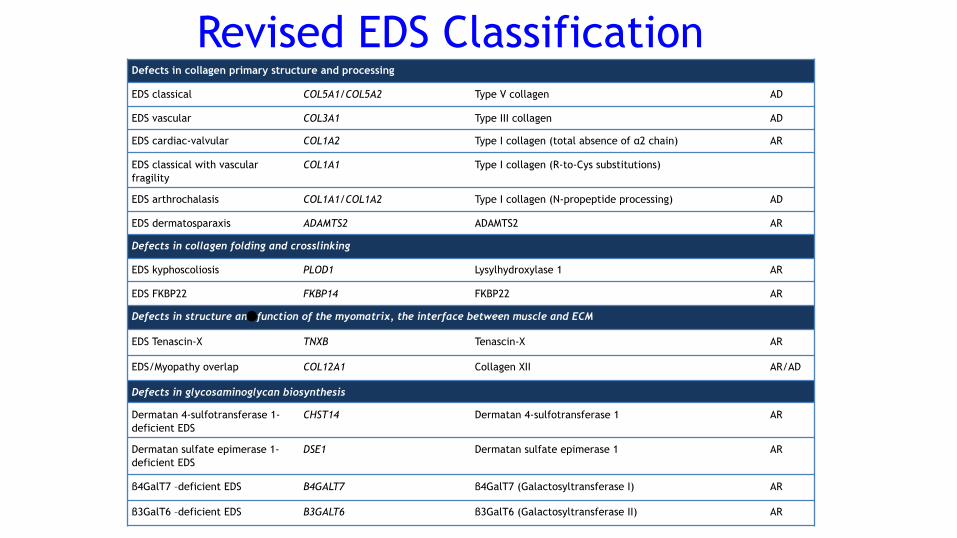
Defects in collagen primary structure and processing
EDS classical COL5A1/COL5A2 Type V collagen AD
EDS vascular COL3A1 Type III collagen AD
EDS cardiac-valvular COL1A2 Type I collagen (total absence of α2 chain) AR
EDS classical with vascular fragility
COL1A1 Type I collagen (R-to-Cys substitutions)
EDS arthrochalasis COL1A1/COL1A2 Type I collagen (N-propeptide processing) AD
EDS dermatosparaxis ADAMTS2 ADAMTS2 AR
Defects in collagen folding and crosslinking
EDS kyphoscoliosis PLOD1 Lysylhydroxylase 1 AR
EDS FKBP22 FKBP14 FKBP22 AR
Defects in structure and function of the myomatrix, the interface between muscle and ECM
EDS Tenascin-X TNXB Tenascin-X AR
EDS/Myopathy overlap COL12A1 Collagen XII AR/AD
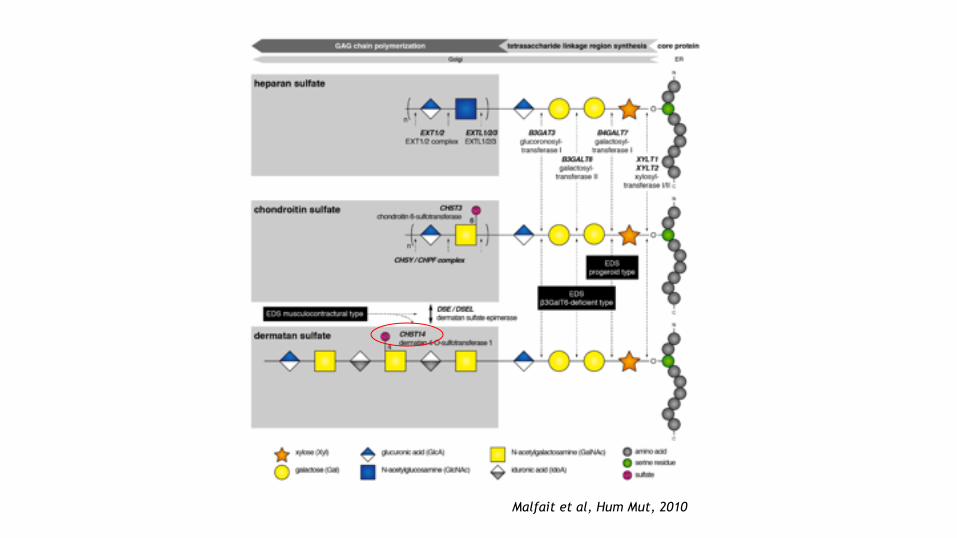
Defects in glycosaminoglycan biosynthesis
Dermatan 4-sulfotransferase 1-deficient EDS
CHST14 Dermatan 4-sulfotransferase 1 AR
Dermatan sulfate epimerase 1-deficient EDS
DSE1 Dermatan sulfate epimerase 1 AR
β4GalT7 –deficient EDS B4GALT7 β4GalT7 (Galactosyltransferase I) AR
β3GalT6 –deficient EDS B3GALT6 β3GalT6 (Galactosyltransferase II) AR
Revised EDS Classification
•
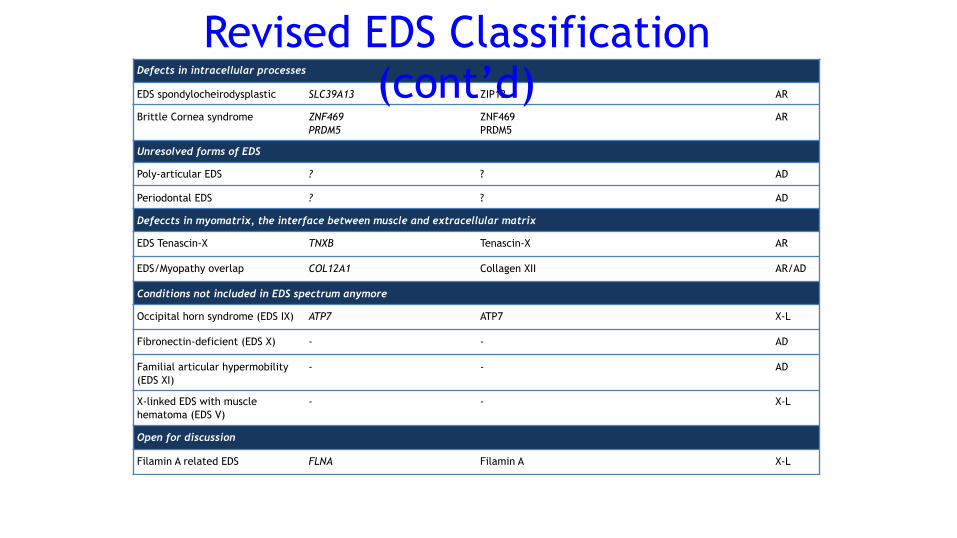
Defects in intracellular processes
EDS spondylocheirodysplastic SLC39A13 ZIP13 AR
Brittle Cornea syndrome ZNF469 PRDM5
ZNF469 PRDM5
AR
Unresolved forms of EDS
Poly-articular EDS ? ? AD
Periodontal EDS ? ? AD
Defeccts in myomatrix, the interface between muscle and extracellular matrix
EDS Tenascin-X TNXB Tenascin-X AR
EDS/Myopathy overlap COL12A1 Collagen XII AR/AD
Conditions not included in EDS spectrum anymore
Occipital horn syndrome (EDS IX) ATP7 ATP7 X-L
Fibronectin-deficient (EDS X) - - AD
Familial articular hypermobility (EDS XI)
- - AD
X-linked EDS with muscle hematoma (EDS V)
- - X-L
Open for discussion
Filamin A related EDS FLNA Filamin A X-L
Revised EDS Classification (cont’d)
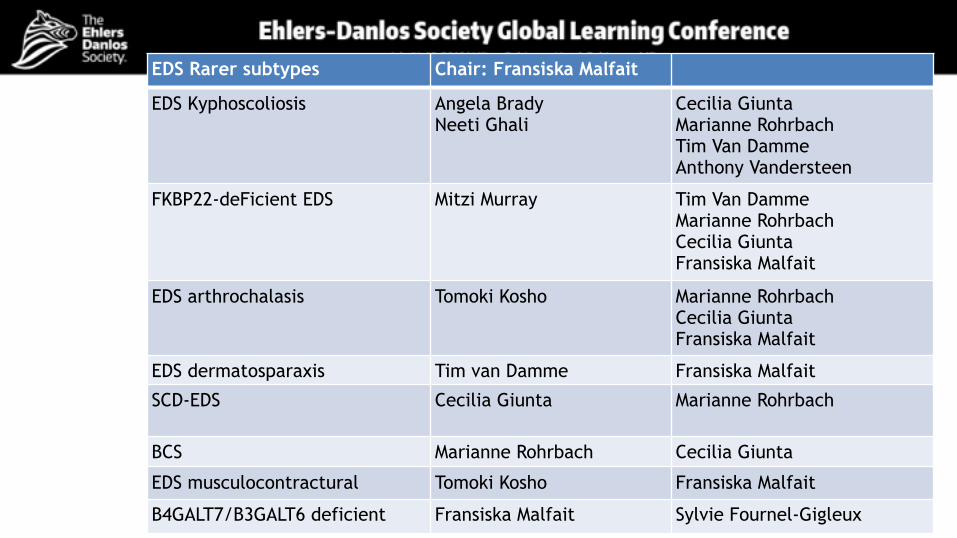
EDS Rarer subtypes Chair: Fransiska Malfait
EDS Kyphoscoliosis Angela Brady Neeti Ghali
Cecilia Giunta Marianne Rohrbach Tim Van Damme Anthony Vandersteen
FKBP22-deFicient EDS Mitzi Murray Tim Van Damme Marianne Rohrbach Cecilia Giunta Fransiska Malfait
EDS arthrochalasis Tomoki Kosho Marianne Rohrbach Cecilia Giunta Fransiska Malfait
EDS dermatosparaxis Tim van Damme Fransiska Malfait
SCD-EDS Cecilia Giunta Marianne Rohrbach
BCS Marianne Rohrbach Cecilia Giunta
EDS musculocontractural Tomoki Kosho Fransiska Malfait
B4GALT7/B3GALT6 deficient Fransiska Malfait Sylvie Fournel-Gigleux

