
Double-lock ratchet mechanism revealing the role of αSER-344 in ...
13
Double-lock ratchet mechanism revealing the role of αSER-344 in F o F 1 ATP synthase Tamás Beke-Somfai a,b,1 , Per Lincoln a , and Bengt Nordén a,1 a Department of Chemical and Biological Engineering, Physical Chemistry, Chalmers University of Technology, SE-412 96 Göteborg, Sweden; and b Protein Modeling Group, Hungarian Academy of Sciences, Institute of Chemistry, Eötvös Loránd University, P.O. Box 32, H-1538 Budapest, Hungary Edited* by Peter H. von Hippel, Institute of Molecular Biology, Eugene, OR, and approved February 8, 2011 (received for review August 5, 2010) In a majority of living organisms, F o F 1 ATP synthase performs the fundamental process of ATP synthesis. Despite the simple net reaction formula, ADP þ P i → ATP þ H 2 O, the detailed step-by-step mechanism of the reaction yet remains to be resolved owing to the complexity of this multisubunit enzyme. Based on quantum mechanical computations using recent high resolution X-ray struc- tures, we propose that during ATP synthesis the enzyme first pre- pares the inorganic phosphate for the γP-O ADP bond-forming step via a double-proton transfer. At this step, the highly conserved αS344 side chain plays a catalytic role. The reaction thereafter progresses through another transition state (TS) having a planar PO 3 − ion configuration to finally form ATP. These two TSs arecon- cluded crucial for ATP synthesis. Using stepwise scans and several models of the nucleotide-bound active site, some of the most important conformational changes were traced toward direction of synthesis. Interestingly, as the active site geometry progresses toward the ATP-favoring tight binding site, at both of these TSs, a dramatic increase in barrier heights is observed for the reverse direction, i.e., hydrolysis of ATP. This change could indicate a “ratchet” mechanism for the enzyme to ensure efficacy of ATP synthesis by shifting residue conformation and thus locking access to the crucial TSs. quantum mechanics ∣ reaction mechanism ∣ molecular motor T he F o F 1 -ATP synthase is vital for cell energy production, being responsible for most of the ATP synthesis in membranes of bacteria, chloroplasts, and mitochondria. This multisubunit en- zyme complex has high structural similarity among different species. It produces ATP as a biological nanomotor driven by an electrochemical potential difference across membranes as part of respiration or photosynthesis (1). The active sites are located in the F 1 domain, which in Escherichia coli consists of α 3 β 3 γδϵ subunits, three αβ-pairs surrounding a central α-helical coiled coil γ-subunit. The ATP synthesis ADP þ P i → ATP þ H 2 O [1] with P i ¼ H 2 PO 4 − , occurs at catalytic sites formed in the three β-subunits at the α/β-interfaces (2). The most prominent model assumes that the ATP-producing reaction occurs due to a rotation of the γ-shaft, inducing conformational changes in the surround- ing αβ-subunit pairs according to the unbinding and rotational coupling mechanism (3–7). It was also demonstrated that excess ATP may be hydrolyzed by isolated F 1 resulting in reverse rota- tion of the γ-subunit (7). In the first crystal structure of F 1 -ATPase (3), the three cata- lytic sites contained a nonhydrolyzable ATP analogue in one site, an ADP in the other, the third site being empty. Because of this observation, also supported by ATP binding affinity mea- surements (8, 9), the sites were named “tight binding site” (β TP ), “loose binding site” (β DP ), and “empty site” (β E ). According to the trisite mechanism model, during ATP synthesis each of the three αβ-subunit pairs changes conformation along the repeating cycle, β E → β DP → β TP → β E , where every step is associated with an approximately 120° rotation of the γ-subunit (2, 10–12). Later, these steps were further refined, and a “half-closed” (β HC ) con- formation was identified, representing the posthydrolysis, prepro- duct release state (12). Single-molecule studies showed that the 120° rotational steps can be further divided into approximately 90° and 30° substeps during hydrolysis (7, 10). A roughly 2-ms waiting time is observed between these substeps, most likely due to two separate 1-ms steps, one corresponding to the hydrolysis or synthesis step, the other to P i release or binding (10). Considering the function of the ATP synthase, structure deter- minations (12–16), mutation studies on the α-, β-, and γ-subunits (1, 17–20), single-molecule studies (6, 7, 10), and simulations (11, 21–27) have contributed to a rather detailed understanding of its mode of mechanical action. However, one of several issues that need to be better characterized (1) is the mechanistic resolution of the chemical reaction steps on the way from ADP to ATP at an atomic level. Performing this reaction efficiently is likely a key biochemical reason for the early evolutionary de- velopment of the enzyme complex, so understanding the detailed catalytic steps is most desirable. Investigation of reaction mechanisms at atomic level can be performed using quantum mechanical (QM) approaches (28), such as QM active site (29–31), and combined quantum me- chanics/molecular mechanics (QM/MM) methods (32–37). These ab initio or density functional theory (DFT) calculations, which in principle could describe reaction profiles with close to chemi- cal accuracy for small molecules, are computationally rather de- manding. Still, the ultimate goal when characterizing enzyme catalysis is to determine free energy changes along the reaction path. During the last two decades, several methods have emerged able to describe such free energy profiles, nevertheless, some challenges remain to be addressed (35–37). For instance, these methods require extensive configurational sampling of the enzy- matic environment which can be achieved by using molecular dynamics (MD). Combined QM/MM methods, which have the computational efficiency to afford MD sampling, usually treat the QM layer at a semiempirical level, and thus heavily rely on the reaction mechanism previously determined by more accurate ab initio or DFT QM methods. Recent advances toward free energy calculations with ab initio or DFT level in the QM layer make application of these rather promising in the future (35, 36). Considering phosphate ester hydrolysis, several alternative me- chanisms were characterized and the preferred reaction mechan- ism could even vary depending on the effect of the environment (38, 39). Partially due to this reason, proper characterization of the phosphoryl bond breaking represents a major challenge even in water, which was addressed successfully only recently (40), em- phasizing the importance of a detailed atomic level description of the reaction steps. Author contributions: T.B.-S., P.L., and B.N. designed research; T.B.-S. performed research; T.B.-S., P.L., and B.N. analyzed data; and T.B.-S. and B.N. wrote the paper. The authors declare no conflict of interest. *This Direct Submission article had a prearranged editor. 1 To whom correspondence may be addressed. E-mail: [email protected] or norden@ chalmers.se. This article contains supporting information online at www.pnas.org/lookup/suppl/ doi:10.1073/pnas.1010453108/-/DCSupplemental. 4828–4833 ∣ PNAS ∣ March 22, 2011 ∣ vol. 108 ∣ no. 12 www.pnas.org/cgi/doi/10.1073/pnas.1010453108
Transcript of Double-lock ratchet mechanism revealing the role of αSER-344 in ...

Double-lock ratchet mechanism revealing therole of αSER-344 in FoF1 ATP synthaseTamás Beke-Somfaia,b,1, Per Lincolna, and Bengt Nordéna,1
aDepartment of Chemical and Biological Engineering, Physical Chemistry, Chalmers University of Technology, SE-412 96 Göteborg, Sweden; andbProtein Modeling Group, Hungarian Academy of Sciences, Institute of Chemistry, Eötvös Loránd University, P.O. Box 32, H-1538 Budapest, Hungary
Edited* by Peter H. von Hippel, Institute of Molecular Biology, Eugene, OR, and approved February 8, 2011 (received for review August 5, 2010)
In a majority of living organisms, FoF1 ATP synthase performs thefundamental process of ATP synthesis. Despite the simple netreaction formula, ADPþ Pi → ATPþ H2O, the detailed step-by-stepmechanism of the reaction yet remains to be resolved owing tothe complexity of this multisubunit enzyme. Based on quantummechanical computations using recent high resolution X-ray struc-tures, we propose that during ATP synthesis the enzyme first pre-pares the inorganic phosphate for the γP-OADP bond-forming stepvia a double-proton transfer. At this step, the highly conservedαS344 side chain plays a catalytic role. The reaction thereafterprogresses through another transition state (TS) having a planarPO3
− ion configuration to finally form ATP. These two TSs are con-cluded crucial for ATP synthesis. Using stepwise scans and severalmodels of the nucleotide-bound active site, some of the mostimportant conformational changes were traced toward directionof synthesis. Interestingly, as the active site geometry progressestoward the ATP-favoring tight binding site, at both of these TSs,a dramatic increase in barrier heights is observed for the reversedirection, i.e., hydrolysis of ATP. This change could indicate a“ratchet” mechanism for the enzyme to ensure efficacy of ATPsynthesis by shifting residue conformation and thus locking accessto the crucial TSs.
quantum mechanics ∣ reaction mechanism ∣ molecular motor
The FoF1-ATP synthase is vital for cell energy production, beingresponsible for most of the ATP synthesis in membranes of
bacteria, chloroplasts, and mitochondria. This multisubunit en-zyme complex has high structural similarity among differentspecies. It produces ATP as a biological nanomotor driven by anelectrochemical potential difference across membranes as partof respiration or photosynthesis (1). The active sites are locatedin the F1 domain, which in Escherichia coli consists of α3β3γδϵsubunits, three αβ-pairs surrounding a central α-helical coiled coilγ-subunit. The ATP synthesis
ADPþ Pi → ATPþH2O [1]
with Pi ¼ H2PO4−, occurs at catalytic sites formed in the three
β-subunits at the α/β-interfaces (2). The most prominent modelassumes that the ATP-producing reaction occurs due to a rotationof the γ-shaft, inducing conformational changes in the surround-ing αβ-subunit pairs according to the unbinding and rotationalcoupling mechanism (3–7). It was also demonstrated that excessATP may be hydrolyzed by isolated F1 resulting in reverse rota-tion of the γ-subunit (7).
In the first crystal structure of F1-ATPase (3), the three cata-lytic sites contained a nonhydrolyzable ATP analogue in onesite, an ADP in the other, the third site being empty. Becauseof this observation, also supported by ATP binding affinity mea-surements (8, 9), the sites were named “tight binding site” (βTP),“loose binding site” (βDP), and “empty site” (βE). According tothe trisite mechanism model, during ATP synthesis each of thethree αβ-subunit pairs changes conformation along the repeatingcycle, βE → βDP → βTP → βE, where every step is associated withan approximately 120° rotation of the γ-subunit (2, 10–12). Later,these steps were further refined, and a “half-closed” (βHC) con-
formation was identified, representing the posthydrolysis, prepro-duct release state (12). Single-molecule studies showed that the120° rotational steps can be further divided into approximately90° and 30° substeps during hydrolysis (7, 10). A roughly 2-mswaiting time is observed between these substeps, most likely dueto two separate 1-ms steps, one corresponding to the hydrolysis orsynthesis step, the other to Pi release or binding (10).
