
Doenças desmielinizantes e degenerativas do...
15
1 Doença de Creutzfeldt-Jakob A doença de Creutzfeldt-Jakob está incluída no grupo das Encefalopatias Espongiformes. É uma doença rara, que se manifesta clinicamente por uma demência rapidamente progressiva. Embora haja diferenças entre as várias encefalopatias espongiformes, todas estão associadas com formas anormais da proteína priónica (PrP). A PrP é uma proteína celular normal presente nos neurónios. A doença ocorre quando esta proteína sofre uma alteração conformacional da sua isoforma normal contendo uma α-hélice (PrP c ) para uma isoforma β anormal em folha pregueada (PrP sc ) resistente a proteases (como a protease K). A acumulação de PrP sc parece ser a causa da patologia nestas doenças, porém, o mecanismo pelo causa morte neuronal é ainda desconhecido. A alteração conformacional ocorre: • Espontaneamente (em 85% dos casos) • Hereditariamente A PrP c é codificada pelo gene PRNP, com locais importantes de mutação (codão 178) e polimorfismo associado com a doença (codão 129). Nos indivíduos normais, o codão 178 codifica Asp e o codão 129 codifica Met ou Val. Em formas familiares da doença, a mutação altera o codão 178 para Asn. Quando o alelo contendo a mutação Asn tem também uma Val no codão 129, o paciente desenvolve doença de Creutzfeldt-Jakob. Pelo contrário, quando o alelo tem um Met no codão 129 o distúrbio clínico é a insónia familiar fatal (outra doença pertencente às encefalopatias enpongiformes). Faculdade de Medicina da Universidade do Porto Seminário de Biopatologia 13 Dezembro 2006 Doenças desmielinizantes e degenerativas do SNC
Transcript of Doenças desmielinizantes e degenerativas do...

1
Doença de Creutzfeldt-Jakob
A doença de Creutzfeldt-Jakob está incluída no grupo das Encefalopatias Espongiformes. É uma doença rara, que se manifesta clinicamente por uma demência rapidamente progressiva.
Embora haja diferenças entre as várias encefalopatias espongiformes, todas estão associadas com formas anormais da proteína priónica (PrP). A PrP é uma proteína celular normal presente nos neurónios. A doença ocorre quando esta proteína sofre uma alteração conformacional da sua isoforma normal contendo uma α-hélice (PrPc) para uma isoforma β anormal em folha pregueada (PrPsc) resistente a proteases (como a protease K). A acumulação de PrPsc parece ser a causa da patologia nestas doenças, porém, o mecanismo pelo causa morte neuronal é ainda desconhecido.
A alteração conformacional ocorre:
• Espontaneamente (em 85% dos casos) • Hereditariamente
A PrPc é codificada pelo gene PRNP, com locais importantes de mutação (codão 178) e polimorfismo associado com a doença (codão 129). Nos indivíduos normais, o codão 178 codifica Asp e o codão 129 codifica Met ou Val. Em formas familiares da doença, a mutação altera o codão 178 para Asn. Quando o alelo contendo a mutação Asn tem também uma Val no codão 129, o paciente desenvolve doença de Creutzfeldt-Jakob. Pelo contrário, quando o alelo tem um Met no codão 129 o distúrbio clínico é a insónia familiar fatal (outra doença pertencente às encefalopatias enpongiformes).