Chair: Fransiska Malfait
Tenascin-X deficient EDS Nicol Voermans Eelco Dulfer Serwet Demirdas
EDS/Myopathy overlap Roberto Mendoza-Londono
EDS periodontal Anthony Vandersteen Ines Kapferer-Seebach Johannes Zschocke Cecilia Giunta Marianne Rohrbach Michael Pope,Mitzi Murray
Filamin A -associated Anthony Vandersteen Eyal Reinstein Steven Robertson
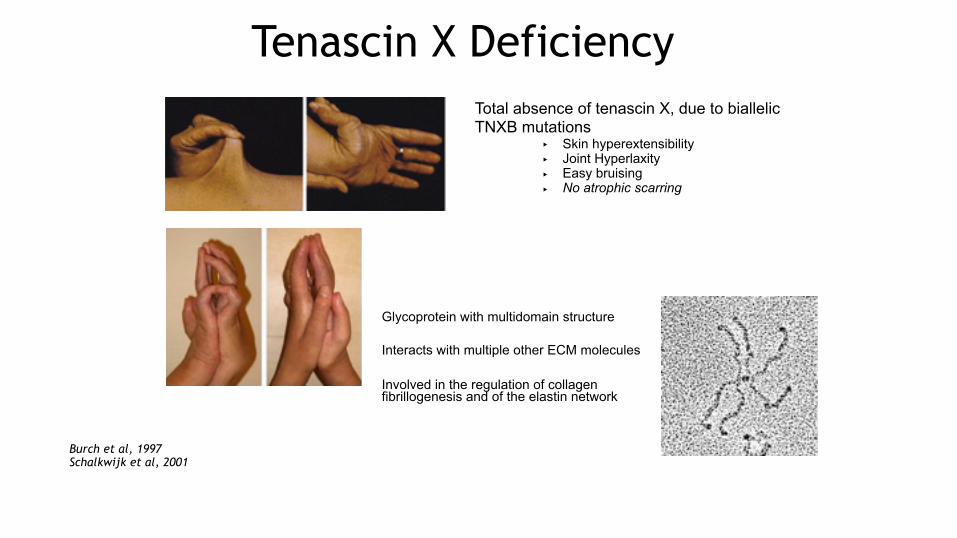
Tenascin X DeficiencyTotal absence of tenascin X, due to biallelic TNXB mutations
‣ Skin hyperextensibility ‣ Joint Hyperlaxity ‣ Easy bruising ‣ No atrophic scarring
Glycoprotein with multidomain structure Interacts with multiple other ECM molecules Involved in the regulation of collagen fibrillogenesis and of the elastin network
Burch et al, 1997 Schalkwijk et al, 2001
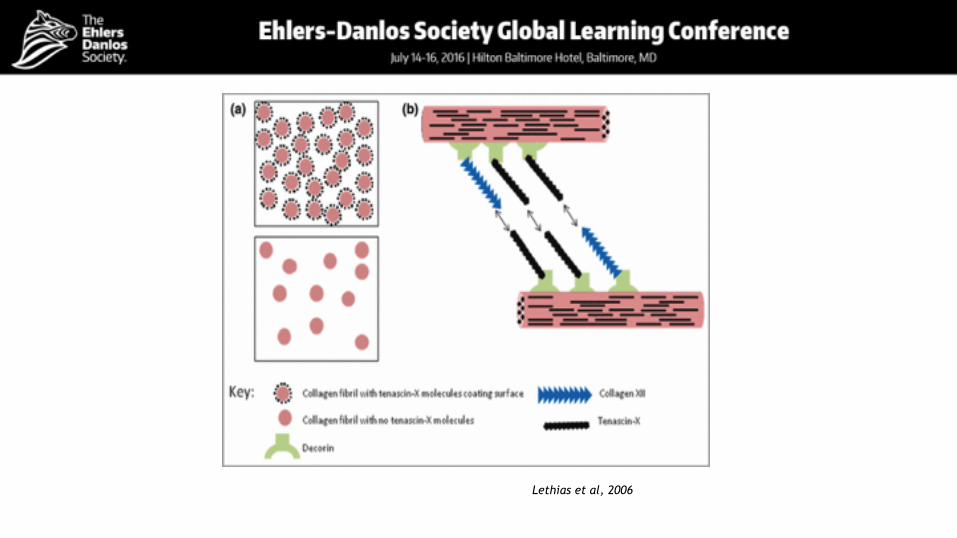
Lethias et al, 2006

Tenascin-X• The presence of joint hypermobility in heterozygote carriers of
TNXB null mutations suggested Tenascin X as a candidate for EDS hypermobility type
• 65% of female carriers presented joint hypermobility (Zweers et al, 2003) • However, wider screening identified mutations in TNXB in only
2.5% of persons with hypermobility (EDS) ! The role of TNXB in EDS hypermobility type remains currently unclear
Malfait et al, Hum Mut 2010
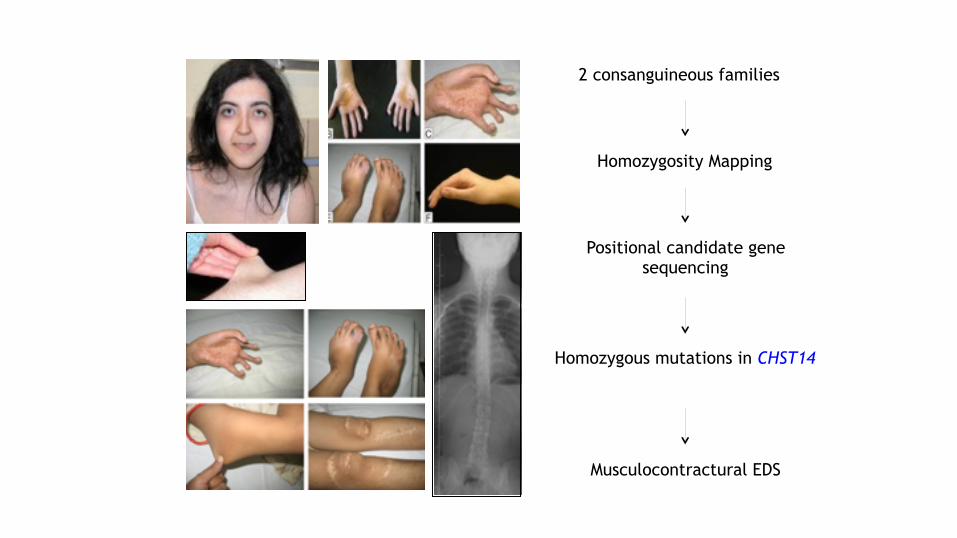
2 consanguineous families
Positional candidate gene sequencing
Homozygous mutations in CHST14
Musculocontractural EDS
Homozygosity Mapping
Malfait et al, Hum Mut, 2010
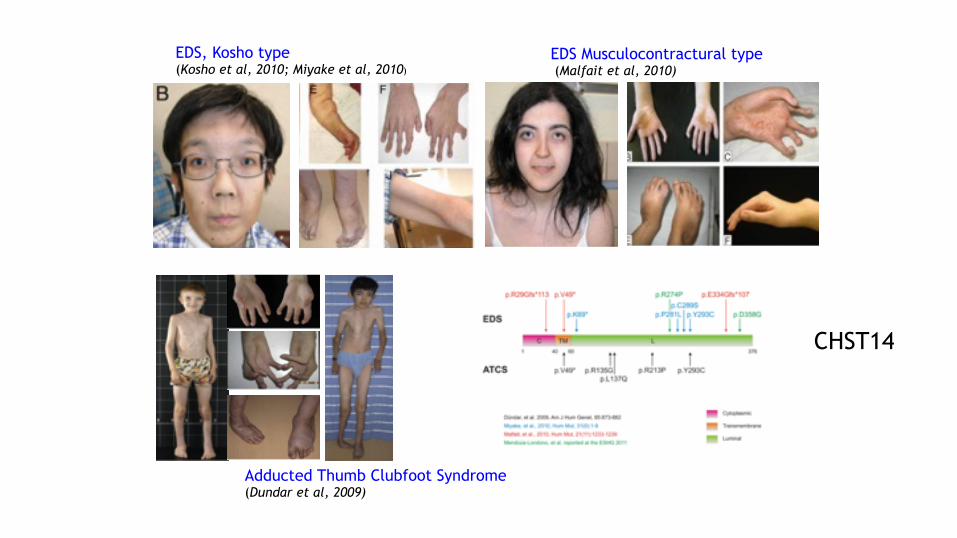
Adducted Thumb Clubfoot Syndrome (Dundar et al, 2009)
EDS Musculocontractural type (Malfait et al, 2010)
EDS, Kosho type (Kosho et al, 2010; Miyake et al, 2010)
CHST14

Mitzi Murray, Tim Van Damme, Fransiska Malfait
FKBP22-deficient Ehlers-Danlos Syndrome

Organ System Involvement
9 patients from 7 independent families reported to date
7 children and 2 adults

Musculoskeletal
Number of patients 9
Joint hypermobility 9
Dislocations 1
Muscle hypotonia/decreased muscle bulk 9
Kyphoscolisosis 8
Club feet 3
Pectus deformity (carinatum) 1
Coxa valga 1
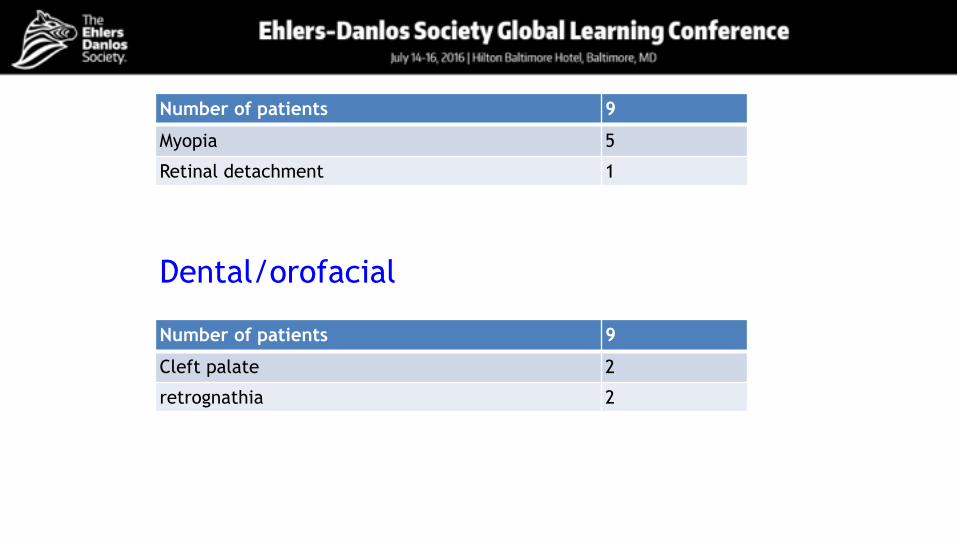
Ocular involvementNumber of patients 9
Myopia 5
Retinal detachment 1
Dental/orofacial
Number of patients 9
Cleft palate 2
retrognathia 2
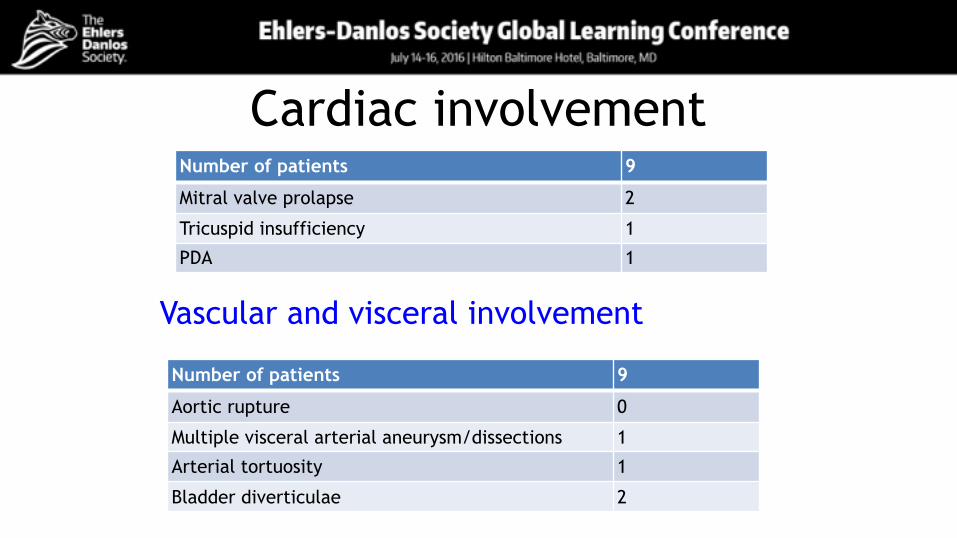
Cardiac involvementNumber of patients 9
Mitral valve prolapse 2
Tricuspid insufficiency 1
PDA 1
Vascular and visceral involvement
Number of patients 9
Aortic rupture 0
Multiple visceral arterial aneurysm/dissections 1
Arterial tortuosity 1
Bladder diverticulae 2