Considering the function of the ATP synthase, structure deter-minations (12–16), mutation studies on the α-, β-, and γ-subunits(1, 17–20), single-molecule studies (6, 7, 10), and simulations(11, 21–27) have contributed to a rather detailed understandingof its mode of mechanical action. However, one of severalissues that need to be better characterized (1) is the mechanisticresolution of the chemical reaction steps on the way from ADPto ATP at an atomic level. Performing this reaction efficientlyis likely a key biochemical reason for the early evolutionary de-velopment of the enzyme complex, so understanding the detailedcatalytic steps is most desirable.
Investigation of reaction mechanisms at atomic level can beperformed using quantum mechanical (QM) approaches (28),such as QM active site (29–31), and combined quantum me-chanics/molecular mechanics (QM/MM)methods (32–37). Theseab initio or density functional theory (DFT) calculations, whichin principle could describe reaction profiles with close to chemi-cal accuracy for small molecules, are computationally rather de-manding. Still, the ultimate goal when characterizing enzymecatalysis is to determine free energy changes along the reactionpath. During the last two decades, several methods have emergedable to describe such free energy profiles, nevertheless, somechallenges remain to be addressed (35–37). For instance, thesemethods require extensive configurational sampling of the enzy-matic environment which can be achieved by using moleculardynamics (MD). Combined QM/MM methods, which have thecomputational efficiency to afford MD sampling, usually treatthe QM layer at a semiempirical level, and thus heavily rely onthe reaction mechanism previously determined by more accurateab initio or DFT QM methods. Recent advances toward freeenergy calculations with ab initio or DFT level in the QM layermake application of these rather promising in the future (35, 36).Considering phosphate ester hydrolysis, several alternative me-chanisms were characterized and the preferred reaction mechan-ism could even vary depending on the effect of the environment(38, 39). Partially due to this reason, proper characterization ofthe phosphoryl bond breaking represents a major challenge evenin water, which was addressed successfully only recently (40), em-phasizing the importance of a detailed atomic level description ofthe reaction steps.
Author contributions: T.B.-S., P.L., and B.N. designed research; T.B.-S. performed research;T.B.-S., P.L., and B.N. analyzed data; and T.B.-S. and B.N. wrote the paper.
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.1To whom correspondence may be addressed. E-mail: [email protected] or [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1010453108/-/DCSupplemental.
4828–4833 ∣ PNAS ∣ March 22, 2011 ∣ vol. 108 ∣ no. 12 www.pnas.org/cgi/doi/10.1073/pnas.1010453108

The complex function of ATP synthase makes this enzymespecial compared to many other enzymes and makes computa-tional investigation challenging: There are three active sites atwhich the reaction may take place and these are subject to con-formational changes during the revolving cycle. Significantvariations of reaction rate constants have been observed: LowATP concentrations are characterized by unisite catalysis (8, 9),whereas at physiological ATP concentrations multisite catalysistakes place (1, 2). An explanation proposed for the latter en-hancement in rate of catalysis is that, at multisite catalysis, thebinding of a second ATP to an empty site initiates such confor-mational changes that can assist the large, approximately 90°rotation substep of the γ-subunit (7, 11). Consequently, this rota-tion would change the conformation of the already closed activesite into a conformation where ATP can be more effectivelyhydrolyzed (i.e., most likely from βTP to βDP). These observationsmake the validation of the computationally determined reactionsteps challenging, because an acceptable mechanism should re-produce both unisite and multisite catalysis reasonably. In orderto understand the effects of the active site conformation along thereaction, models using both the βDP and βTP active sites as well asconformational changes between βHC, βDP, and βTP active siteswere considered. This approach was inspired by the pioneeringstudies by Dittrich et al. (21, 22) and Štrajbl et al. (27) on ATPasereaction mechanism. In the former, the authors used a lowerlevel of theory on a 70 atom QM part and a crystal structure withADP and not ATP in the active sites (15). In the latter, theauthors used a linear response approximation and the empiricalvalence bond (EVB) approach with parameters from the GTPhydrolysis mechanism of RasGap complex to evaluate the freeenergy profile of the ATP synthesis in the different conforma-tional states. They used structures which had a transition statemimic substrate, Mg2þ·ADP and aluminum fluoride in theiractive sites (12, 41).
We have here applied atomic positions obtained from a recenthigh resolution (1.9 Å) structure for the “ground state” of F1-ATPase (16) (ground F1), where βDP and βTP are occupied byan ATP analogue, adenosine 5'-[β;γ-imido]triphosphate (AMP-PNP). In this structure, the two sites show relatively small struc-tural variations for the involved residues and substrates, wherethe largest difference between the two sites is the approximate1.1-Å relative shift of αR373. Thus, along the minimum energypathway of [1], the same reaction steps were obtained in bothactive sites. Nevertheless, the relatively small shift of αR373 posi-tion had dramatic mechanistic consequences, as the γP-OADP
bond-forming or breaking step resulted in an approximate40 kJ∕mol difference in barrier between βDP and βTP (42). Oursimulations indicate that there are two crucial transition statesin the occupied sites: (i) the bond formation through a planarmetaphosphate ion (PO3
−) and (ii) a double-proton transfer,or transprotonation, on Pi, preparing the appropriate configura-tion for the γP-OADP bond formation (Fig. 1). In the latter,the highly conserved serine residue (αS344) is actively involved.Recent mutation studies focusing on this residue indicate thatit has a direct role in Pi binding and stabilization of the P-Obond-forming transition state (TS) (43). The obtained barrierheights can be compared to free energy values derived backfrom experimental rate constants of the partial reactions (9, 10,18–20) using classical transition state theory. The contribution ofthese two residues to TS stabilization was tested by performingstepwise scans on their positions along the βHC → βDP → βTPcycle using the structural information from available X-ray struc-tures. These calculations showed a monotonic increase of bothbarrier heights toward ATP hydrolysis. Considering results ofboth these systematic scans and the calculations in the βDP andβTP sites, the proposed minimum energy pathway qualitativelyreproduces the change in rate of catalysis between unisite andmultisite conditions.
Results and DiscussionInitial Steps: ADPþ Pi. To find the initial reactant state, out ofsix total possible spatial orientations of H2PO4
− in the active site,the four more probable were optimized. Their relative energiesindicate that the conformation with the Pi hydrogen bonded tothe ADP is the most stable one, which was also concluded byKarplus and coworkers (26). All energetic and structural proper-ties are detailed according to our largest model with 189 QMatoms (QM189), if not otherwise stated. Note also that althoughthe reaction steps are described toward synthesis, most of the ex-perimental kinetic data are available for the reverse reaction,ATP hydrolysis, thus energetic properties are mostly discussedtoward direction of hydrolysis.
In the initial state, I, the OγP⋯OADP distance is 2.6 Å in βDP,which suggests that the hydrogen bond between Pi and ADP isstrong (Fig. 1). As ATP synthesis is, in chemical terms, a dehy-dration, the apparent problem with such a configuration is thatthe proton between Pi and ADP not only separates the two ne-gatively charged fragments, but also makes a one-step formationof water unlikely. To achieve the product state ATPþH2O (IV),the proton between the two fragments needs to be transferred to
Fig. 1. Scheme showing critical points along minimum energy pathway toward ATP synthesis. Important atoms and nomenclature shown at initial state, I.Protons with active roles in the reaction are colored. The αSer344 side chain is colored red in Seron states. Critical point III is shown in Seron state (IIIon).
Beke-Somfai et al. PNAS ∣ March 22, 2011 ∣ vol. 108 ∣ no. 12 ∣ 4829
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
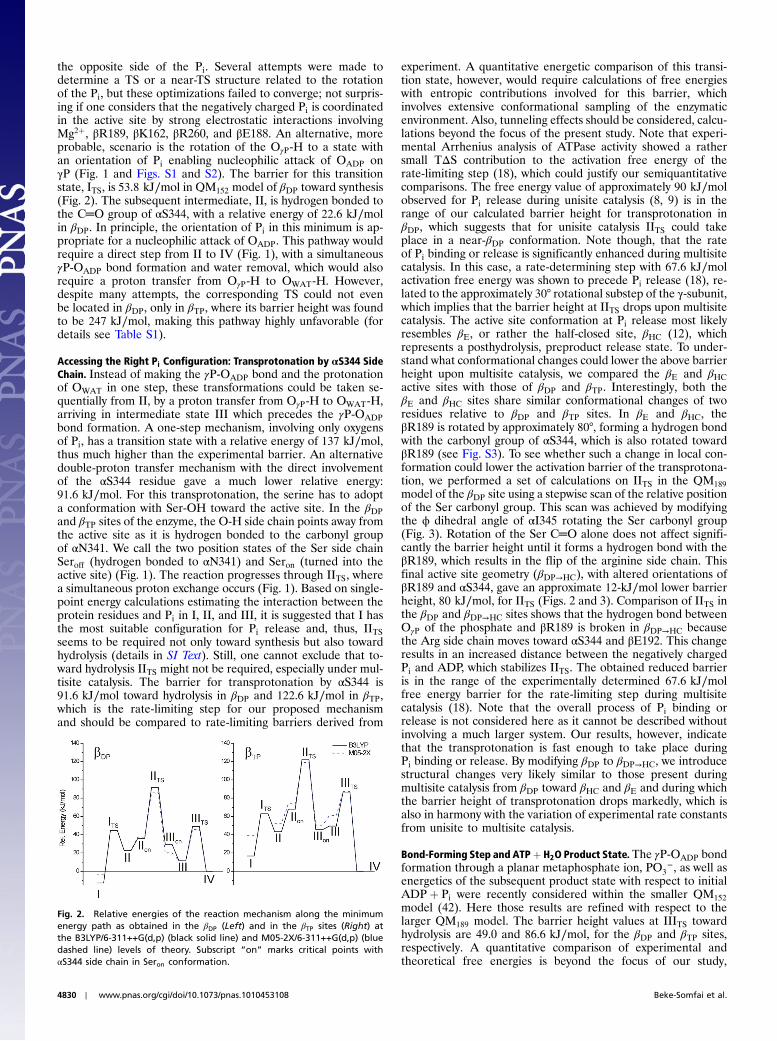
the opposite side of the Pi. Several attempts were made todetermine a TS or a near-TS structure related to the rotationof the Pi, but these optimizations failed to converge; not surpris-ing if one considers that the negatively charged Pi is coordinatedin the active site by strong electrostatic interactions involvingMg2þ, βR189, βK162, βR260, and βE188. An alternative, moreprobable, scenario is the rotation of the OγP-H to a state withan orientation of Pi enabling nucleophilic attack of OADP onγP (Fig. 1 and Figs. S1 and S2). The barrier for this transitionstate, ITS, is 53.8 kJ∕mol in QM152 model of βDP toward synthesis(Fig. 2). The subsequent intermediate, II, is hydrogen bonded tothe C═O group of αS344, with a relative energy of 22.6 kJ∕molin βDP. In principle, the orientation of Pi in this minimum is ap-propriate for a nucleophilic attack of OADP. This pathway wouldrequire a direct step from II to IV (Fig. 1), with a simultaneousγP-OADP bond formation and water removal, which would alsorequire a proton transfer from OγP-H to OWAT-H. However,despite many attempts, the corresponding TS could not evenbe located in βDP, only in βTP, where its barrier height was foundto be 247 kJ∕mol, making this pathway highly unfavorable (fordetails see Table S1).