Faculdade de Medicina da Universidade do Porto
Seminário de Biopatologia
13 Dezembro 2006
Doenças desmielinizantes e degenerativas do SNC
2
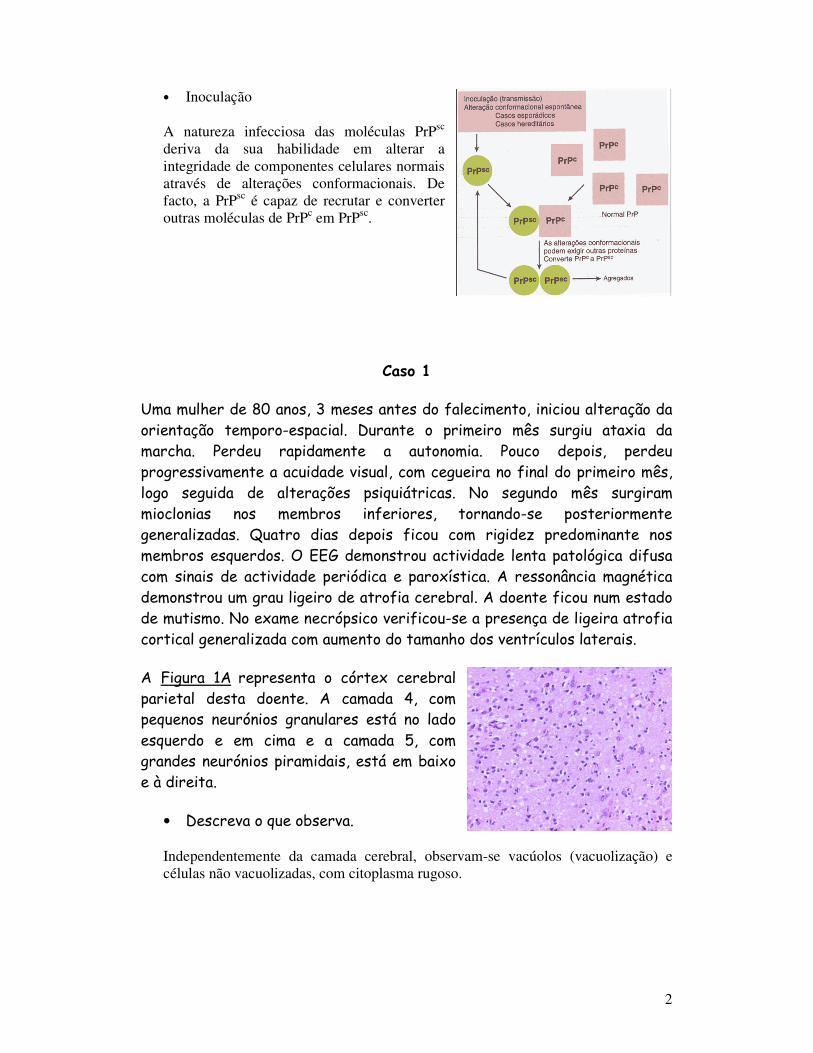
• Inoculação
A natureza infecciosa das moléculas PrPsc deriva da sua habilidade em alterar a integridade de componentes celulares normais através de alterações conformacionais. De facto, a PrPsc é capaz de recrutar e converter outras moléculas de PrPc em PrPsc.
Caso 1
Uma mulher de 80 anos, 3 meses antes do falecimento, iniciou alteração da orientação temporo-espacial. Durante o primeiro mês surgiu ataxia da marcha. Perdeu rapidamente a autonomia. Pouco depois, perdeu progressivamente a acuidade visual, com cegueira no final do primeiro mês, logo seguida de alterações psiquiátricas. No segundo mês surgiram mioclonias nos membros inferiores, tornando-se posteriormente generalizadas. Quatro dias depois ficou com rigidez predominante nos membros esquerdos. O EEG demonstrou actividade lenta patológica difusa com sinais de actividade periódica e paroxística. A ressonância magnética demonstrou um grau ligeiro de atrofia cerebral. A doente ficou num estado de mutismo. No exame necrópsico verificou-se a presença de ligeira atrofia cortical generalizada com aumento do tamanho dos ventrículos laterais.
A Figura 1A representa o córtex cerebral parietal desta doente. A camada 4, com pequenos neurónios granulares está no lado esquerdo e em cima e a camada 5, com grandes neurónios piramidais, está em baixo e à direita.
• Descreva o que observa.
Independentemente da camada cerebral, observam-se vacúolos (vacuolização) e células não vacuolizadas, com citoplasma rugoso.

3
• Que tipo de células são as que têm citoplasma vermelho e núcleo pálido?
Astrócitos, as células mais frequentes da glia, reagem em muitos processos de lesão do SNC (gliose).
A gliose é o indicador histopatológico mais importante de lesão no SNC, independentemente da etiologia.
Animais transgénicos, em que a proteína priónica foi abolida, crescem normalmente. O que acontece quando estes animais transgénicos são injectados com priões no encéfalo?
O PrPsc é capaz de recrutar e converter outras moléculas de PrPc para a forma anormal da proteína. Quando se administra a PrPsc aos ratos transgénicos (sem PrPc), esta interacção não ocorre. Para além disso, a quantidade de inoculado é demasiado pequena. Não há acumulação e portanto, não se desenvolve doença.