Skin and integumentNumber of patients 9
Hyperextensible 6
Soft, doughy 7
Hyperkeratosis follicularis 5
Easy bruising 3
Hernia (Inguinal/umbilical) 4
Excessive periumbilical skin 1
Number of patients
Sensorineural and conductive hearing loss 6
Hypotonia 9
Decreased endurance 9
Intellectual disability with white matter atrophy 1
Subdural hygroma 1
Nervous system

Clinical phenotype
Baumann et al, AJHG, 2012

Murray et al, AJMG, 2014
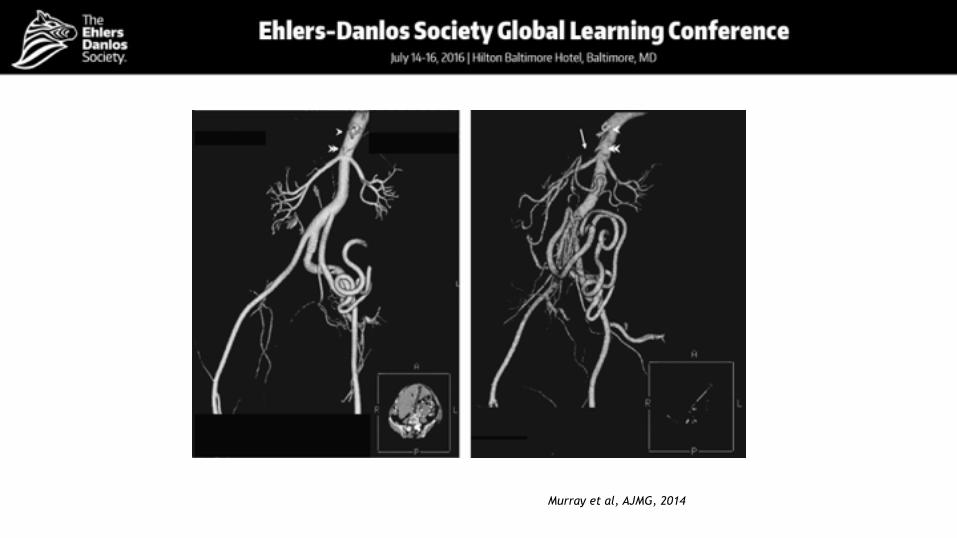
Murray et al, AJMG, 2014

Allelic heterogeneity
Mutations
p.(Glu122Argfs*7) p.(Thr15*)c.197+5_197+8delGTAA
Gene: FKBP14 Gene locus: 7p14.3; 6 exons
Protein: FKBP22 Function: - ER-resident peptidyl-prolyl cis-trans isomerase (Prolyl isomerisation is rate-limiting step during triple helix formation of collagen) - Catalyzes folding of type III collagen - interacts with collagen III, VI and X, but not with collagen I and V (Ishikawa et al, 2014)
Inheritance pattern: Autosomal recessive

Diagnostic criteriaClinical criteria
Kyphoscoliosis
Joint hypermobility (large and small joints)
Muscle atrophy, hypotonia
Club feet
Osteopenia
Hearing loss
Myopia
Soft, hyperextensible skin
Bladder diverticulae

Differential Diagnosis
▪ Kyphoscoliotic Ehlers-Danlos syndrome ▪ D4ST1-deficient Ehlers-Danlos syndrome ▪ Ullrich congenital muscular dystrophy ▪ Bethlem myopathy ▪ Other neuromuscular disorders ▪ Arterial tortuosity syndrome, other arterial fragility syndromes

Confirmation of diagnosisMolecular diagnosis: sequencing of FKBP14 Methods:
• Sanger or NGS • Deletion/duplication analysis
No biochemical test; normal urinary pyridinolines

ManagementEvaluations following initial diagnosis
▪ Clinical evaluation for kyphoscoliosis ▪ Hearing evaluation ▪ Pulmonary function tests ▪ Developmental evaluation in children ▪ Functional assessment in adults ▪ Consider ophthalmologic exam ▪ Consider sleep study to assess for
nocturnal hypoxemia ▪ Consider bone mineral density
Treatment of Manifestations ▪ Management of kyphoscoliosis as indicated ▪ Therapy for motor development and functionality
as indicated ▪ Surgical management of hernias, bladder
diverticuli, kyphoscolisois etc, as indicated
Surveillance ▪ Hearing surveillance (annual?) ▪ Pulmonary function tests (optimal schedule
unknown) ▪ Sleep study (optimal schedule unknown) ▪ Consider vascular surveillance (optimal mode or
schedule unknown) ▪ Consider ophthalmologic surveillance

Tim Van Damme, MD PhD candidate Fransiska Malfait, MD PhD
Center for Medical Genetics Ghent University & Ghent University Hospital
Belgium
www.cmgg.be
Ehlers-Danlos Syndrome Dermatosparaxis type

History of dermatosparaxis
Nusgens et al, Nat Gen. 1992 Malfait et al, AJMG. 2004
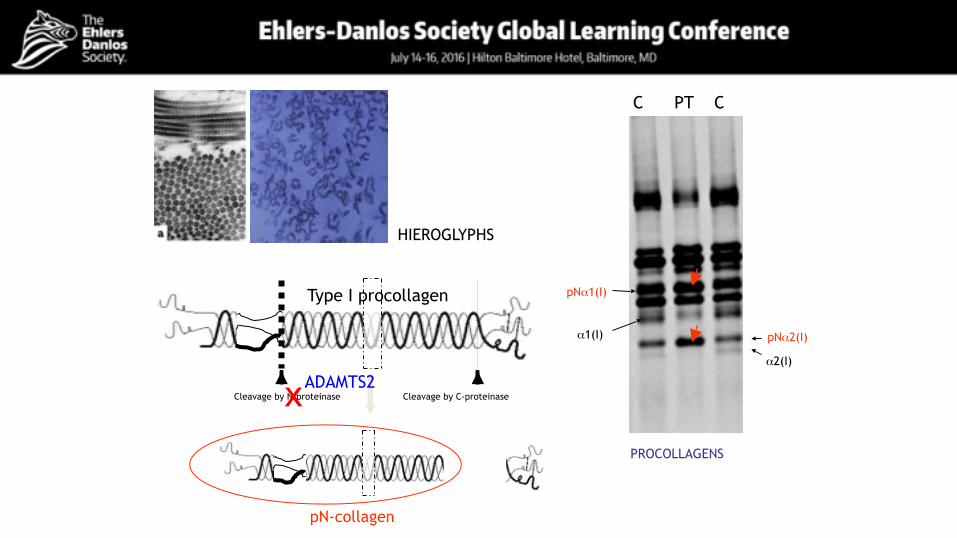
Cleavage by N-proteinase Cleavage by C-proteinase
pN-collagen
x
HIEROGLYPHS
pNα2(I)
pNα1(I)
α1(I)
α2(I)
C PT C
PROCOLLAGENS
Type I procollagen
ADAMTS2

Literature Review
Number of patients 15
Male 11
Female 4
Parental Consanguinity 3
Age at diagnosis Mean 62 months (range birth- 13 yrs)
Oldest patient 21 yrs
15 patients from 14 independent families reported to date
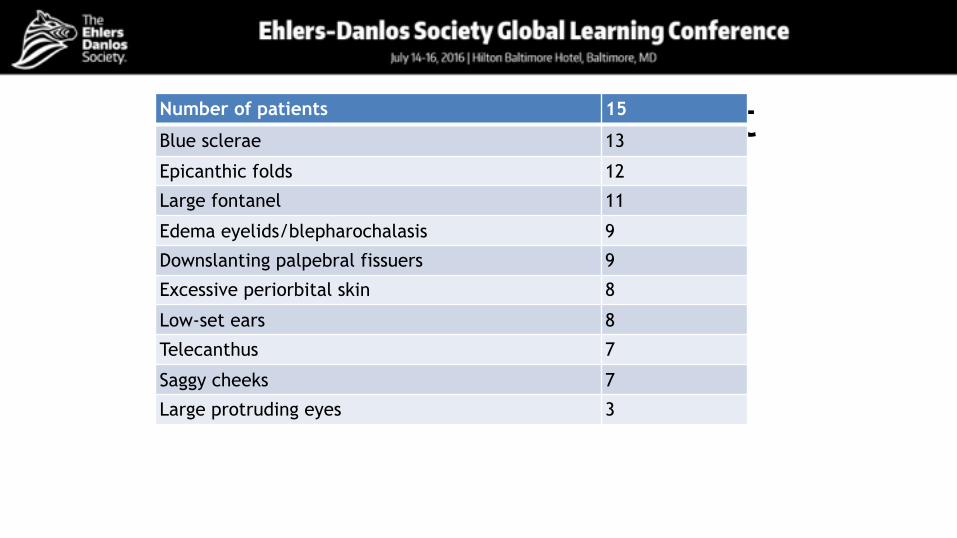
Craniofacial involvementNumber of patients 15
Blue sclerae 13
Epicanthic folds 12
Large fontanel 11
Edema eyelids/blepharochalasis 9
Downslanting palpebral fissuers 9
Excessive periorbital skin 8
Low-set ears 8
Telecanthus 7
Saggy cheeks 7
Large protruding eyes 3