Accessing the Right Pi Configuration: Transprotonation by αS344 SideChain. Instead of making the γP-OADP bond and the protonationof OWAT in one step, these transformations could be taken se-quentially from II, by a proton transfer from OγP-H to OWAT-H,arriving in intermediate state III which precedes the γP-OADPbond formation. A one-step mechanism, involving only oxygensof Pi, has a transition state with a relative energy of 137 kJ∕mol,thus much higher than the experimental barrier. An alternativedouble-proton transfer mechanism with the direct involvementof the αS344 residue gave a much lower relative energy:91.6 kJ∕mol. For this transprotonation, the serine has to adopta conformation with Ser-OH toward the active site. In the βDPand βTP sites of the enzyme, the O-H side chain points away fromthe active site as it is hydrogen bonded to the carbonyl groupof αN341. We call the two position states of the Ser side chainSeroff (hydrogen bonded to αN341) and Seron (turned into theactive site) (Fig. 1). The reaction progresses through IITS, wherea simultaneous proton exchange occurs (Fig. 1). Based on single-point energy calculations estimating the interaction between theprotein residues and Pi in I, II, and III, it is suggested that I hasthe most suitable configuration for Pi release and, thus, IITSseems to be required not only toward synthesis but also towardhydrolysis (details in SI Text). Still, one cannot exclude that to-ward hydrolysis IITS might not be required, especially under mul-tisite catalysis. The barrier for transprotonation by αS344 is91.6 kJ∕mol toward hydrolysis in βDP and 122.6 kJ∕mol in βTP,which is the rate-limiting step for our proposed mechanismand should be compared to rate-limiting barriers derived from
experiment. A quantitative energetic comparison of this transi-tion state, however, would require calculations of free energieswith entropic contributions involved for this barrier, whichinvolves extensive conformational sampling of the enzymaticenvironment. Also, tunneling effects should be considered, calcu-lations beyond the focus of the present study. Note that experi-mental Arrhenius analysis of ATPase activity showed a rathersmall TΔS contribution to the activation free energy of therate-limiting step (18), which could justify our semiquantitativecomparisons. The free energy value of approximately 90 kJ∕molobserved for Pi release during unisite catalysis (8, 9) is in therange of our calculated barrier height for transprotonation inβDP, which suggests that for unisite catalysis IITS could takeplace in a near-βDP conformation. Note though, that the rateof Pi binding or release is significantly enhanced during multisitecatalysis. In this case, a rate-determining step with 67.6 kJ∕molactivation free energy was shown to precede Pi release (18), re-lated to the approximately 30° rotational substep of the γ-subunit,which implies that the barrier height at IITS drops upon multisitecatalysis. The active site conformation at Pi release most likelyresembles βE, or rather the half-closed site, βHC (12), whichrepresents a posthydrolysis, preproduct release state. To under-stand what conformational changes could lower the above barrierheight upon multisite catalysis, we compared the βE and βHCactive sites with those of βDP and βTP. Interestingly, both theβE and βHC sites share similar conformational changes of tworesidues relative to βDP and βTP sites. In βE and βHC, theβR189 is rotated by approximately 80°, forming a hydrogen bondwith the carbonyl group of αS344, which is also rotated towardβR189 (see Fig. S3). To see whether such a change in local con-formation could lower the activation barrier of the transprotona-tion, we performed a set of calculations on IITS in the QM189
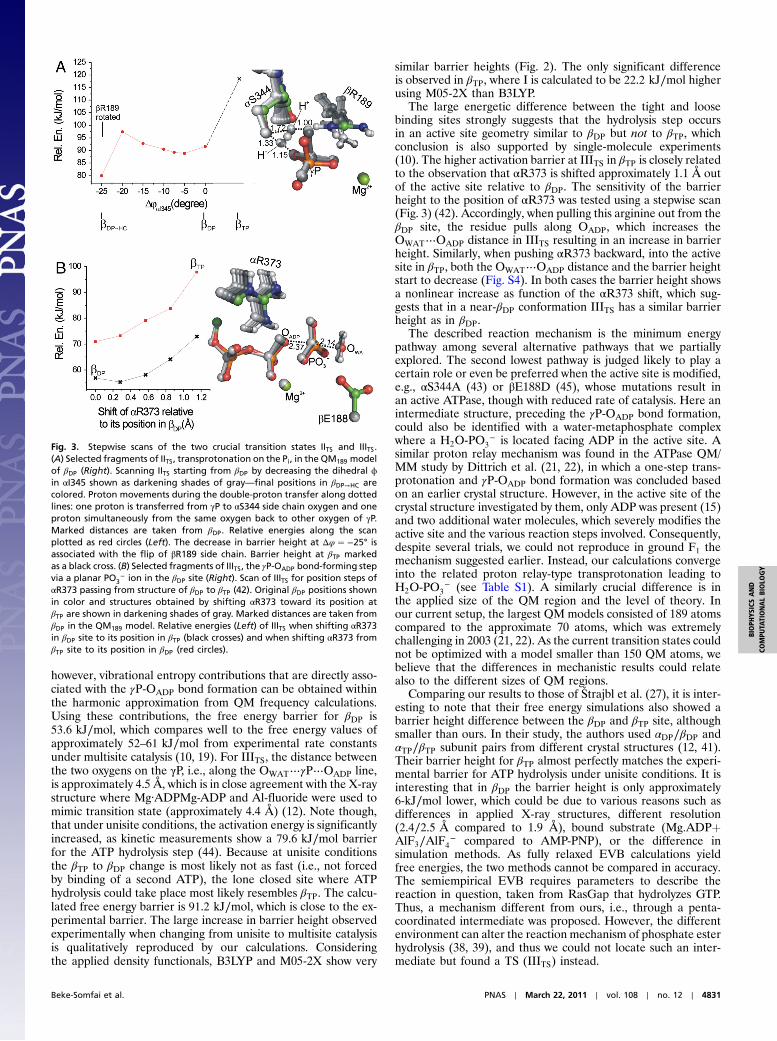
model of the βDP site using a stepwise scan of the relative positionof the Ser carbonyl group. This scan was achieved by modifyingthe ϕ dihedral angle of αI345 rotating the Ser carbonyl group(Fig. 3). Rotation of the Ser C═O alone does not affect signifi-cantly the barrier height until it forms a hydrogen bond with theβR189, which results in the flip of the arginine side chain. Thisfinal active site geometry (βDP→HC), with altered orientations ofβR189 and αS344, gave an approximate 12-kJ∕mol lower barrierheight, 80 kJ∕mol, for IITS (Figs. 2 and 3). Comparison of IITS inthe βDP and βDP→HC sites shows that the hydrogen bond betweenOγP of the phosphate and βR189 is broken in βDP→HC becausethe Arg side chain moves toward αS344 and βE192. This changeresults in an increased distance between the negatively chargedPi and ADP, which stabilizes IITS. The obtained reduced barrieris in the range of the experimentally determined 67.6 kJ∕molfree energy barrier for the rate-limiting step during multisitecatalysis (18). Note that the overall process of Pi binding orrelease is not considered here as it cannot be described withoutinvolving a much larger system. Our results, however, indicatethat the transprotonation is fast enough to take place duringPi binding or release. By modifying βDP to βDP→HC, we introducestructural changes very likely similar to those present duringmultisite catalysis from βDP toward βHC and βE and during whichthe barrier height of transprotonation drops markedly, which isalso in harmony with the variation of experimental rate constantsfrom unisite to multisite catalysis.
Bond-Forming Step and ATPþ H2O Product State.The γP-OADP bondformation through a planar metaphosphate ion, PO3
−, as well asenergetics of the subsequent product state with respect to initialADPþ Pi were recently considered within the smaller QM152
model (42). Here those results are refined with respect to thelarger QM189 model. The barrier height values at IIITS towardhydrolysis are 49.0 and 86.6 kJ∕mol, for the βDP and βTP sites,respectively. A quantitative comparison of experimental andtheoretical free energies is beyond the focus of our study,
Fig. 2. Relative energies of the reaction mechanism along the minimumenergy path as obtained in the βDP (Left) and in the βTP sites (Right) atthe B3LYP/6-311++G(d,p) (black solid line) and M05-2X/6-311++G(d,p) (bluedashed line) levels of theory. Subscript “on” marks critical points withαS344 side chain in Seron conformation.
4830 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1010453108 Beke-Somfai et al.
however, vibrational entropy contributions that are directly asso-ciated with the γP-OADP bond formation can be obtained withinthe harmonic approximation from QM frequency calculations.Using these contributions, the free energy barrier for βDP is53.6 kJ∕mol, which compares well to the free energy values ofapproximately 52–61 kJ∕mol from experimental rate constantsunder multisite catalysis (10, 19). For IIITS, the distance betweenthe two oxygens on the γP, i.e., along the OWAT⋯γP⋯OADP line,is approximately 4.5 Å, which is in close agreement with the X-raystructure where Mg·ADPMg-ADP and Al-fluoride were used tomimic transition state (approximately 4.4 Å) (12). Note though,that under unisite conditions, the activation energy is significantlyincreased, as kinetic measurements show a 79.6 kJ∕mol barrierfor the ATP hydrolysis step (44). Because at unisite conditionsthe βTP to βDP change is most likely not as fast (i.e., not forcedby binding of a second ATP), the lone closed site where ATPhydrolysis could take place most likely resembles βTP. The calcu-lated free energy barrier is 91.2 kJ∕mol, which is close to the ex-perimental barrier. The large increase in barrier height observedexperimentally when changing from unisite to multisite catalysisis qualitatively reproduced by our calculations. Consideringthe applied density functionals, B3LYP and M05-2X show very
similar barrier heights (Fig. 2). The only significant differenceis observed in βTP, where I is calculated to be 22.2 kJ∕mol higherusing M05-2X than B3LYP.