A Figura 1B é do putámen. O caudato tinha aspecto idêntico. Descreva o que observa.
Transformação espongiforme – vacuolização microscópica
A transformação espongiforme é uma característica comum a todas as encefalopatias espongiformes.
A Figura 1C é do cerebelo. Descreva o que observa. Se não há evidência de perda neuronal, pode sugerir uma explicação?
Na camada molecular há espongificação. A densidade nuclear é normal, não havendo perda neuronal.
A história clínica é muito curta (3 meses) para que tenha ocorrido morte neuronal. Neste caso a história demencial não é devida à morte de células nervosas, mas sim à vacuolização (transformação espongiforme).
4
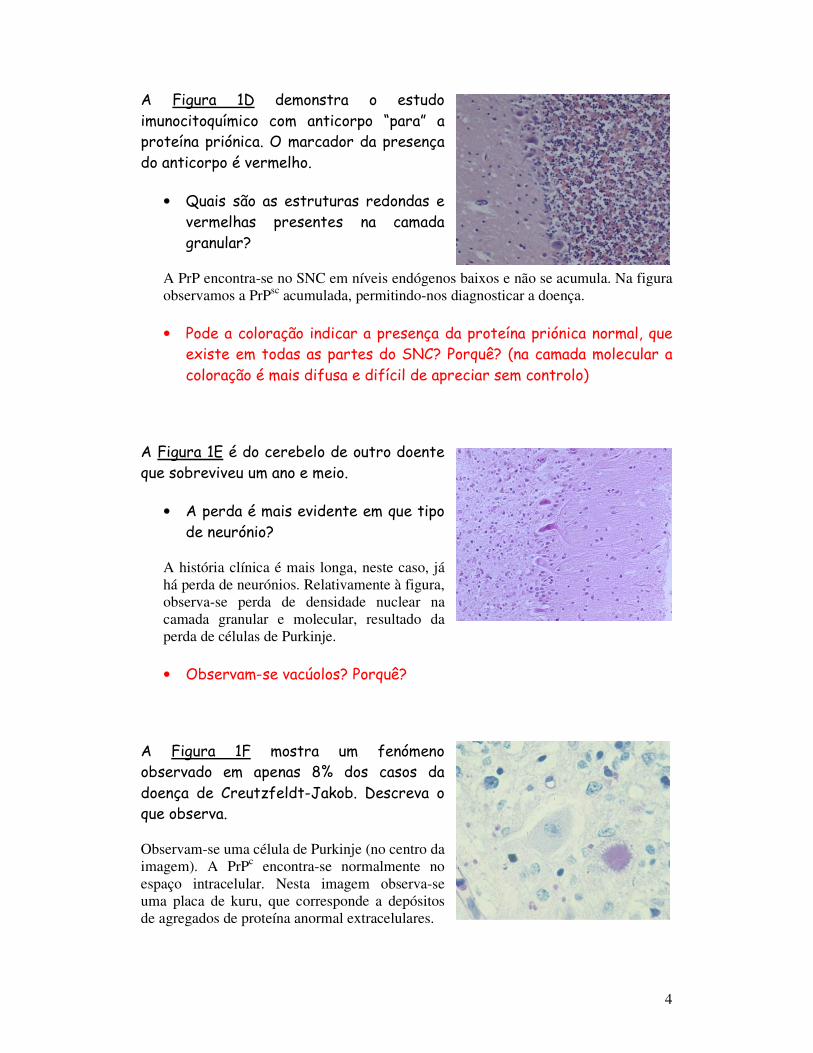
A Figura 1D demonstra o estudo imunocitoquímico com anticorpo “para” a proteína priónica. O marcador da presença do anticorpo é vermelho.
• Quais são as estruturas redondas e vermelhas presentes na camada granular?
A PrP encontra-se no SNC em níveis endógenos baixos e não se acumula. Na figura observamos a PrPsc acumulada, permitindo-nos diagnosticar a doença.
• Pode a coloração indicar a presença da proteína priónica normal, que existe em todas as partes do SNC? Porquê? (na camada molecular a coloração é mais difusa e difícil de apreciar sem controlo)
A Figura 1E é do cerebelo de outro doente que sobreviveu um ano e meio.
• A perda é mais evidente em que tipo de neurónio?
A história clínica é mais longa, neste caso, já há perda de neurónios. Relativamente à figura, observa-se perda de densidade nuclear na camada granular e molecular, resultado da perda de células de Purkinje.