Craniofacial involvement
Malfait et al, AJMG. 2004
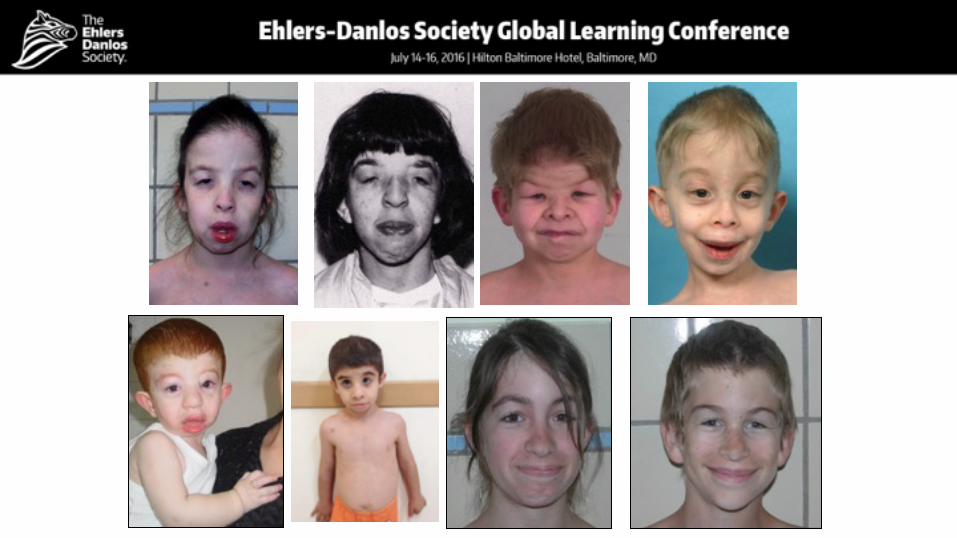

Dental
Number of patients 15
Gingival Hyperplasia/hyperkeratosis 6
Tooth dysplasia 6
Gingival bleeding 4
Hypodontia/oligodontia 3
Tooth discoloration 3
Frontal open bite 3

Dental/orofacial
Malfait et al, AJMG. 2004

Skin and integumentNumber of patients 15
Severe skin fragility 14
Umbilical hernia 14
Skin tears 14
Soft, doughy 12
Excessive bruising 11
Sagging, redundant skin 11
Thin, translucent 6
Increased palmar wrinkling 6
Hypertrichosis 6
Hyperextensible skin 5
Atrophic scarring 5
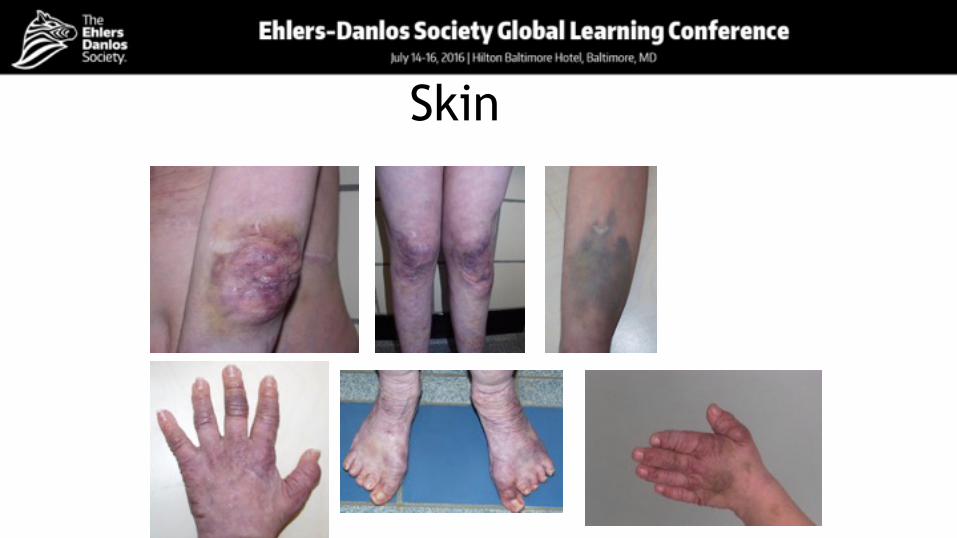
Skin
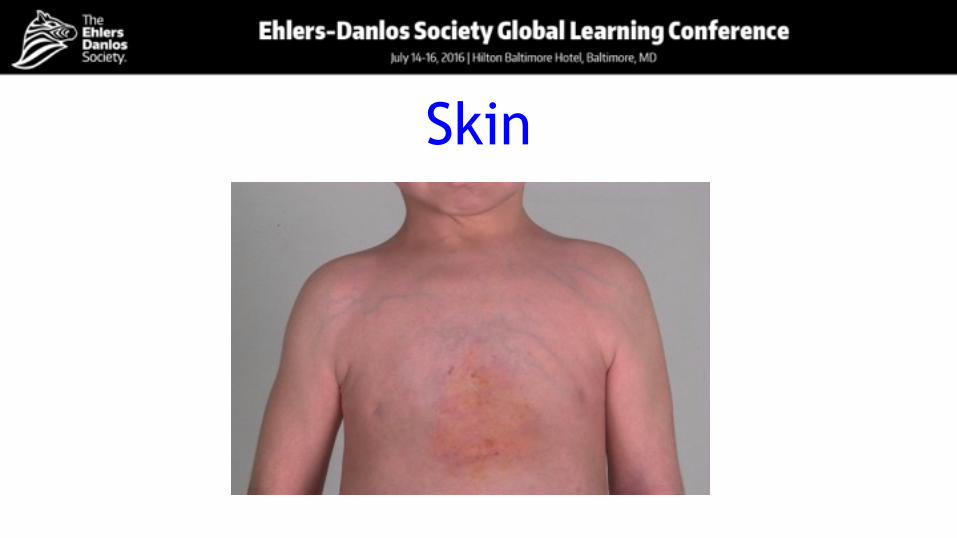
Skin
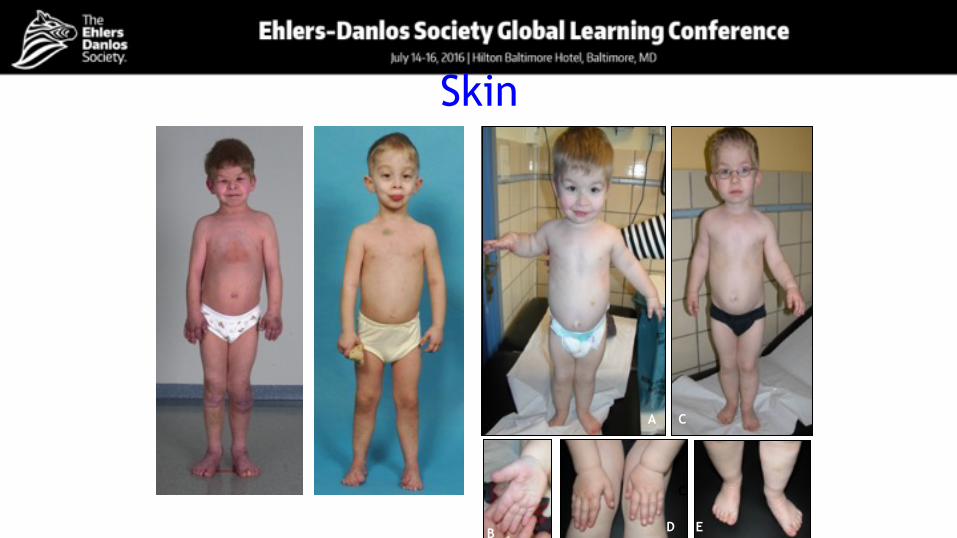
C
D E
CA
B
Skin
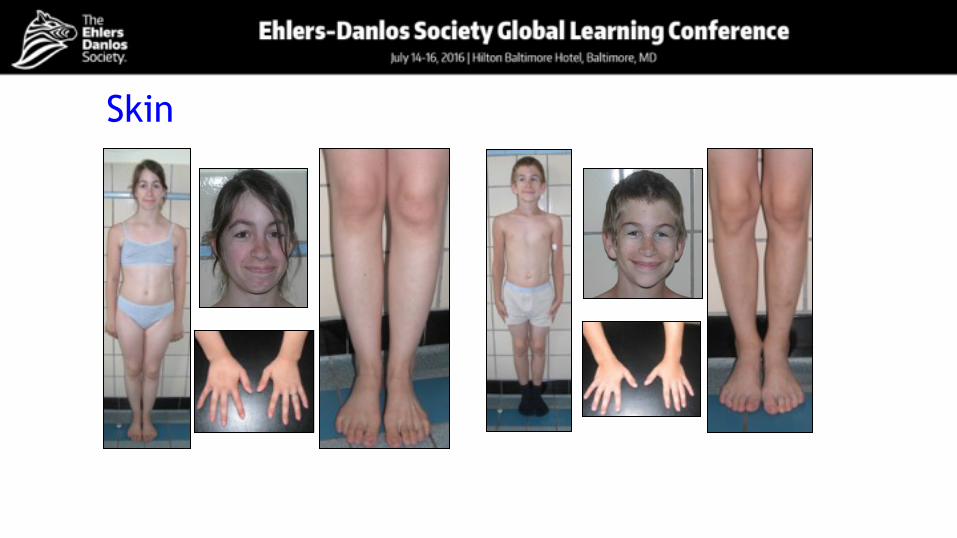
Skin

MusculoskeletalNumber of patients 15
Postnatal growth retardation 13
Joint hypermobility 11
Short limbs, hands and feet 11
Motor delay 8
Fractures 4
Congenital skull fractures 2

OtherNumber of patients 15
Bleeding complications (epistaxis, gum bleeding, (congenital) cerebral hemorrhage
7
Complications of visceral Fragility 5
Strabismus 5
Myopia 5
Astigmatism 3
Severe glaucoma at young age 1
Poor breast development 1
Visceral complications include: - Rupture of diaphragm (1) - Bladder diverticulae (2) with bladder rupture (1) - Rectal prolapse with profuse bleeding (2) - No vascular problems reported

Perinatal complicationsNumber of patients 15
PPROM 6
Gestational age 28-41 weeks Mean: 34 w 4 d
Friable umbilical cord 2
Multiple skull fractures and subgaleal hemorrhage (died) 1
Cerebral hemorrhage 2
Pneumothorax 1
Hydronephrosis 1
Hypoglycemia, hypocalcemia and hypothyroidism 1

Diagnostic criteriaMajor Diagnostic criteria
Characteristic craniofacial features
Extreme skin fragility
Soft and doughy skin
Redundant, almost lax skin with excessive skin folds around wrist, ankles
Increased palmar creases
Excessive bruising
Umbilical hernia
Postnatal growth retardation
Short limbs, hands and feet
Perinatal complications due to connective tissue fragility
Complications of visceral fragility (bladder diverticulae, rectal prolapse, …)

Diagnostic criteria
Minor Diagnostic criteria
skin hyperextensibility, atrophic scars
hypertrichosis
dental abnormalities
Joint hypermobility
delayed motor development
osteopenia
refractive errors (myopia, astigmatism)
strabismus

Differential Diagnosis
▪ Classical EDS ▪ Cutis laxa syndromes ▪ Achondroplasia ▪ Osteogenesis imperfecta
Inheritance PatternAutosomal recessive