The large energetic difference between the tight and loosebinding sites strongly suggests that the hydrolysis step occursin an active site geometry similar to βDP but not to βTP, whichconclusion is also supported by single-molecule experiments(10). The higher activation barrier at IIITS in βTP is closely relatedto the observation that αR373 is shifted approximately 1.1 Å outof the active site relative to βDP. The sensitivity of the barrierheight to the position of αR373 was tested using a stepwise scan(Fig. 3) (42). Accordingly, when pulling this arginine out from theβDP site, the residue pulls along OADP, which increases theOWAT⋯OADP distance in IIITS resulting in an increase in barrierheight. Similarly, when pushing αR373 backward, into the activesite in βTP, both the OWAT⋯OADP distance and the barrier heightstart to decrease (Fig. S4). In both cases the barrier height showsa nonlinear increase as function of the αR373 shift, which sug-gests that in a near-βDP conformation IIITS has a similar barrierheight as in βDP.
The described reaction mechanism is the minimum energypathway among several alternative pathways that we partiallyexplored. The second lowest pathway is judged likely to play acertain role or even be preferred when the active site is modified,e.g., αS344A (43) or βE188D (45), whose mutations result inan active ATPase, though with reduced rate of catalysis. Here anintermediate structure, preceding the γP-OADP bond formation,could also be identified with a water-metaphosphate complexwhere a H2O-PO3
− is located facing ADP in the active site. Asimilar proton relay mechanism was found in the ATPase QM/MM study by Dittrich et al. (21, 22), in which a one-step trans-protonation and γP-OADP bond formation was concluded basedon an earlier crystal structure. However, in the active site of thecrystal structure investigated by them, only ADP was present (15)and two additional water molecules, which severely modifies theactive site and the various reaction steps involved. Consequently,despite several trials, we could not reproduce in ground F1 themechanism suggested earlier. Instead, our calculations convergeinto the related proton relay-type transprotonation leading toH2O-PO3
− (see Table S1). A similarly crucial difference is inthe applied size of the QM region and the level of theory. Inour current setup, the largest QM models consisted of 189 atomscompared to the approximate 70 atoms, which was extremelychallenging in 2003 (21, 22). As the current transition states couldnot be optimized with a model smaller than 150 QM atoms, webelieve that the differences in mechanistic results could relatealso to the different sizes of QM regions.
Comparing our results to those of Štrajbl et al. (27), it is inter-esting to note that their free energy simulations also showed abarrier height difference between the βDP and βTP site, althoughsmaller than ours. In their study, the authors used αDP∕βDP andαTP∕βTP subunit pairs from different crystal structures (12, 41).Their barrier height for βTP almost perfectly matches the experi-mental barrier for ATP hydrolysis under unisite conditions. It isinteresting that in βDP the barrier height is only approximately6-kJ∕mol lower, which could be due to various reasons such asdifferences in applied X-ray structures, different resolution(2.4∕2.5 Å compared to 1.9 Å), bound substrate (Mg:ADPþAlF3∕AlF4
− compared to AMP-PNP), or the difference insimulation methods. As fully relaxed EVB calculations yieldfree energies, the two methods cannot be compared in accuracy.The semiempirical EVB requires parameters to describe thereaction in question, taken from RasGap that hydrolyzes GTP.Thus, a mechanism different from ours, i.e., through a penta-coordinated intermediate was proposed. However, the differentenvironment can alter the reaction mechanism of phosphate esterhydrolysis (38, 39), and thus we could not locate such an inter-mediate but found a TS (IIITS) instead.
Fig. 3. Stepwise scans of the two crucial transition states IITS and IIITS.(A) Selected fragments of IITS, transprotonation on the Pi, in the QM189 modelof βDP (Right). Scanning IITS starting from βDP by decreasing the dihedral ϕin αI345 shown as darkening shades of gray—final positions in βDP→HC arecolored. Proton movements during the double-proton transfer along dottedlines: one proton is transferred from γP to αS344 side chain oxygen and oneproton simultaneously from the same oxygen back to other oxygen of γP.Marked distances are taken from βDP. Relative energies along the scanplotted as red circles (Left). The decrease in barrier height at Δφ ¼ −25° isassociated with the flip of βR189 side chain. Barrier height at βTP markedas a black cross. (B) Selected fragments of IIITS, the γP-OADP bond-forming stepvia a planar PO3
− ion in the βDP site (Right). Scan of IIITS for position steps ofαR373 passing from structure of βDP to βTP (42). Original βDP positions shownin color and structures obtained by shifting αR373 toward its position atβTP are shown in darkening shades of gray. Marked distances are taken fromβDP in the QM189 model. Relative energies (Left) of IIITS when shifting αR373in βDP site to its position in βTP (black crosses) and when shifting αR373 fromβTP site to its position in βDP (red circles).
Beke-Somfai et al. PNAS ∣ March 22, 2011 ∣ vol. 108 ∣ no. 12 ∣ 4831
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY

Role of αS344 Residue: Theoretical Results Compared to Experimental.As noticed recently by Li et al. (43) the serine residue is highlyconserved and thus may have a direct role in the reactionmechanism. They show that point mutations of αS347Q andαS347A have dramatic effects on ATP synthase function bothfor synthesis and hydrolysis. The inactivity of the 347Q mutantmay be understood because a larger side chain may stericallydistort the orientation of Pi. By contrast, in the correspondingalanine mutant there is no such sterical hindrance. Also, the smal-ler side chain could allow the presence of additional water mo-lecules in the active site. Thus, the reduced, albeit still somehowretained activity might be due to an alternative parallel, slowerpathway with a water-aided proton relay mechanism (21, 22)(Table S1). Clarification of such a mutation by theoretical meth-ods is beyond our current focus.
According to the ground F1 structure, the side chain of theserine residue, AMP-PNP, and the nucleophilic water are or-iented along the line of the γP-OADP bond-forming or breakingstep, and the Ser side chain is close enough to reach the activesite. In both the βDP and βTP sites of ground F1 this side chain inthe Seroff state is forming a strong hydrogen bond with the car-bonyl group of αN341. In βE, the OH side chain occupies both theSeroff and Seron conformations. Superimposition and backbonedihedral analyses of the α341NVISITD347 helical regions fromthe three α-subunits indicate that in αE the helical motif is elon-gated because both αS344 and αI345 are closer to the optimalhelical conformations (16) (Fig. S3). Thus, the peptide group be-tween αS344 and αI345 most likely competes with the Ser sidechain to form a hydrogen bond with αN341. X-ray structuresof F1-ATPases show that the corresponding serine side chainin the α-subunit is mostly found in the Seroff state (12, 15, 16)(Table S2). However, this serine is in the Seron state in two crystalstructures of rat liver F1-ATPase: one crystallized with ADPand Pi in the active site (13) and the other one crystallized witha transition state mimicked by vanadate and ADP in the activesite (14). Although the preferred state of the Ser side chainshould be investigated by free energy calculations, and thusremains to be settled, the structural results discussed above in-deed support that the αS344 side chain can be located in boththe Seroff and Seron states in the enzyme.
Rotatory Function and Reaction Mechanism. Considering the twoATP-containing sites in ground F1, the same reaction steps canbe envisaged to occur in both βDP and βTP, however, with differ-ent structural and energetic properties (Fig. 2 and Table S1). Therelative energy difference between I and IV shows a 12.8 kJ∕molpreference for hydrolysis in βDP and a 16.7 kJ∕mol preferencefor synthesis in βTP (Fig. 2). This observation is in harmonywith the preference of ATP in the tight binding site (3) and alsowith single-molecule experiments which indicate that hydrolysistakes place after an approximately 90° rotation of the γ-subunitwhen the occupied site is in a near-βDP conformation (7). Towardsynthesis, IITS and IIITS show very similar barrier heights in bothsites (for IITS 104.4 and 105.9 kJ∕mol, for IIITS 61.8 and69.9 kJ∕mol for βDP and βTP, respectively). In sharp contrast,however, in direction toward hydrolysis the barriers are muchhigher for βTP than for βDP. The difference for the γP-OADPbond-forming or breaking step, IIITS, is approximately 40 kJ∕mol(42) (Figs. 2 and 3). The activation barrier difference is approxi-mately 30 kJ∕mol for IITS between βDP and βTP, which becomeseven larger, approximately 43 kJ∕mol, if βDP→HC and βTP arecompared.
In conclusion, our results compared with experimental kineticdata suggest that, upon multisite catalysis, IIITS is located in anear-βDP conformation somewhere between changing from βTPto βDP, whereas IITS is in a conformation between βDP andβHC, closely related to Pi release or binding. Because the se-quence of the four active site conformations considered here
is most likely βTP → βDP → βHC → βE toward direction of hydro-lysis (12, 25), we conclude that IIITS and IITS are most likelysomewhat separated in time by the conformational changes ofthe active site. We notice that hydrolysis of ATP takes place afteran approximately 90° substep of the γ-subunit (10) and Pi releasealso takes place following an approximately 90° γ-subunit substep(10, 20). In view of our current results, it thus seems logical that,in a selected active site, ATP hydrolysis and Pi release requiretwo separate 90° γ-subunit steps or, in other words, during the2-ms waiting dwells these occur approximately in synchrony,but in different active sites. The concept of the two steps occur-ring in different sites during the 2-ms dwell between approxi-mately 90° and 30° substeps was earlier discussed by Adachi et al.(10) although disapproved due to its complexity.
Considering the progress of the reaction toward synthesis,along the βE → βHC → βDP conformational cycle of the activesite, Pi and ADP are bound, and the αS344-involved transproto-nation occurs. When arriving at the βDP conformation, the posi-tioning of the αS344 (and the βR189) is changed, resulting inan increase of the barrier by approximately 12 kJ∕mol towardthe reverse direction, partially locking IITS. Along the changefrom βDP to βTP, IIITS, the P-O bond-forming step, takes placenear βDP, resulting in ATPþH2O in the active site. As the posi-tion of αR373 approaches that in βTP, the barrier height for thebackward hydrolysis step monotonically increases. At βTP, bothIITS and IIITS are effectively locked toward hydrolysis as thebarrier height at IITS increases by an overall of approximately43 kJ∕mol, and at IIITS by approximately 40 kJ∕mol duringthe conformational changes of the active site.