• Observam-se vacúolos? Porquê?
A Figura 1F mostra um fenómeno observado em apenas 8% dos casos da doença de Creutzfeldt-Jakob. Descreva o que observa.
Observam-se uma célula de Purkinje (no centro da imagem). A PrPc encontra-se normalmente no espaço intracelular. Nesta imagem observa-se uma placa de kuru, que corresponde a depósitos de agregados de proteína anormal extracelulares.

5
Caso 2
Um homem de 17 anos começou com alterações do comportamento, mudanças abruptas de disposição, insónia, apatia e ilusões não mantidas. Dez meses depois desenvolveu dificuldade na coordenação, dores nas mãos e nos pés e sabor desagradável na boca. Quatro meses depois manifestou rigidez, disfagia, incontinência e movimentos espasmódicos sem controlo. Três meses depois evidenciou demência. Sofria de disestesias e de neuropatia sensitiva. Morreu após dois anos.
A Figura 2 mostra o córtex cerebral.
• Descreva o que observa.
Transformação espongiforme em torno das placas de kuru
• Como se chama esta doença?
Variante da doença de Creutzfeldt-Jakob – Doença das vacas loucas.
As alterações descritas na alínea anterior são características desta variante. Ao contrário da doença de Creutzfeldt-Jakob, esta variante afecta sobretudo adultos jovens, os distúrbios do comportamento ocorrem proeminentemente nos estágios precoces da doença e a síndrome neurológica progride mais lentamente.
• Como pode um prião ter propriedades diferentes de outros priões?
Na variante da doença não foram encontradas alterações no gene PRNP e todos os casos estudados eram homozigóticos Met/Met no codão 129. Por outro lado surgiram evidências que ligam a variante da doença a uma encefalopatia espongiforme bovina.
Doença de Alzheimer
A doença de Alzheimer é a principal doença degenerativa cortical. A sua principal manifestação clínica é a demência, isto é, uma perda progressiva da função cognitiva independentemente do estado de atenção. Esta doença compromete também as estruturas subcorticais, porém muitos dos sintomas clínicos estão relacionados com as alterações no córtex cerebral. A amiloide β (Aβ) é uma molécula critica na patogénese desta demência. Este peptídeo agrega-se rapidamente, forma lâminas β pregueadas e liga-se ao vermelho de Congo. É relativamente resistente à degradação, desencadeia uma resposta dos astrócitos e da microglia e é directamente neurotóxico. Os peptídeos Aβ são derivados do processamento da proteína amilóide precursora (APP).

6
A APP é uma proteína transmembranar, com locais potenciais de clivagem para três enzimas distintas (α, β, γ-secretases). Quando a APP é clivada pela α-secretase, a clivagem subsequente pela γ-secretase não origina Aβ. Pelo contrário, a clivagem pela β-secretase seguida pela γ-secretase resulta na produção de Aβ então se agregar e forma fibrilas.
Caso 3
Homem de 73 anos que começou a ter dificuldade em lembrar-se de coisas recentes. Perdeu o caminho perto de casa várias vezes. Alguns meses depois tinha dificuldade em manusear objectos familiares (ex: talheres). Não reconhecia pessoas com quem costumava relacionar-se. Começou a ter dificuldade em usar palavras. Desenvolveu incontinência. Ficou na cama quase sem falar. Morreu 5 anos depois do início da doença. A doença de Alzheimer é a forma mais comum de demência nos idosos. Casos antes dos 60 anos de idade são raros e sobretudo hereditários.
A Figura 3A demonstra um hemisfério de dois encéfalos, um com doença de Alzheimer, o outro normal.
• Qual é o anormal? Quais as alterações que são visíveis?
Direito. Atrofia cortical generalizada com alargamento dos sulcos cerebrais (mais pronunciada nos lobos frontais, temporais e parietais). Com atrofia significativa, há um alargamento compensatório dos ventrículos secundário à perda de parênquima.
• Que estrutura específica está alterada, e que sintomas podiam ser relacionados com essa estrutura?
Córtex – demência; Hipocampo – perda da memória e da capacidade de formar novas memórias.
Na Figura 3B vê-se uma das alterações características da doença de Alzheimer - uma placa neurítica em grande ampliação e corada pela prata (coloração de Bielschowsky). As estruturas pretas arredondadas são prolongamentos neuronais anormais.