Confirmation of diagnosisMolecular diagnosis: sequencing of ADAMTS2 gene Methods:
• Sanger or NGS • Deletion/duplication analysis
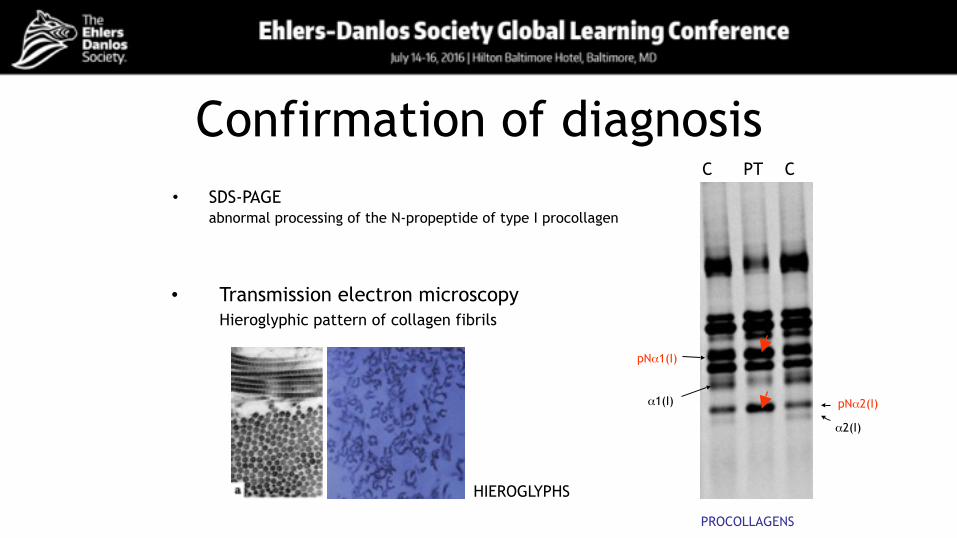
Confirmation of diagnosis• SDS-PAGE
abnormal processing of the N-propeptide of type I procollagen
pNα2(I)
pNα1(I)
α1(I)
α2(I)
C PT C
PROCOLLAGENS
• Transmission electron microscopy Hieroglyphic pattern of collagen fibrils
HIEROGLYPHS

Management• Skin
– Follow advice for classical EDS – Routine examination for umbilical/inguinal hernia
• Musculoskeletal – At diagnosis: whole body skeletal survey – Physical and occupational therapy, orthotic management – Avoid contact sports
• Cardiovascular screening • Ophthalmological screening • Pregnancy management: consider pregnancy as high risk

Research needed• Studies to determine the prevalence of EDS dermatosparaxis type
• Create a register of patients with prospective collection of detailed information regarding phenotype. This information should help address:
• genotype-phenotype correlations • clinical variability (inter- and intrafamilial) • age-dependent organ-system involvement • pregnancy outcome • The association of other signs and symptoms, eg pain, autonomic
dysfunction, fatigue, mast cell activation syndrome, psychological problems, neurological involvement etc
• Collection of tissue samples (Biobank) for future pathogenic and electron-microscopic studies
• Quality of Life studies

Research needed• In view of the role of ADAMTS in cancer: increased risk for cancer?
• Male ADAMTS2 knock-out mice show reduced spermatogenesis and sterility. Other animals with dermatosparaxis (cattle, dogs, cats, sheep), usually do not survive until the reproductive age, and so far human males with EDS dermatosparaxis type have not reached the reproductive age, so follow-up of these patients is needed to evaluate whether the sterility is also seen in human males.
• What is contribution of deficient processing of other ADAMTS2 substrates to pathogenesis the severity of the disorder, such as the extreme skin fragility and laxity (type V collagen) and the propensity for bruising and bleeding (type III collagen).
Li et al, 2001

Tim Van Damme, MD PhD candidate Fransiska Malfait, MD PhD
Center for Medical Genetics Ghent University & Ghent University Hospital, Belgium
B4GALT7- and B3GALT6-associated Ehlers-Danlos Syndrome
Sylvie Fournel-Gigleux Research Director INSERM
UMR 7365 CNRS-Université de Lorraine, MolCelTEG Team
Vandœuvre-les-Nancy, France

History of Progeroid EDS• Hernandez et al (1979, 1981, 1986) reported 5 patients with EDS-features and
features of early aging • Wrinkled facies • Significant growth failure • Fine/curly hair • Peridontitis • Bilateral cryptorchidism • Apparent intellectual deficit • Classical features of EDS
No molecular diagnosis! • 4 additional patients showing resemblance to these 5 patients were further
identified (Kresse et al, 1987, Faiyaz-Ul-Haque et al, 2004, Guo et al, 2103)
• Defective glycosaminoglycan addition to several proteoglycan core proteins (Kresse et al, 1987)
• Shown to result from bi-allelic mutations in B4GALT7, encoding galactosyltransferase I (Quentin et al, 1990)
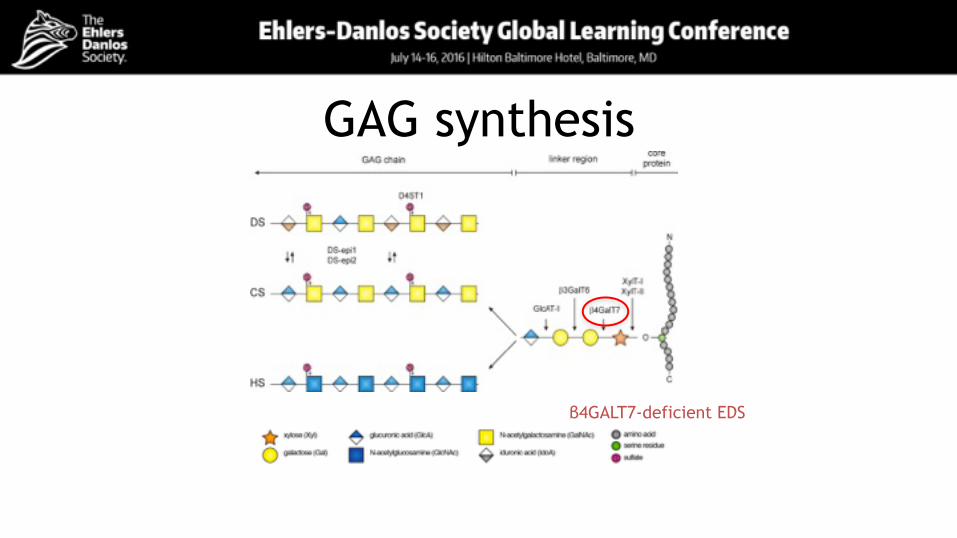
GAG synthesis
β4GALT7-deficient EDS

Organ System Involvement
Number of patients 6
Age at diagnosis 2 yrs-33 yrs
Oldest reported patient 33 yrs
6 independent EDS patients with bi-allelic B4GALT7 mutations reported to date
In addition: 22 patients reported with Larsen of Reunion Island Syndrome with bi-allelic B4GALT7 mutations
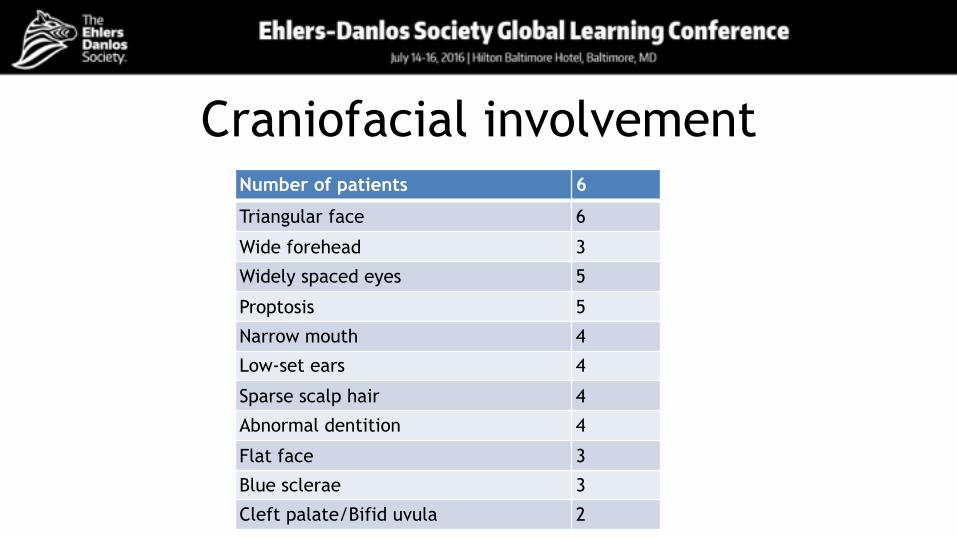
Craniofacial involvementNumber of patients 6
Triangular face 6
Wide forehead 3
Widely spaced eyes 5
Proptosis 5
Narrow mouth 4
Low-set ears 4
Sparse scalp hair 4
Abnormal dentition 4
Flat face 3
Blue sclerae 3
Cleft palate/Bifid uvula 2

MusculoskeletalNumber of patients 6
Short stature (severe) 6
Muscle Hypotonia 6
Joint hypermobility 6
Radio-Ulnar synostosis 6
Bowing of limbs 5
Osteopenia 4
Fractures (vertebral fractures) 1
Swedish key features of femur 1
Number of patients 6
Cognitive delay 5
Motor delay 5
Development