The reaction steps obtained in an active site from high-resolu-tion structures are often useful as a first approach to interpretthe mechanism of an enzyme. However, in order to understandthe mechanism of ATP synthase, it is necessary to resolve alsothe conformational changes of the active sites coupled to theγ-subunit rotation. Accordingly, by following the changes inthe active site geometry and performing stepwise scans of thetwo crucial TSs, we have shown how the positions of αS344 andαR373 may drastically influence the rate and, in this way, attenu-ate the reversal of these reaction steps. To have a quantitativedescription, it is crucial to characterize free energy profiles ofthe reaction mechanism proposed here. The role of the serineside chain could be similar in other motor ATPases, such asRecA-DNA where crystal structures show a side-chain conforma-tional change upon ATP binding.
MethodsComputational Details. All calculations on the active site models wereperformed using the Gaussian 09 software package (46). The QM modelsof βDP and βTP active sites (QM) involving 106 (QM106), 128 (QM128), 152(QM152), and 189 atoms (QM189) were submitted to geometry optimizationsat the B3LYP/6-31G(d) level of theory (For details see Fig. S5 and SI Text). En-ergies were corrected using point energy calculations at the B3LYP/6-311++G(d,p) level of theory. Polarization effects of the protein environment wereconsidered using the integral equation formalism for the polarizable conti-nuummodel continuummodel, with a dielectric constant of four and solventradius of 2.5 Å. Zero point energy (ZPE) correction and thermal corrections toenergy were acquired from frequency calculations on the QM152 models,which were also used to confirm the nature of the critical points. ForQM189, the ZPE and thermal correction terms were assumed to be similaras in QM152. Note that, due to several constrained Cartesian coordinatesin the larger active site models, some additional imaginary frequenciesoccurred, which were low and thus not contributing significantly to ZPE.However, they would render harmonic entropy contributions unreliable, thusthese were estimated for IIITS and IV from QM128 models. The active siteswere also investigated in the larger protein environment by models usingthe ONIOM (our own N-layered integrated molecular orbitalþmolecularmechanics) integrated QM/MM method (47) with a B3LYP/6-31G(d):AM-BER setup.
The smallest QM models, QM106, were compared with the ONIOM QM/MM models (QM106∕MM), which had the same QM atoms as in QM106.QM106 does not include the P loop and has a net one positive charge. For
4832 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1010453108 Beke-Somfai et al.

the QM106∕MMmodel, residues within 20-Å distance from theMg2þ ion wereincluded in the MM layer, and all atoms which were farther than 15 Å fromthe Mg2þ ion were kept frozen. The QM106∕MM model consisted of 4,626atoms for βTP and 4,829 atoms for βDP site. The protonation state of theresidues was investigated in a similar structure in previous studies, whereusual residue pKa values were found (26). The initial state of Pi was supposedto be a doubly protonated, H2PO4
−, as indicated earlier (26). Hydrogenswere added using the tool PROTONATE of the AMBER software package(48). Finally, the protein-Pi interactions were estimated in I, II, and III bysingle-point calculations [B3LYP/6-311++G(d,p)] in a modified QM152 (seeSI Text). Parts of Fig. 3 were made using PyMOL (49).
Precision and Accuracy. B3LYP is one of the most used method in QM regionsfor enzymatic catalysis (28, 31, 35). Nevertheless, its performance on phos-phate ester hydrolysis was tested earlier (42) against higher level theoreticalapproaches which showed excellent agreement regarding the applied basisset and level of theory. Quality of the current results was also estimated herewith calculations varying model size and the larger environment. In addition,the obtained critical points were also calculated using the M05-2X functional(50), with the 6-311++G(d,p) basis set, which resulted a similar reaction pro-file as with B3LYP (Fig. 2 and Table S1). Thus only B3LYP values are discussedunless noted otherwise.
These were supplemented here by calculating hydrolysis of ATPγS, whichhydrolyzes approximately 70 times slower than ATP. This relative barrierheight difference was reproduced with nearly chemical accuracy. Furtheron, structural and energetic differences between a QM106 active site modeland QM106∕MM model were investigated in detail. Both systems with 106QM atoms showed severe limitations in describing the reaction mechanism,which, comparing results to those obtained with QM152 and QM189, suggeststhat the presence of the P loop in the QM region is crucial. The main reasonfor incorporating QM/MM (ONIOM) calculations is to determine structuralshifts in the active site during a fully relaxed optimization, as well as to assessthe small differences between βTP and βDP. The two sites show no large var-iation and the differences between them remain almost the same as in theoriginal X-ray structure, which validates our larger QM models (see SI Textand Table S3).
ACKNOWLEDGMENTS. This work is funded by King Abdullah University of
Science and Technology (Grant KUK-11-008-23). The Eötvös Universitycomputer facility, the High Performance Computing Group (University ofSzeged), and the Swedish National Infrastructure for Computing resourceswere used for calculations.
1. Junge W, Sielaff H, Engelbrecht S (2009) Torque generation and elastic power trans-mission in the rotary F0F1-ATPase. Nature 459:364–370.
2. Senior AE, Nadanaciva S, Weber J (2002) The molecular mechanism of ATP synthesis byF1F0-ATP synthase. Biochim Biophys Acta, Bioenerg 1553:188–211.
3. Abrahams JP, Leslie AGW, Lutter R, Walker JE (1994) Structure at 2.8-angstrom resolu-tion of F1-ATPase from bovine heart-mitochondria. Nature 370:621–628.
4. Boyer PD (2000) Catalytic site forms and controls in ATP synthase catalysis. BiochimBiophys Acta, Bioenerg 1458:252–262.
5. Cox GB, Jans DA, Fimmel AL, Gibson F, Hatch L (1984) Hypothesis—the mechanism ofATP synthase—conformational change by rotation of the B-subunit. Biochim BiophysActa 768:201–208.
6. Noji H, Yasuda R, Yoshida M, Kinosita K (1997) Direct observation of the rotation ofF1-ATPase. Nature 386:299–302.
7. Yasuda R, Noji H, Yoshida M, Kinosita K, Itoh H (2001) Resolution of distinct rotationalsubsteps by submillisecond kinetic analysis of F1-ATPase. Nature 410:898–904.
8. Penefsky HS (1986) Rate constants and equilibrium-constants for the elementary stepsof ATP hydrolysis by beef-heart mitochondrial ATPase.Methods Enzymol 126:608–618.
9. Senior AE (1992) Catalytic sites of Escherichia coli F1-ATPase. J Bioenerg Biomembr24:479–484.
10. Adachi K, et al. (2007) Coupling of rotation and catalysis in F1-ATPase revealed bysingle-molecule imaging and manipulation. Cell 130:309–321.
11. Gao YQ, Yang W, Karplus M (2005) A structure-based model for the synthesis andhydrolysis of ATP by F1-ATPase. Cell 123:195–205.
12. Menz RI, Walker JE, Leslie AGW (2001) Structure of bovine mitochondrial Fl-ATPasewith nucleotide bound to all three catalytic sites: Implications for the mechanismof rotary catalysis. Cell 106:331–341.
13. Bianchet MA, Hullihen J, Pedersen PL, Amzel LM (1998) The 2.8-angstrom structure ofrat liver F1-ATPase: Configuration of a critical intermediate in ATP synthesis/hydrolysis.Proc Natl Acad Sci USA 95:11065–11070.
14. Chen C, et al. (2006) Mitochondrial ATP synthase—crystal structure of the catalyticF1 unit in a vanadate-induced transition-like state and implications for mechanism.J Biol Chem 281:13777–13783.
15. Gibbons C, MontgomeryMG, Leslie AGW,Walker JE (2000) The structure of the centralstalk in bovine F1-ATPase at 2.4 angstrom resolution. Nat Struct Biol 7:1055–1061.
16. Bowler MW, Montgomery MG, Leslie AGW, Walker JE (2007) Ground state structureof F1-ATPase from bovine heart mitochondria at 19 A resolution. J Biol Chem282:14238–14242.
17. Mnatsakanyan N, Hook JA, Quisenberry L, Weber J (2009) ATP synthase with itsgamma subunit reduced to the N-terminal helix can still catalyze ATP synthesis. J BiolChem 284:26519–26525.
18. Mnatsakanyan N, Krishnakumar AM, Suzuki T, Weber J (2009) The role of the betaDELSEED-loop of ATP synthase. J Biol Chem 284:11336–11345.
19. Scanlon JAB, Al-Shawi MK, Le NP, Nakamoto RK (2007) Determination of the partialreactions of rotational catalysis in F1-ATPase. Biochemistry 46:8785–8797.
20. Scanlon JAB, Al-Shawi MK, Nakamoto RK (2008) A rotor-stator cross-link in the F1-AT-Pase blocks the rate-limiting step of rotational catalysis. J Biol Chem 283:26228–26240.
21. Dittrich M, Hayashi S, Schulten K (2003) On the mechanism of ATP hydrolysis inF1-ATPase. Biophys J 85:2253–2266.
22. Dittrich M, Hayashi S, Schulten K (2004) ATP hydrolysis in the beta(TP) and beta(DP)catalytic sites of F1-ATPase. Biophys J 87:2954–2967.
23. Koga N, Takada S (2006) Folding-based molecular simulations reveal mechanisms ofthe rotary motor F1-ATPase. Proc Natl Acad Sci USA 103:5367–5372.
24. Ma JP, et al. (2002) A dynamic analysis of the rotation mechanism for conformationalchange in F1-ATPase. Structure 10:921–931.
25. Pu JZ, Karplus M (2008) How subunit coupling produces the gamma-subunit rotarymotion in F1-ATPase. Proc Natl Acad Sci USA 105:1192–1197.
26. Yang W, Gao YQ, Cui Q, Ma J, Karplus M (2003) The missing link between thermo-dynamics and structure in F1-ATPase. Proc Natl Acad Sci USA 100:874–879.
27. Štrajbl M, Shurki A, Warshel A (2003) Converting conformational changes to electro-static energy in molecular motors: The energetics of ATP synthase. Proc Natl Acad SciUSA 100:14834–14839.
28. Friesner RA, Guallar V (2005) Ab initio quantum chemical and mixed quantummechanics/molecular mechanics (QM/MM) methods for studying enzymatic catalysis.Annu Rev Phys Chem 56:389–427.
29. Noodleman L, Lovell T, Han WG, Li J, Himo F (2004) Quantum chemical studies ofintermediates and reaction pathways in selected enzymes and catalytic syntheticsystems. Chem Rev 104:459–508.