7
• Qual é a substância no centro da placa?
Placas neuríticas são colecções esférica focais de expansões neuríticas dilatadas e tortuosas, frequentemente em torno de um núcleo central de amilóide. O componente dominande do núcleo é o β-amilóide, mas outras proteínas podem estar presentes em menor quantidade, incluindo componentes da cascata do complemento, citocinas pró-inflamatórias, α1-antiquimiotripsina e apolipotroteínas.
Na Figura 3C observa-se em cima uma placa neurítica marcada com um anticorpo “para” a proteína tau.
• Quais as estruturas que são positivas?
A figura A é positiva para a proteína tau e para a substância amilóide – placa neurítica.
• Em baixo observa-se outro tipo de placa marcada pelo mesmo anticorpo. Como se chama este tipo de placa?
A figura B não cora “para” tau mas cora “para” a substância amilóide – placa difusa. Não há neuritos degradados (logo não é uma placa neurítica), mas sim uma placa difusa. A placa difusa ou placa senil não traduz a existência de doença de Alzheimer, podendo a sua presença dever-se à idade.
A Figura 3D é de baixa ampliação e abrange a profundidade do córtex parietal de um doente. A coloração usada é com prata, que dá uma ideia da quantidade de placas neuríticas, num doente que faleceu com doença de Alzheimer típica.
Na Figura 3E observa-se um neurónio que tem um corpo alongado no citoplasma (coloração com vermelho de Congo).
• Para que serve esta coloração?

8
O vermelho de Congo cora todas as formas de amilóide graças à sua conformação β-pregueada.
• Come se chama este corpo?
• Qual é a relação deste corpo com as placas neuríticas?
Na Figura 3F (coloração com prata) observa-se outro corpo mais alongado num neurónio do hipocampo. Como se designa?
Tranças neurofibrilares, feixes de filamentos no citoplasma dos neurónios que deslocam ou circundam o núcleo.
A Figura 3G é de microscopia electrónica. A maior parte desta figura demonstra o citoplasma e uma pequena parte do núcleo de um neurónio. Identificam-se mitocôndrias, retículo rugoso, e algum pigmento normal para a idade (lipofuscínio).
• Como se chama o feixe que atravessa o citoplasma da esquerda para a direita (em cima)? E de que são compostos?
Trata-se de feixes helicoidais emparelhados e de alguns filamentos rectos que parecem ter composição semelhante. Os filamentos helicoidais emparelhados são constituídos principalmente pela da proteína tau (na sua forma hiperfosforilada e insolúvel), uma proteína associada ao microtúbulo axonal que reforça a montagem de microtúbulos. Outros antigénios incluem MAP2 (outra proteína associada ao microtúbulo) e a ubiquitina (molécula que se liga a proteínas, sinalizando-as para a degradação proteossómica).
• Qual é a relação entre a acumulação da proteína tau nos neurónios e da beta-amilóide nas placas?

9
Caso 4
Uma mulher de 62 anos com sinais ligeiros de deterioração mental, desenvolveu subitamente dor de cabeça, paralisia do braço direito e dificuldade em falar. Depois entrou em coma e morreu. Na autópsia demonstrou-se uma grande hemorragia no córtex parietal esquerdo, com extensão à substância branca.
A Figura 4A demonstra um vaso no córtex com coloração de vermelho de Congo. A Figura 4B é do mesmo vaso observado com luz polarizada.
• Qual é a substância que está a ocupar a parede do vaso?
β-amilóide (angiopatia amiloidótica cerebral – acompanhante quase invariável da doença de Alzheimer, no entanto pode ser encontrada em indivíduos sem a doença).
• Quais são os cores que aparecem com a polarização desta substância corada pelo vermelho do Congo?
Sob luz polarizada, o vermelho de Congo apresenta uma birrefrigerância verde.
Doença de Parkinson
A Doença de Parkinson está incluída no grupo de doenças degenerativas dos núcleos da base e do tronco cerebral. As doenças que comprometem estas regiões do cérebro estão frequentemente associadas com distúrbios dos movimentos.
Caso 5
Um homem de 56 anos desenvolveu gradualmente lentidão dos movimentos, dificuldade em iniciar movimentos e tremor dos dedos em repouso. A cara perdeu expressividade. A frequência do pestanejar diminuiu. Ao andar, os passos eram mais curtos. Depois destes sintomas piorarem durante 3 anos, foi diagnosticado com doença de Parkinson. Notou-se naquela altura rigidez
10
dos membros, mas a força era normal. Sofria de insónia. O intelecto parecia normal.