Skin and integumentNumber of patients 6
Hyperextensible skin 6
Delayed wound healing 3
Atrophic scarring 3
Hands and Feet
Number of patients 6
Syndactyly 2
Long fingers 4
Single Transverse Palmar crease 4
Pes planus 5
Salter et al, AJMG 2015
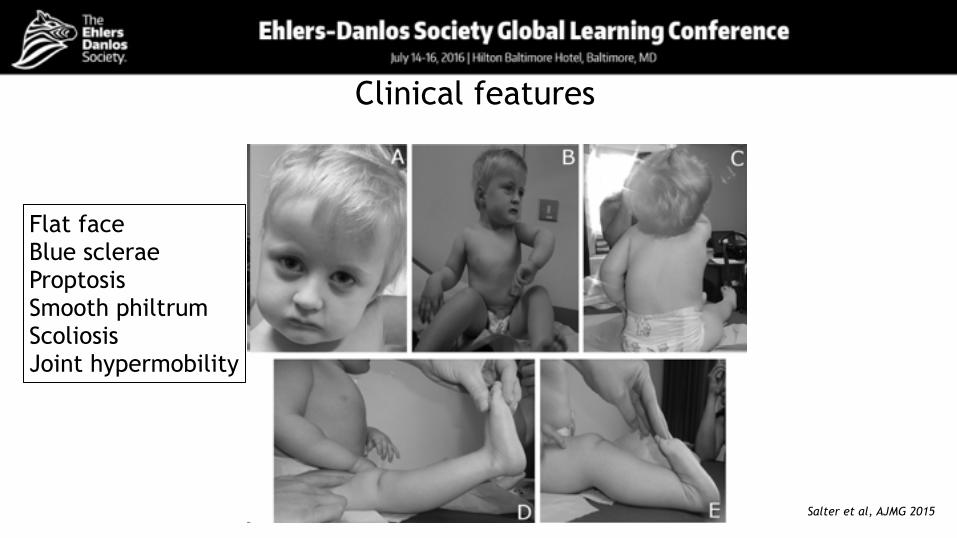
Clinical features
Flat face Blue sclerae Proptosis Smooth philtrum Scoliosis Joint hypermobility
Salter et al, AJMG 2015
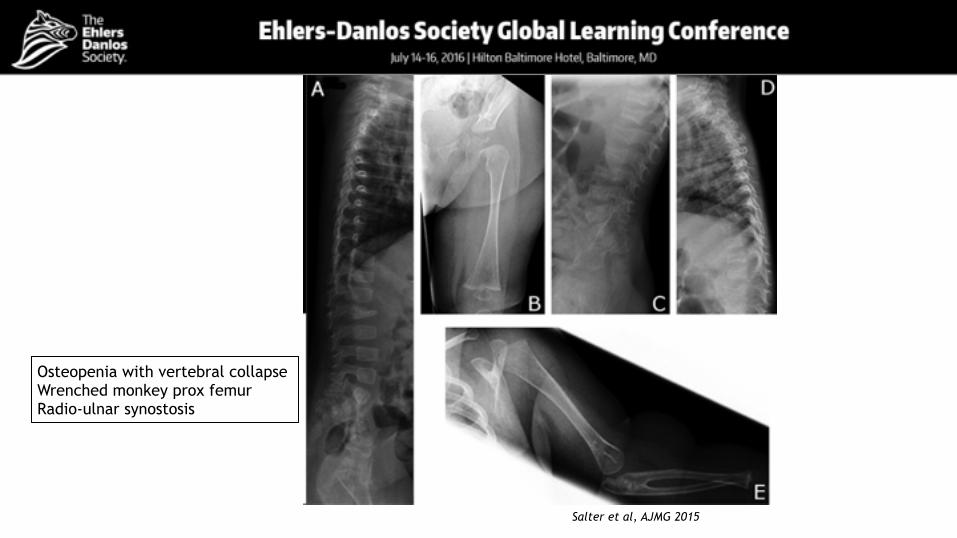
Osteopenia with vertebral collapse Wrenched monkey prox femur Radio-ulnar synostosis
Salter et al, AJMG 2015
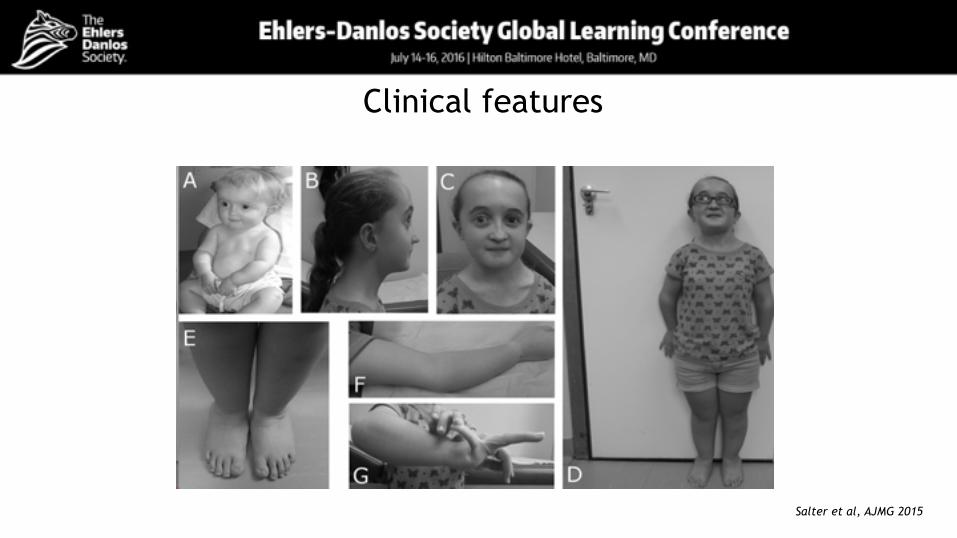
Clinical features
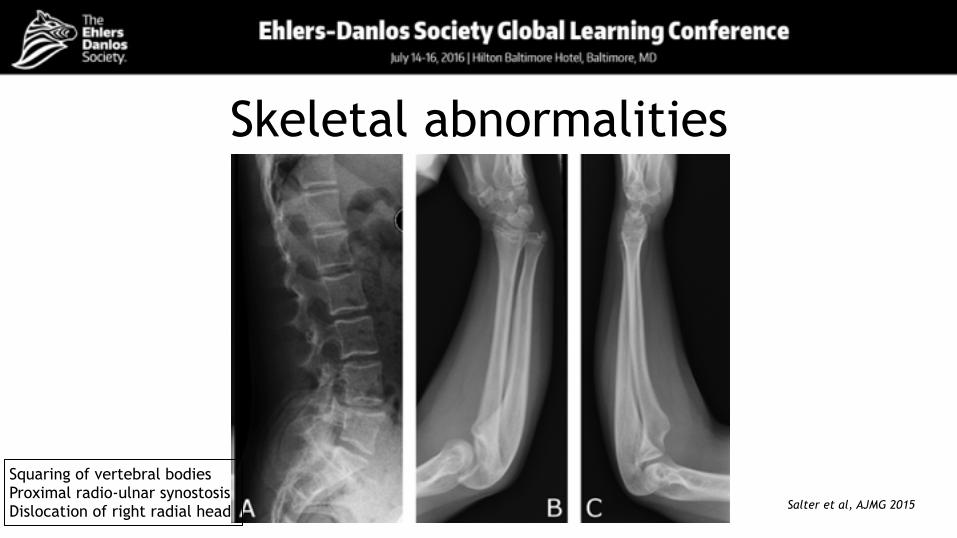
Skeletal abnormalities
Salter et al, AJMG 2015
Squaring of vertebral bodies Proximal radio-ulnar synostosis Dislocation of right radial head
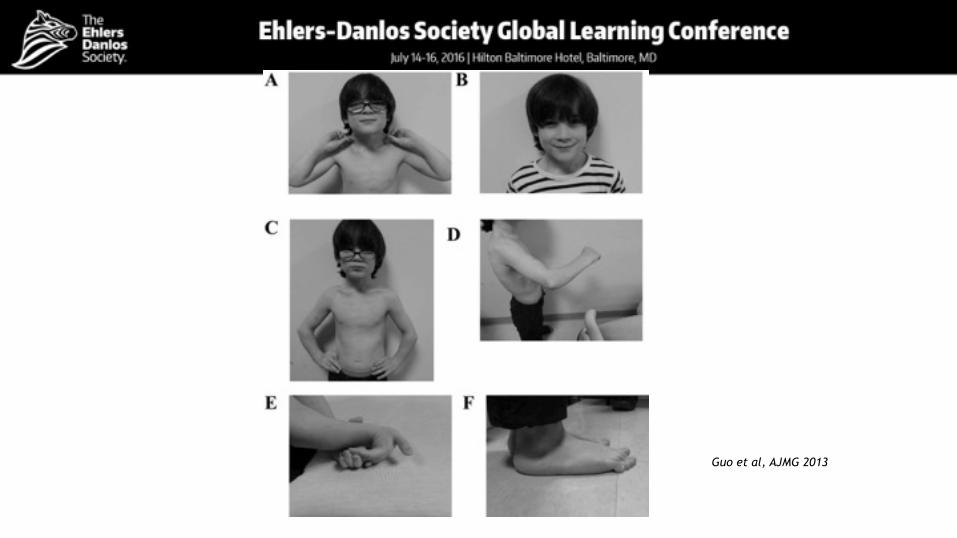
Guo et al, AJMG 2013

Allelic heterogeneity
Gene: B4GALT7 Gene locus: 5q35.3; 6 exons
Protein: Galactosyltransferase I

Allelic condition: Larsen of Reunion Island syndrome
Large Joint Dislocation Ligamentous laxity/ Joint hypermobility Significant short stature (-6 SD) Radio-ulnar synostosis Swedish key appearance proximal femur Patella dislocation Characteristic facial appearance flat face wide forehead proptosis narrow mouth under-eye shadows Mutation: homozygous p.Arg270Cys
Cartault et al, EJHG, 2015

Approach to DiagnosisClinical Criteria
Radio-ulnar synostosis
Short Stature
Joint Hypermobility
Muscle Hypotonia
Hyperelastic Skin
Bowing of Limbs
Osteopenia
Hypermetropia
Craniofacial: Triangular face, proptosis, widely spaced eyes
Of note: No progressive premature aging features observed in molecularly confirmed patients!

Differential Diagnosis
▪ β3GALT6-deficient EDS ▪ Ehlers-Danlos syndrome kyphoscoliotic type ▪ Musculo-contractural EDS ▪ (FKBP22-deficient EDS) ▪ Skeletal dysplasias
Inheritance patternAutosomal recessive

Confirmation of diagnosisMolecular diagnosis: sequencing of B4GALT7 Methods:
• Sanger or NGS • Deletion/duplication analysis
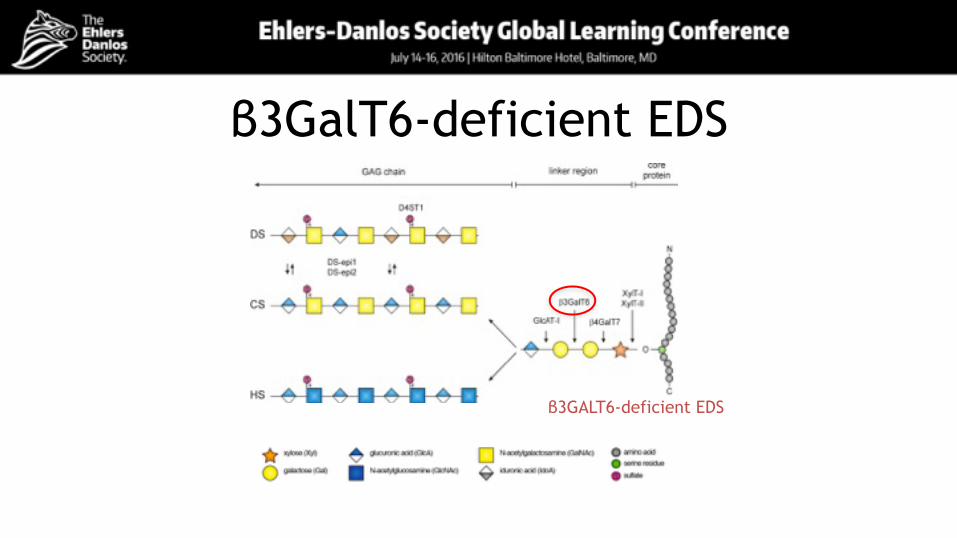
β3GalT6-deficient EDS
β3GALT6-deficient EDS