30. Ramos MJ, Fernandes PA (2008) Computational enzymatic catalysis. Acc Chem Res41:689–698.
31. Siegbahn PEM, Himo F (2009) Recent developments of the quantum chemical clusterapproach for modeling enzyme reactions. J Biol Inorg Chem 14:643–651.
32. Klahn M, Braun-Sand S, Rosta E, Warshel A (2005) On possible pitfalls in ab initioquantum mechanics/molecular mechanics minimization approaches for studies ofenzymatic reactions. J Phys Chem B 109:15645–15650.
33. Lin H, Truhlar DG (2007) QM/MM:What have we learned, where are we, and where dowe go from here? Theor Chem Acc 117:185–199.
34. Lundberg M, Kawatsu T, Vreven T, Frisch MJ, Morokuma K (2009) Transition states in aprotein environment—ONIOM QM:MM modeling of isopenicillin N synthesis. J ChemTheory Comput 5:222–234.
35. Senn HM, Thiel W (2009) QM/MMmethods for biological systems. Angew Chem Int Ed48:1198–1229.
36. Rosta E, Klähn M, Warshel A (2006) Towards accurate ab initio QM/MM free-energyprofiles of enzymatic reactions. J Phys Chem B 110:2934–2941.
37. Hu H, Yang W (2008) Free energies of chemical reactions in solution and in enzymeswith ab initio quantum mechanics/molecular mechanics methods. Annu Rev PhysChem 59:573–601.
38. Klähn M, Rosta E, Warshel A (2006) On the mechanism of hydrolysis of phosphatemonoesters dianions in solutions and proteins. J Am Chem Soc 128:15310–15323.
39. Marcos E, Crehuet R, Anglada JM (2008) Inductive and external electric field effects inpentacoordinated phosphorous compounds. J Chem Theory Comput 4:49–63.
40. Kamerlin SCL, Warshel A (2009) On the energetics of ATP hydrolysis in solution. J PhysChem B 113:15692–15698.
41. Braig K, Menz RI, Montgomery MG, Leslie AGW, Walker JE (2000) Structure of bovinemitochondrial F1-ATPase inhibited by Mg2þADP and aluminium fluoride. Structure8:567–573.
42. Beke-Somfai T, Lincoln P, Nordén B (2010) Mechanical control of ATP synthase func-tion: Activation energy difference between tight and loose binding sites. Biochemistry49:401–403.
43. Li WZ, Brudecki LE, Senior AE, Ahmad Z (2009) Role of alpha-subunit VISIT-DGsequence residues Ser-347 and Gly-351 in the catalytic sites of Escherichia coli ATPsynthase. J Biol Chem 284:10747–10754.
44. Al-Shawi MK, Parsonage D, Senior AE (1990) Thermodynamic analyses of the catalyticpathway of F1-ATPase from Escherichia coli. J Biol Chem 265:4402–4410.
45. Masaike T, Muneyuki E, Noji H, Kinosita K, Yoshida M (2002) F1-ATPase changes itsconformations upon phosphate release. J Biol Chem 277:21643–21649.
46. Frisch MJ, et al. (2009) Gaussian 09, Rev. A.1 (Gaussian, Inc, Wallingford, CT).47. Vreven T, et al. (2006) Combining quantum mechanics methods with molecular
mechanics methods in ONIOM. J Chem Theory Comput 2:815–826.48. Case DA, et al. (2010) AMBER 11 (Univ of California, San Francisco).49. DeLano WL (2002) The PyMOL Molecular Graphics System (DeLano Scientific, Palo
Alto, CA).50. Zhao Y, Schultz NE, Truhlar DG (2006) Design of density functionals by combining
the method of constraint satisfaction with parametrization for thermochemistry, the-mochemical kinetics, and noncovalent interactions. J Chem Theory Comput 2:364–382.
Beke-Somfai et al. PNAS ∣ March 22, 2011 ∣ vol. 108 ∣ no. 12 ∣ 4833
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY

Supporting InformationBeke-Somfai et al. 10.1073/pnas.1010453108SI TextEstimating the Pi-Protein Interactions. In principle, not all stepsseem necessary toward hydrolysis, as Pi release could occur be-fore transprotonation, i.e., in intermediate states II or III already.In order to obtain a rough estimate of which Pi configurationwould be preferred for Pi release, we considered that Pi releasehas been shown to take place before release of ADP (1), mostlikely in a conformation similar to the half-closed binding site(βHC). In the latter, ADP and Pi (SO4
2− in the crystal structure)are already significantly separated from one another, the γP⋯βPdistance being approximately 5.4 Å and γP⋯Mg2þ 5 Å (2). It isreasonable that Pi release in this conformation depends mainlyon its interactions with the active site residues. The protein-Piinteractions were estimated in I, II, and III by single-pointcalculations [B3LYP/6-311++G(d,p)] in a modified quantummechanical QM152, from which ADP and Mg2þ were deleted. Piconfigurations in II and III showed 38 and 123.5 kJ∕mol extrastabilization relative to I, suggesting that I is the most appropriatestate for Pi release, but more importantly the very low energy ofthe protein-Pi configuration in III indicates that toward directionof hydrolysis the Pi release would take place after transprotona-tion by αS344, (i.e., in either I or II).
Precision and Accuracy. In general, the quality of the computationalresults in a mechanistic study depends on several parameters, forexample, on the quality and size of the models used to describethe active site geometry, on the accuracy of the chosen theoreticalapproach on the reaction mechanism, on the effect of the largerenzymatic environment, or on the enzyme’s conformational fluc-tuations along the reaction. Calculations varying these factors cangive a reasonable guess for the optimal approach and help toavoid severe artifacts which would influence conclusions. There-fore, a comparison was performed on the accuracy of the differ-ent models and theoretical approaches applied here. Resultsfrom test calculations obtained earlier (3) are also describedbriefly.
Basis Sets, Density Functional Theory Methods.The basis set used foroptimization, 6-31G(d), was tested by calculations made on theATP and ADPþ Pi states in the loose binding site (βDP) at theB3LYP/6-31G(d,p) level of theory and the structural differenceswere found to be negligible. Single-point energies obtained at theB3LYP/6-311++G(d,p) level of theory in QM152, were comparedto energetic properties calculated using the M05-2X functionaland the same basis set. This functional was described to performbetter than B3LYP in estimating barrier heights for bond disso-ciation. Relative energies for the γP-OADP bond-forming orbreaking TS in βDP and in the tight binding site (βTP) usingthe two above functionals were within 5 kJ∕mol.
In addition, the M05-2X was applied for the QM189 models aswell to describe the reaction steps in both βDP and βTP (Fig. 2).
B3LYP on Phosphate-Ester Hydrolysis. Further on, to see the accu-racy of hybrid density functional B3LYP applied to the magne-sium-coordinated phosphate-ester hydrolysis, a smaller neutralmodel was built, ðMg2þ½H2O�4½H2P2O7
2−�Þ þH2O, and criticalpoints along the reaction coordinate were calculated at theB3LYP/6-31+G(d,p) andMP2/6-31+G(d,p) levels of theory. Thestructural and energetic properties obtained with the B3LYPfunctional showed an excellent correlation with second-orderMøller–Plesset (MP2) calculations (3).
Relative Accuracy with ATPγS. The previously obtained barrierheight for ATP hydrolysis is here compared to hydrolysis ofATPγS. ATPγS is known to hydrolyze about 70 times slower thanATP in F1-ATPase (4), and thus calculating the barrier heightfor the γP-OADP bond-breaking step could further validate theapplied theoretical model. Accordingly, QM152 was modified tohave ATPγS instead of ATP, where the sulfur replacing one oxy-gen on γP was placed to the most likely position suggested byavailable crystal structures (5, 6). Residual positions in the origi-nal site, however, are not exactly suitable for such a modification,e.g., P-S bond is 2.02 Å, compared to 1.52 Å of the P-O bond. Freeenergy calculations, which would require sampling of the confor-mational space of the environment and would thus consider struc-tural changes due to the inserted sulfur, are however beyondthe scope of the present study. Nevertheless, because the samereaction steps are calculated, the zero point energy contributionswere assumed to be similar for ATP and ATγS. The obtainedenergies showed an activation energy barrier of 74.8 kJ∕mol.Based on the barrier obtained for the ATP hydrolysis and suppos-ing a 70 times slower hydrolysis, the ideal value should beapproximately 67 kJ∕mol. The calculated barrier height is within8 kJ∕mol of the optimal value, which is close to chemical accu-racy. Thus, the difference between barrier heights of ATP andATPγS hydrolysis correlates well in relative terms with the 70times slower hydrolysis rate for the latter observed experi-mentally.
Structural Comparison QuantumMechanics/Molecular Mechanics (QM/MM). To assess structural differences between a QM active sitemodel and a QM/MM model, the initial state toward synthesisADPþ Pi (I), the final state ATPþH2O (IV), and an intermedi-ate (III) preceding the γP-OADP bond formation was obtained forboth βDP and βTP in QM106 and also in QM106∕MM. This changein active site geometry was measured by several important differ-ences between the two active sites, of which some are detailedhere. The most important difference in this respect is the approx-imate 1.1 Å relative shift of αR373, which means that in βTP thisresidue is pulled somewhat out of the active site, resulting in acrucial difference in the barrier height of ATP hydrolysis step(3). This relative shift of αR373 in the QM106∕MM model showsonly slight deviations in the investigated critical points, which in-dicates that the appearance of the charged Pi does not pull theαR373 closer to the active site in βTP (Table S3). When comparingthe QM regions of the βDP and βTP active sites in QM106∕MM, therelative structural difference remains similar between the twosites as in the X-ray structure: Important distances do not deviatesignificantly from those of the X-ray structure, and rms value forthe same atoms is 0.444 (0.274 without αR373) for QM106∕MM,and 0.417 (0.164 without αR373) for the original X-ray structure(Table S3). Superposition of the QM106 model and the QM partof the QM106∕MM model shows smaller rms values for I and IV,but somewhat higher for III in βDP (Table S3). The latter is mainlybecause of the flipped relative orientation of the α-phosphate inADP. Within the βTP site, the largest difference between theQM106 and QM106∕MM models is the position of the αS344carbonyl oxygen. As the QM106 model is relatively small, to keepthe active site geometry additional constraints were introducedon this residue, which prohibited its movement during optimiza-tion. In the case of βDP, the 1.5-Å relative shift of the βR189 ni-trogen coordinating γP arises because of the somewhat differentorientation of γP and the water molecules coordinating to Mg2þ.Nevertheless, these structural differences do not result in a qua-
Beke-Somfai et al. www.pnas.org/cgi/doi/10.1073/pnas.1010453108 1 of 7

litatively different hydrogen bond pattern or coordination prefer-ence among the residues included in the model. Therefore theactive sites described by QM106 and QM106∕MM do not havelarge differences, which is also represented by the qualitativelysimilar relative energy distributions of the critical points(Table S3). (Note that QM106 lacks the effects of the protein en-vironment.) As shown above, in principle the difference betweenthe loose and tight binding site geometries remains in theQM106∕MM model almost to the same extent as in the originalX-ray structure. Thus we conclude that the differences in the re-action steps obtained by active site models in the βDP and βTP sitesmainly arise due to the small, but apparently significant structuraldistortions between the two active site geometries.
Note that an appropriate energetic description of the reactionmechanism was not possible with a QM region including 106atoms due to several limitations: Both for βDP and for βTP site,III shows very high relative energy with values above 100 kJ∕molin QM106 and also in the QM106∕MM model. Further on, incontrast to QM152 and QM189 models, in the βDP site, the Pi andADP configuration in I is not the most stable for QM106 andQM106∕MM models. Instead, a modified structure is found,which is referred to as Ialt. In this critical point, the proton whichis on OγP in I is transferred to OADP, and thus H-OADP is shiftedfrom its original position to form a hydrogen bond with an otheroxygen of Pi. This geometry, Ialt, is approximately 30 kJ∕mol lessstable at the B3LYP/6-31G(d) level of theory in QM152 than I. InQM106, Ialt is preferred by more than 40 kJ∕mol over I and usingQM106∕MM we could not obtain I in βDP as it converged to Ialt(Table S3). Finally, despite several trials, IIITS could be obtainedneither in QM106 nor in QM106∕MM. All these results suggestthat a model with 106 atoms in the QM region is not able toaccurately describe the reaction mechanism, in contrast to thelarger QM models, which indicates that the lack of the larger en-vironment, especially the P loop, in the QM part significantlychanges the energetic preference in intermediates and thus intransition states. Thus the smaller QM setup (with 106 atoms)
is most likely not able to satisfactorily depolarize ADP and avoidthe large electrostatic repulsion between the negative HPO4
2−
and ½ADP�2− in III.
Considering Enzymatic Fluctuations. Enzymatic fluctuations duringthe intermediate steps and transition states could affect the over-all mechanism. In this respect it was important to assess whetherthe currently used Ground F1 structure would be suitable for thedescription of the different substeps. In particular, as detailedabove, the flexible P-loop region has an important contributionto the active site near the γP-OADP bond-forming or breakingsteps. Therefore, the βDP site of two crystal structures, theGround F1 and the Menz et al. structure (2) having a transitionstate analog in its active sites, were superimposed. The two activesites do not show significant variation in either the position of theimportant side chains or in the P-loop conformation, suggestingthat the βDP site obtained from Ground F1 is indeed suitable todescribe most of the reaction steps.
To validate the modified βDP active site, βDP→HC, I and II inSeron state were also obtained in this model at the B3LYP/6-311++G(d,p)//B3LYP/6-31G(d) level of theory. I has a relativeenergy of −12.8 kJ∕mol in βDP and −11.2 kJ∕mol βDP→HC. ForIIðSeronÞ the values are 36.1 and 46.4 kJ∕mol for βDP andβDP→HC, respectively, indicating that in I the modificationsbetween βDP and βDP→HC have in principle no effect on relativestability, whereas for IIðSeronÞ, these result in a 10-kJ∕mol desta-bilization. These variations confirm that stabilization of IITS inβDP→HC is not because the geometry of βDP→HC is in generalpreferred over βDP. Comparison of IITS in the βDP andβDP→HC sites shows that the hydrogen bond between OγP ofthe phosphate and βR189 is broken in βDP→HC because theArg side chain moves toward αS344 and βE192. This changeresults in a more relaxed position of the Pi, e.g., OγP⋯OADP dis-tance increases from approximately 2.8 to 2.91 Å, which reducesthe repulsion between the two oxygens and thus stabilizes IITS.
1. Masaike T, Muneyuki E, Noji H, Kinosita K, Yoshida M (2002) F1-ATPase changes itsconformations upon phosphate release. J Biol Chem 277:21643–21649.
2. Menz RI, Walker JE, Leslie AGW (2001) Structure of bovine mitochondrial F1-ATPasewith nucleotide bound to all three catalytic sites: Implications for the mechanism ofrotary catalysis. Cell 106:331–341.
3. Beke-Somfai T, Lincoln P, Norden B (2010) Mechanical control of ATP synthase func-tion: Activation energy difference between tight and loose binding sites. Biochemistry49:401–403.
4. Adachi K, et al. (2007) Coupling of rotation and catalysis in F1-ATPase revealed by
single-molecule imaging and manipulation. Cell 130:309–321.
5. Woo JS, et al. (2009) Structural studies of a bacterial condensin complex reveal
ATP-dependent disruption of intersubunit interactions. Cell 136:85–96.
6. Gulick AM, Bauer CB, Thoden JB, Rayment I (1997) X-ray structures of the MgADP,
MgATP gamma S, andMgAMPPNP complexes of the Dichyostelium discoideummyosin
motor domain. Biochemistry 36:11619–11628.
Beke-Somfai et al. www.pnas.org/cgi/doi/10.1073/pnas.1010453108 2 of 7

Fig. S1. Schematic diagram describing the geometric properties of critical points I, ITS, II, and IITS obtained in the βDP and βTP sites as calculated at the B3LYP/6-31G(d) level of theory using the QM189 model. Besides hydrogen bond interactions, OWAT⋯γP and γP⋯OADP distances are also shown by dashed lines. Bridgeatom distances (in Ångströms) are given for βDP and in parenthesis for βTP. The initial state is I with the most stable orientation of Pi in the active site (UpperLeft). The transition state, ITS, of the OγP-H rotation (Upper Right). Intermediate II, with Pi hydrogen bonded to 344SC═O (Lower Left). Note that while in βDPH2PO4
− is present, in βTP Pi is HPO42− as a spontaneous protontransfer occurs from OWAT in Pi to OGlu in βE188 during optimization. IITS is the transprotonation
on the Pi by the αS344 OH side chain (Lower Right).
Beke-Somfai et al. www.pnas.org/cgi/doi/10.1073/pnas.1010453108 3 of 7

Fig. S2. Schematic diagram describing the geometric properties of critical points III, IIITS, IVobtained in the βDP and βTP sites as calculated at the B3LYP/6-31G(d)level of theory using the QM189 model. Besides hydrogen bond interactions, OWAT—γP and γP—OADP distances are also shown by dashed lines. Bridge atomdistances (in Ångströms) are given for βDP and in parenthesis for βTP. Intermediate III preceding the formation of ATP and H2O (Upper Left). In both active sites,HPO4
2− is present, where the proton is on OWAT hydrogen bonded to αS344C═O. The other proton required to formation of the nucleophilic water is on OGlu,hydrogen bonded to OWAT. In βDP the relative position of γP and OADP is much more favorable for the P-O bond formation. The γP-OADP bond formation occursthrough IIITS, with a planar PO3
− ion (3) (Upper Right). The final state, IV, has ATP and the nucleophilic water molecule in the active site (Lower).
Beke-Somfai et al. www.pnas.org/cgi/doi/10.1073/pnas.1010453108 4 of 7
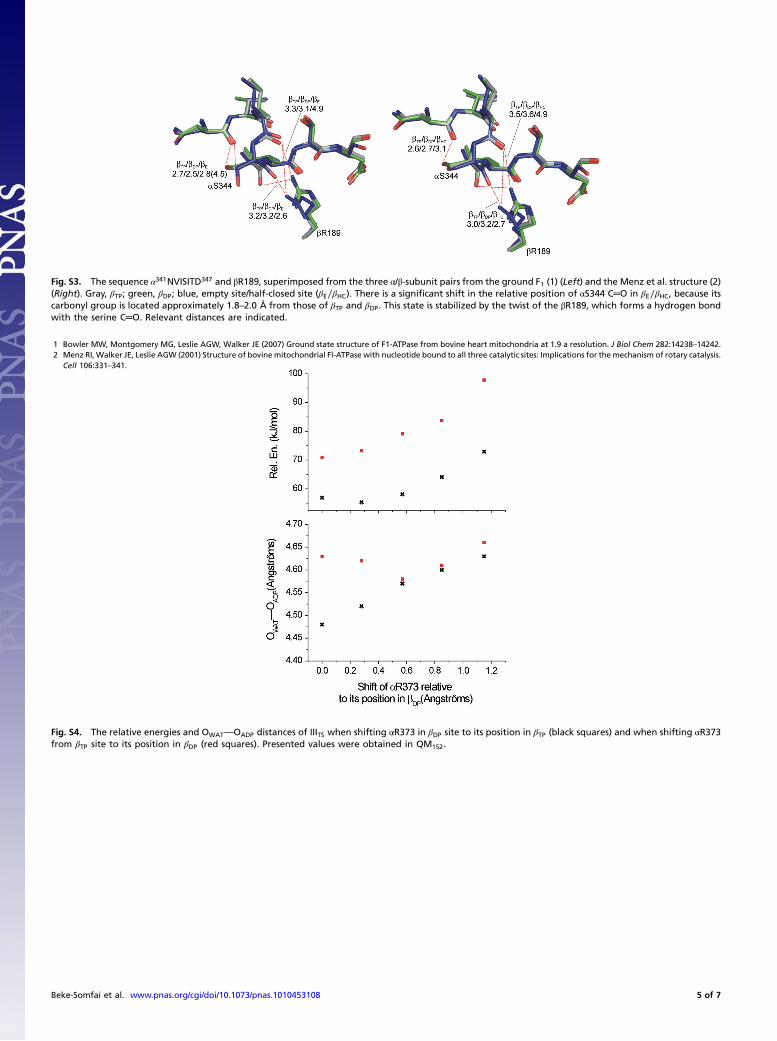
Fig. S3. The sequence α341NVISITD347 and βR189, superimposed from the three α/β-subunit pairs from the ground F1 (1) (Left) and the Menz et al. structure (2)(Right). Gray, βTP; green, βDP; blue, empty site/half-closed site (βE∕βHC). There is a significant shift in the relative position of αS344 C═O in βE∕βHC, because itscarbonyl group is located approximately 1.8–2.0 Å from those of βTP and βDP. This state is stabilized by the twist of the βR189, which forms a hydrogen bondwith the serine C═O. Relevant distances are indicated.