Organ system involvement14 EDS patients from 11 families have been reported with bi-allelic B3GALT6 mutations Clinical data available from 10 patients from 8 families
Number of patients 10
Age at assessment 1 month-26 yrs
adults 3
Children 7
Oldest reported patient 26 yrs
In addition: patients with Spondyloepimethaphyseal Dysplasia with Joint Hyperlaxity type 1 (SEMD-JL) reported with bi-allelic B3GALT6 mutations (Nakajima et al, 2013, Vorster et al, 2015)

Craniofacial involvement14 EDS patients from 11 families have been reported with bi-allelic B3GALT6 mutations Clinical data available from 10 patients from 8 families
Number of patients 11
Flat face with wide forehead 11
Blue Sclerae 10
Sparse scalp hair 8
Abnormal dentition 6
Prognathism 2
Micrognathia 1
Cleft palate/Bifid uvula 1

MusculoskeletalNumber of patients 11
Short stature 11
Muscle Hypotonia 11
Joint Hypermobility 11
Dislocations 4
Club feet/severe foot deformities 9
Kyphoscoliosis 8
Congenital bilateral hip dislocation 7
Pectus deformity 6
Osteopenia 6
Multiple low-impact fractures 5
Radio-ulnar synostosis 0

SkinNumber of patients 11
Hyperextensibility 10
Doughy skin 10
Thin, translucent skin 6
Atrophic scarring 2
Number of patients 11
Broad spatulate fingers 11
Increased palmar wrinkling 6
Finger contractures 7
Hands
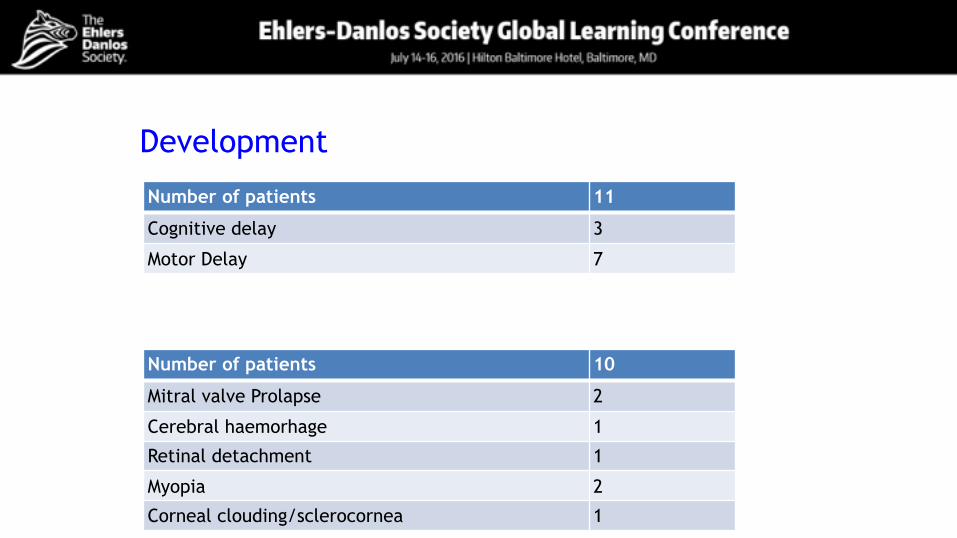
Development
Number of patients 11
Cognitive delay 3
Motor Delay 7
Number of patients 10
Mitral valve Prolapse 2
Cerebral haemorhage 1
Retinal detachment 1
Myopia 2
Corneal clouding/sclerocornea 1

Clinical presentation• congenital muscle hypotonia
• mild delay in motor and cognitive development
• thin, hyperextensible and transparent skin
• generalized joint hypermobility
• repetitive shoulder dislocations
• broad, flat feet
• low bone mineral density (DEXA Z score -2.5)
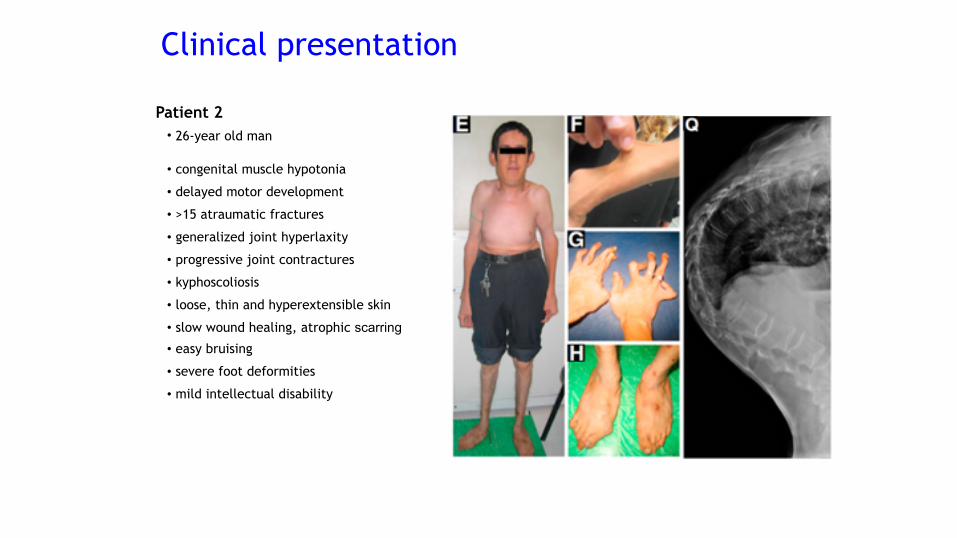
Patient 2• 26-year old man
• congenital muscle hypotonia
• delayed motor development
• >15 atraumatic fractures
• generalized joint hyperlaxity
• progressive joint contractures
• kyphoscoliosis
• loose, thin and hyperextensible skin
• slow wound healing, atrophic scarring• easy bruising
• severe foot deformities
• mild intellectual disability
Clinical presentation
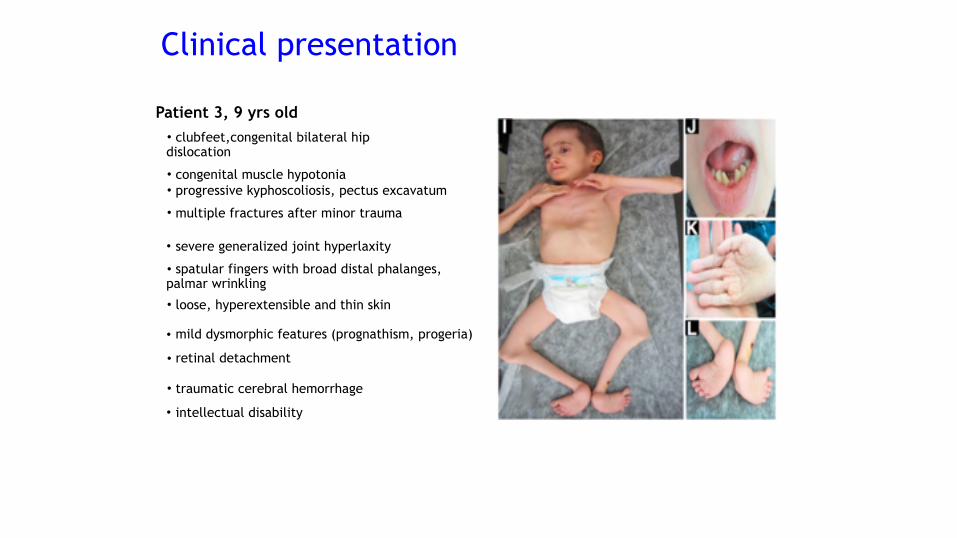
Patient 3, 9 yrs old• clubfeet,congenital bilateral hip dislocation
• congenital muscle hypotonia • progressive kyphoscoliosis, pectus excavatum
• multiple fractures after minor trauma
• mild dysmorphic features (prognathism, progeria)
• severe generalized joint hyperlaxity
• loose, hyperextensible and thin skin
• spatular fingers with broad distal phalanges, palmar wrinkling
• retinal detachment
• traumatic cerebral hemorrhage
• intellectual disability
Clinical presentation
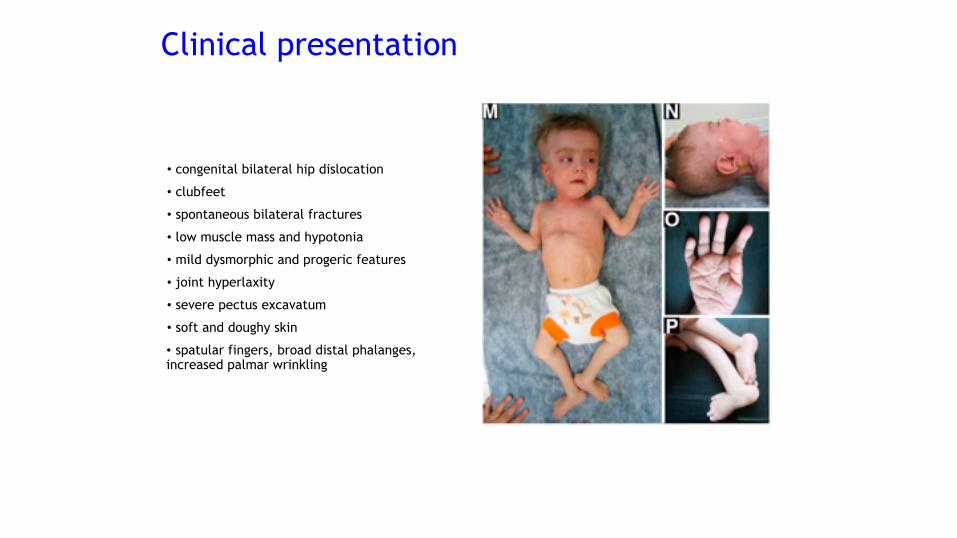
• congenital bilateral hip dislocation
• clubfeet
• spontaneous bilateral fractures
• low muscle mass and hypotonia
• mild dysmorphic and progeric features
• joint hyperlaxity
• severe pectus excavatum
• soft and doughy skin
• spatular fingers, broad distal phalanges, increased palmar wrinkling
Clinical presentation
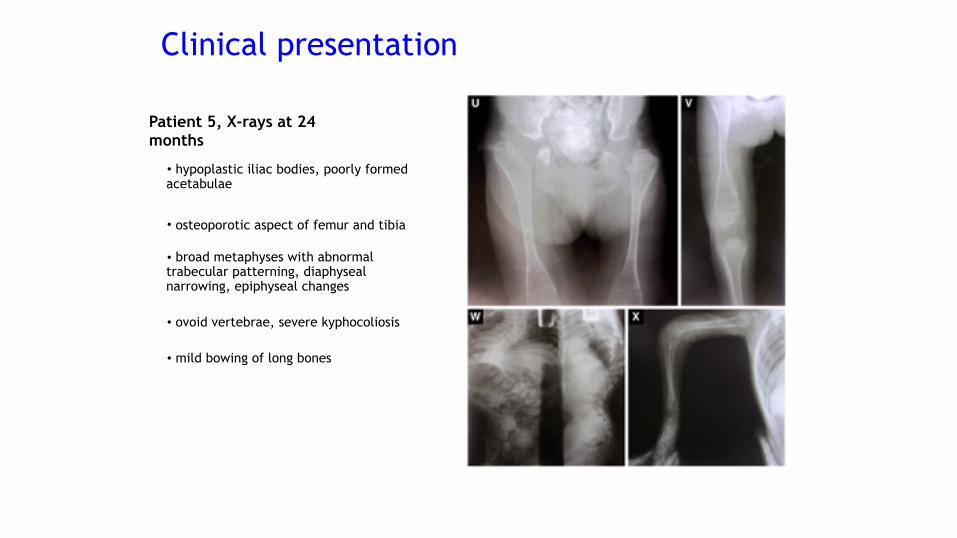
• hypoplastic iliac bodies, poorly formed acetabulae
• osteoporotic aspect of femur and tibia
• broad metaphyses with abnormal trabecular patterning, diaphyseal narrowing, epiphyseal changes
• ovoid vertebrae, severe kyphocoliosis
• mild bowing of long bones
Patient 5, X-rays at 24 months
Clinical presentation
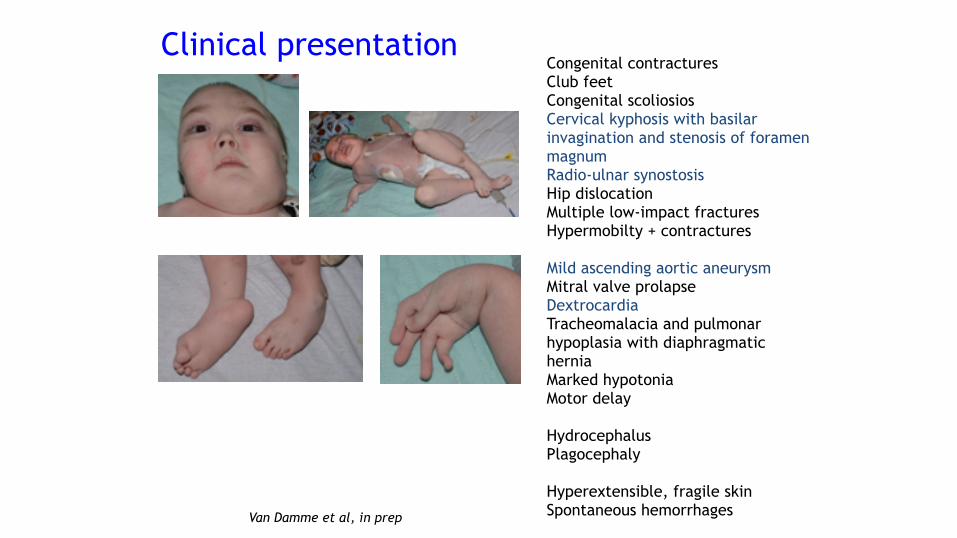
Congenital contractures Club feet Congenital scoliosios Cervical kyphosis with basilar invagination and stenosis of foramen magnum Radio-ulnar synostosis Hip dislocation Multiple low-impact fractures Hypermobilty + contractures
Mild ascending aortic aneurysm Mitral valve prolapse Dextrocardia Tracheomalacia and pulmonar hypoplasia with diaphragmatic hernia Marked hypotonia Motor delay
Hydrocephalus Plagocephaly
Hyperextensible, fragile skin Spontaneous hemorrhages
Clinical presentation
Van Damme et al, in prep

Allelic heterogeneity
Gene: B3GALT6 Gene locus: 1p36.33; single-exon Protein: Galactosyltransferase II
p.(Arg6Trp)
p.(Ser65Gly)
p.(Pro67Leu)
p.(Ile76Thrfs*202)
p.(Ala108Glyfs*163)
p.(Asp118Alafs*160)
p.(Met139Ala141del)
p.(Asp156Asn)
p.(Ser159Tyr)
p.(Arg197Alafs*81)
p.(Asp207His)
p.(Gly217Ser)
p.(Arg232Cys)
p.(Arg256Trp)
p.(Glu265Asp)
p.(Cys300Ser)
p.(Ser309Thr)

Approach to DiagnosisClinical Criteria
Short Stature
Joint Hypermobility with joint contractures
Muscle Hypotonia
Congenital hip dislocation
Club feet
Kyphoscoliosis
Hyperelastic Skin
Doughy skin
Osteopenia
Multiple low-impact fractures
Broad spatulate fingers
Increased palmar wrinkling
Craniofacial: Flat face with prominent forehead, blue sclerae
Spondylo-epimetaphyseal dysplasia
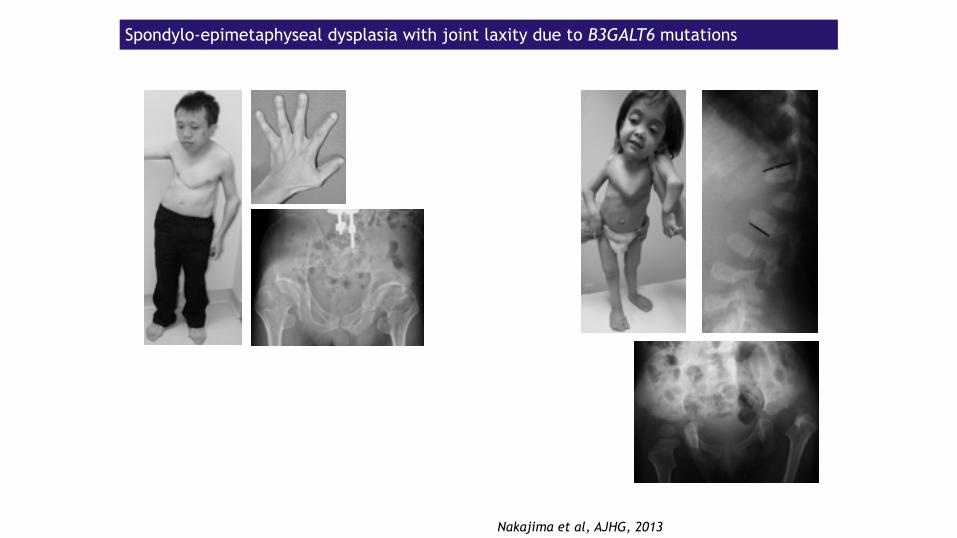
Spondylo-epimetaphyseal dysplasia with joint laxity due to B3GALT6 mutations
Nakajima et al, AJHG, 2013
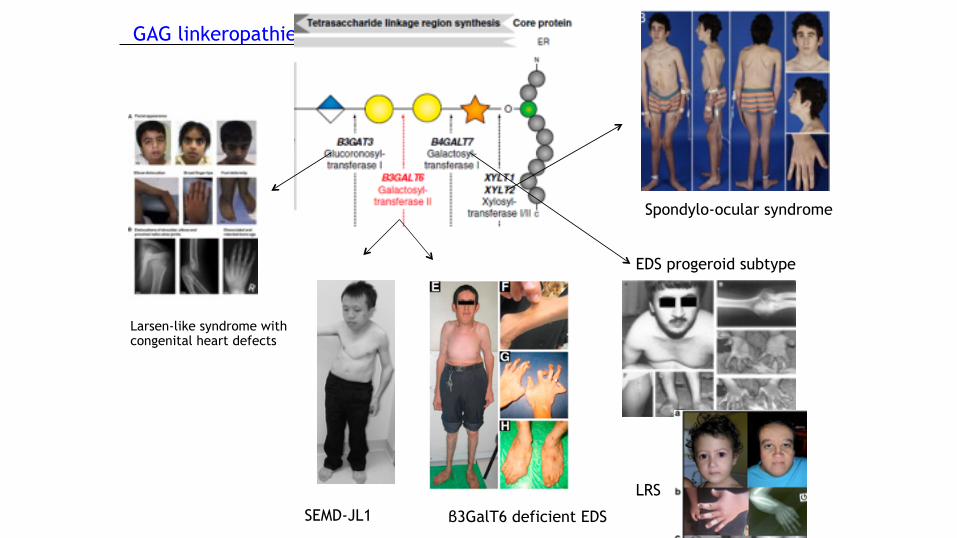
GAG linkeropathies
Larsen-like syndrome with congenital heart defects
SEMD-JL1 β3GalT6 deficient EDS
EDS progeroid subtype
LRS
Spondylo-ocular syndrome

Differential Diagnosis
▪ Other “Linkeropathies” ▪ Osteogenesis imperfecta ▪ Larsen syndrome ▪ Ehlers-Danlos syndrome kyphoscoliotic type ▪ Musculo-contractural EDS ▪ (FKBP22-deficient EDS) ▪ Skeletal dysplasias
Inheritance patternAutosomal recessive

Confirmation of diagnosisMolecular diagnosis: sequencing of B3GALT6 Methods:
• Sanger or NGS • Deletion/duplication analysis

Management• Musculoskeletal
– At diagnosis: • Whole body skeletal survey • Bone densitometry
– Physical and occupational therapy, orthotic management – Avoid contact sports – Consider bisphosphonate therapy
• Cardiovascular work-up • Neurological work up • Dental examination • Ophtalmological work-up • Skin
– Cfr advice for classical EDS – Routine examination for umbilical/inguinal hernia

Research needed• Studies to determine the prevalence of these EDS types • Create a register of patients with prospective collection of detailed
information regarding phenotype. This information should help address: • genotype-phenotype correlations • clinical variability (inter- and intrafamilial) • age-dependent organ-system involvement • pregnancy outcome • The association of other signs and symptoms, eg pain, autonomic
dysfunction, fatigue, mast cell activation syndrome, psychological problems, neurological involvement etc
• Quality of Life studies • Collection of tissue samples (Biobank) for future pathogenic and electron-
microscopic studies • Development of animal models for study of pathogenetic basis and
preclinical studies

Thanks
• Fransiska Malfait and the Entire Rare Disorders Committee
• EDS and EDS-UK • Lara Bloom and Shane Robinson





![Clinical Characteristics to Differentiate · Asthma-COPD overlap syndrome (ACOS) [a description] Asthma-COPD overlap syndrome (ACOS) is characterized by persistent airflow limitation](https://static.fdocument.org/doc/165x107/5f0914d17e708231d4252460/clinical-characteristics-to-differentiate-asthma-copd-overlap-syndrome-acos-a.jpg)








![Embedding Session Types in Haskelljgmorrs/pubs/lindley-hs2016-gvhs.pdfmessage body. Session types [6, 7, 20] capture such protocols in the types of communication channels. Session](https://static.fdocument.org/doc/165x107/5f0294ef7e708231d404fa6a/embedding-session-types-in-haskell-jgmorrspubslindley-hs2016-gvhspdf-message.jpg)