1 Bowler MW, Montgomery MG, Leslie AGW, Walker JE (2007) Ground state structure of F1-ATPase from bovine heart mitochondria at 1.9 a resolution. J Biol Chem 282:14238–14242.2 Menz RI, Walker JE, Leslie AGW (2001) Structure of bovinemitochondrial Fl-ATPase with nucleotide bound to all three catalytic sites: Implications for the mechanism of rotary catalysis.
Cell 106:331–341.
Fig. S4. The relative energies and OWAT—OADP distances of IIITS when shifting αR373 in βDP site to its position in βTP (black squares) and when shifting αR373from βTP site to its position in βDP (red squares). Presented values were obtained in QM152.
Beke-Somfai et al. www.pnas.org/cgi/doi/10.1073/pnas.1010453108 5 of 7

Fig. S5. The βDP site with ATP bound in the QMmodels used in this study. QM106 and QM part of QM106∕MM is shown as ball and stick, QM152 includes QM106
and the regions shown as stick (i.e., the P loop:βG159-βT163 and βE192), whereas QM189 consists of all the atoms plotted here. The original residues forming theactive site in F1-ATPase are labeled. QM128 can be derived from QM152: It does not contain αS344, βE192, and the C-terminal peptide bond of βT163. The atomswhich were constrained in QM189 during the optimizations are marked with an asterisk.
Table S1. Energetic properties of the critical points obtained in the two active sites, βDP and βTP, by using the QM152 and the QM189
models
Critical points Rel. energy,* kJ∕mol
βDP βTP
QM152 QM189 QM152 QM189
B3LYP B3LYP M05-2X B3LYP B3LYP M05-2XI −9.1 −12.8 −3.9 3.3 16.7 38.9ITS 44.7 — — 63.5 — —II 23.4 22.6 23.1 54.1 43.3 52.0
Seron : 36.1 Seron : 37.4 Seron : 68.0 Seron : 73.9IITS 91.6 85.7 122.6 119.1III 16.3 Seron : 29.5 Seron : 20.1 54.3 Seron : 45.5 Seron : 50.5
11.5 11.7 36.7 60.9IIITS 56.9 49.0 45.8 97.8 86.6 87.3IV 0.0 0.0 0.0 0.0 0.0 0.0
Critical points in alternative pathwaysβDP βTP
QM152 Description QM152 DescriptionTSII→IV 247.0 direct transprotonation
and bond formationTSII→III 137.1 direct transprotonationTSP:rel 118.0 proton relay transition state:
ADPþ Pi − Alt: → H2O-PO3−
complexH2O-PO3
− complex 104.7
*Relative energy values were obtained at the B3LYP/6-311++G(d,p) and M05-2X/6-311++G(d,p) levels of theory, including zero point energy and thermalcorrections, as well as polarization effects of the environment using the integral equation formalism for the polarizable continuum model. See Methodsfor more details.
Beke-Somfai et al. www.pnas.org/cgi/doi/10.1073/pnas.1010453108 6 of 7
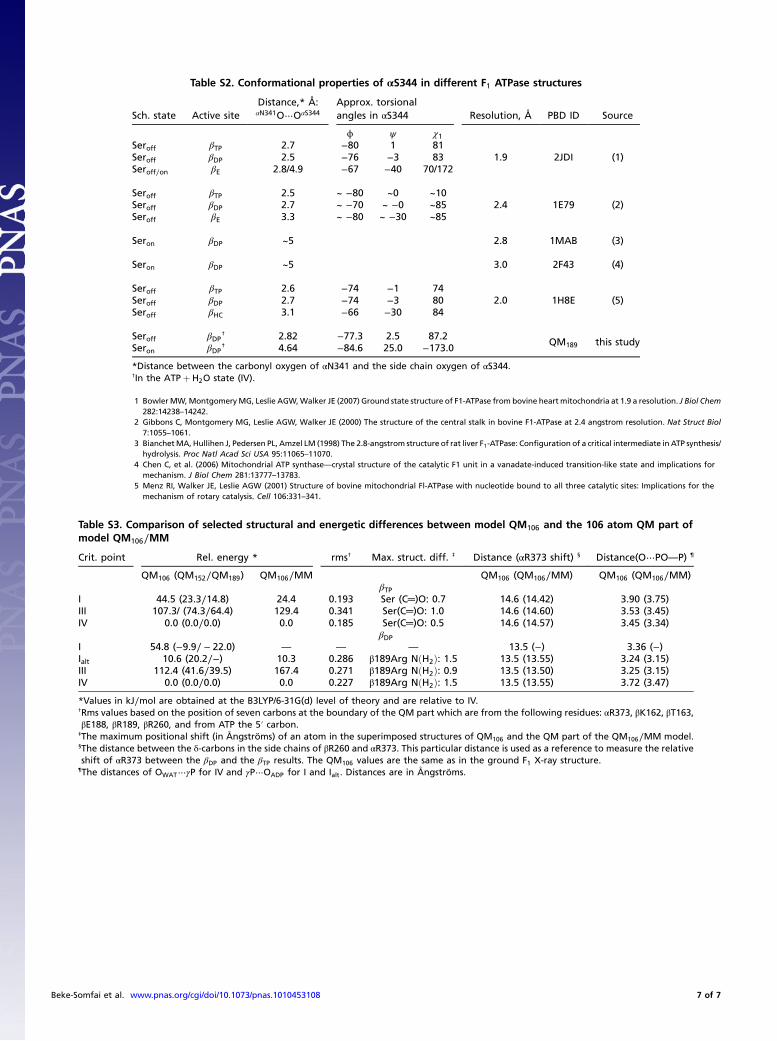
Table S2. Conformational properties of αS344 in different F1 ATPase structures
Sch. state Active siteDistance,* Å:αN341O⋯OαS344
Approx. torsionalangles in αS344 Resolution, Å PBD ID Source
ϕ ψ χ1Seroff βTP 2.7 −80 1 81
1.9 2JDI (1)Seroff βDP 2.5 −76 −3 83Seroff∕on βE 2.8/4.9 −67 −40 70/172
Seroff βTP 2.5 ~ −80 ~0 ~102.4 1E79 (2)Seroff βDP 2.7 ~ −70 ~ −0 ~85
Seroff βE 3.3 ~ −80 ~ −30 ~85
Seron βDP ~5 2.8 1MAB (3)
Seron βDP ~5 3.0 2F43 (4)
Seroff βTP 2.6 −74 −1 742.0 1H8E (5)Seroff βDP 2.7 −74 −3 80
Seroff βHC 3.1 −66 −30 84
Seroff βDP† 2.82 −77.3 2.5 87.2
QM189 this studySeron βDP
† 4.64 −84.6 25.0 −173.0
*Distance between the carbonyl oxygen of αN341 and the side chain oxygen of αS344.†In the ATPþ H2O state (IV).
1 BowlerMW,MontgomeryMG, Leslie AGW,Walker JE (2007) Ground state structure of F1-ATPase from bovine heart mitochondria at 1.9 a resolution. J Biol Chem282:14238–14242.
2 Gibbons C, Montgomery MG, Leslie AGW, Walker JE (2000) The structure of the central stalk in bovine F1-ATPase at 2.4 angstrom resolution. Nat Struct Biol7:1055–1061.
3 BianchetMA, Hullihen J, Pedersen PL, Amzel LM (1998) The 2.8-angstrom structure of rat liver F1-ATPase: Configuration of a critical intermediate in ATP synthesis/hydrolysis. Proc Natl Acad Sci USA 95:11065–11070.
4 Chen C, et al. (2006) Mitochondrial ATP synthase—crystal structure of the catalytic F1 unit in a vanadate-induced transition-like state and implications formechanism. J Biol Chem 281:13777–13783.
5 Menz RI, Walker JE, Leslie AGW (2001) Structure of bovine mitochondrial Fl-ATPase with nucleotide bound to all three catalytic sites: Implications for themechanism of rotary catalysis. Cell 106:331–341.
Table S3. Comparison of selected structural and energetic differences between model QM106 and the 106 atom QM part ofmodel QM106∕MM
Crit. point Rel. energy * rms† Max. struct. diff. ‡ Distance (αR373 shift) § Distance(O⋯PO—P) ¶
QM106 (QM152∕QM189) QM106∕MM QM106 (QM106∕MM) QM106 (QM106∕MM)βTP
I 44.5 (23.3∕14.8) 24.4 0.193 Ser (C═)O: 0.7 14.6 (14.42) 3.90 (3.75)III 107.3/ (74.3∕64.4) 129.4 0.341 Ser(C═)O: 1.0 14.6 (14.60) 3.53 (3.45)IV 0.0 (0.0∕0.0) 0.0 0.185 Ser(C═)O: 0.5 14.6 (14.57) 3.45 (3.34)
βDPI 54.8 (−9.9∕ − 22.0) — — — 13.5 (−) 3.36 (−)Ialt 10.6 (20.2∕−) 10.3 0.286 β189Arg NðH2Þ: 1.5 13.5 (13.55) 3.24 (3.15)III 112.4 (41.6∕39.5) 167.4 0.271 β189Arg NðH2Þ: 0.9 13.5 (13.50) 3.25 (3.15)IV 0.0 (0.0∕0.0) 0.0 0.227 β189Arg NðH2Þ: 1.5 13.5 (13.55) 3.72 (3.47)
*Values in kJ∕mol are obtained at the B3LYP/6-31G(d) level of theory and are relative to IV.†Rms values based on the position of seven carbons at the boundary of the QM part which are from the following residues: αR373, βK162, βT163,βE188, βR189, βR260, and from ATP the 5′ carbon.
‡The maximum positional shift (in Ångströms) of an atom in the superimposed structures of QM106 and the QM part of the QM106∕MM model.§The distance between the δ-carbons in the side chains of βR260 and αR373. This particular distance is used as a reference to measure the relativeshift of αR373 between the βDP and the βTP results. The QM106 values are the same as in the ground F1 X-ray structure.
¶The distances of OWAT⋯γP for IV and γP⋯OADP for I and Ialt. Distances are in Ångströms.
Beke-Somfai et al. www.pnas.org/cgi/doi/10.1073/pnas.1010453108 7 of 7














![Präzisionsexperimente zur Untersuchung der Gravitationeisele/lehrer/gravitations... · 1080 1152 1224 1296 1368 1440 328 330 332 334 336 338 340 342 344 amplitude [µrad] diff/sum;](https://static.fdocument.org/doc/165x107/5b0d0c127f8b9a685a8d9445/przisionsexperimente-zur-untersuchung-der-gravitation-eiselelehrergravitations1080.jpg)