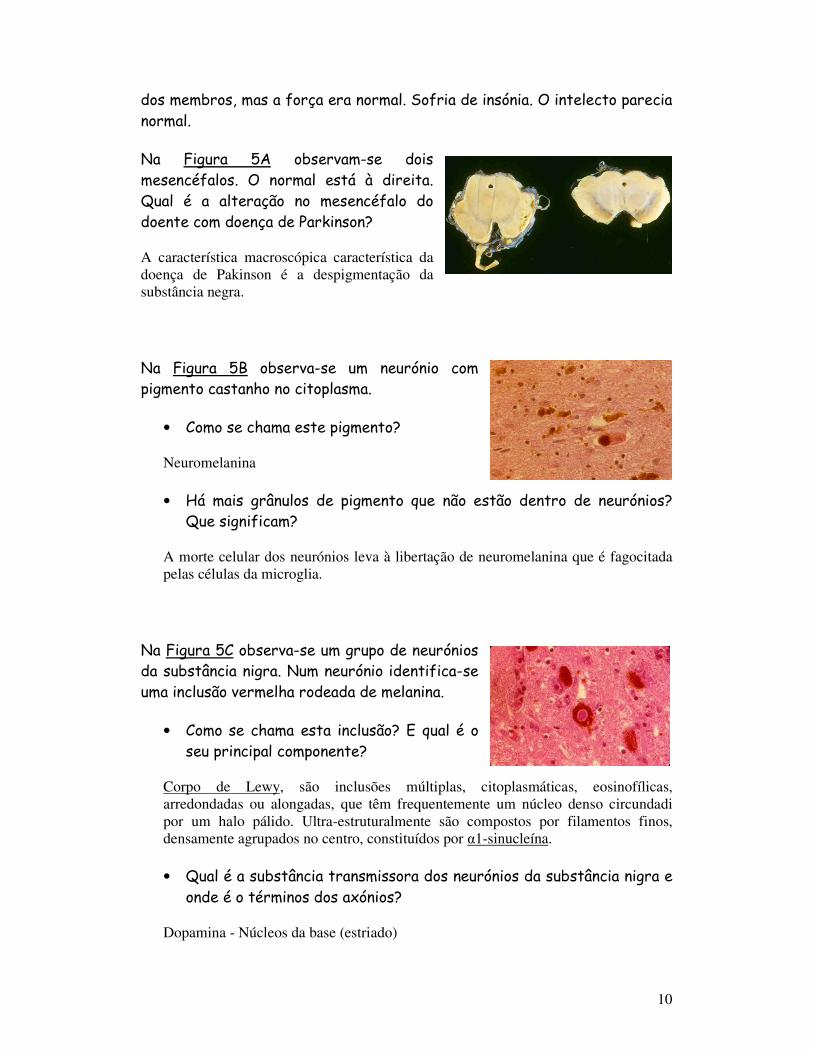
Na Figura 5A observam-se dois mesencéfalos. O normal está à direita. Qual é a alteração no mesencéfalo do doente com doença de Parkinson?
A característica macroscópica característica da doença de Pakinson é a despigmentação da substância negra.
Na Figura 5B observa-se um neurónio com pigmento castanho no citoplasma.
• Como se chama este pigmento?
Neuromelanina
• Há mais grânulos de pigmento que não estão dentro de neurónios? Que significam?
A morte celular dos neurónios leva à libertação de neuromelanina que é fagocitada pelas células da microglia.
Na Figura 5C observa-se um grupo de neurónios da substância nigra. Num neurónio identifica-se uma inclusão vermelha rodeada de melanina.
• Como se chama esta inclusão? E qual é o seu principal componente?
Corpo de Lewy, são inclusões múltiplas, citoplasmáticas, eosinofílicas, arredondadas ou alongadas, que têm frequentemente um núcleo denso circundadi por um halo pálido. Ultra-estruturalmente são compostos por filamentos finos, densamente agrupados no centro, constituídos por α1-sinucleína.
• Qual é a substância transmissora dos neurónios da substância nigra e onde é o términos dos axónios?
Dopamina - Núcleos da base (estriado)
11
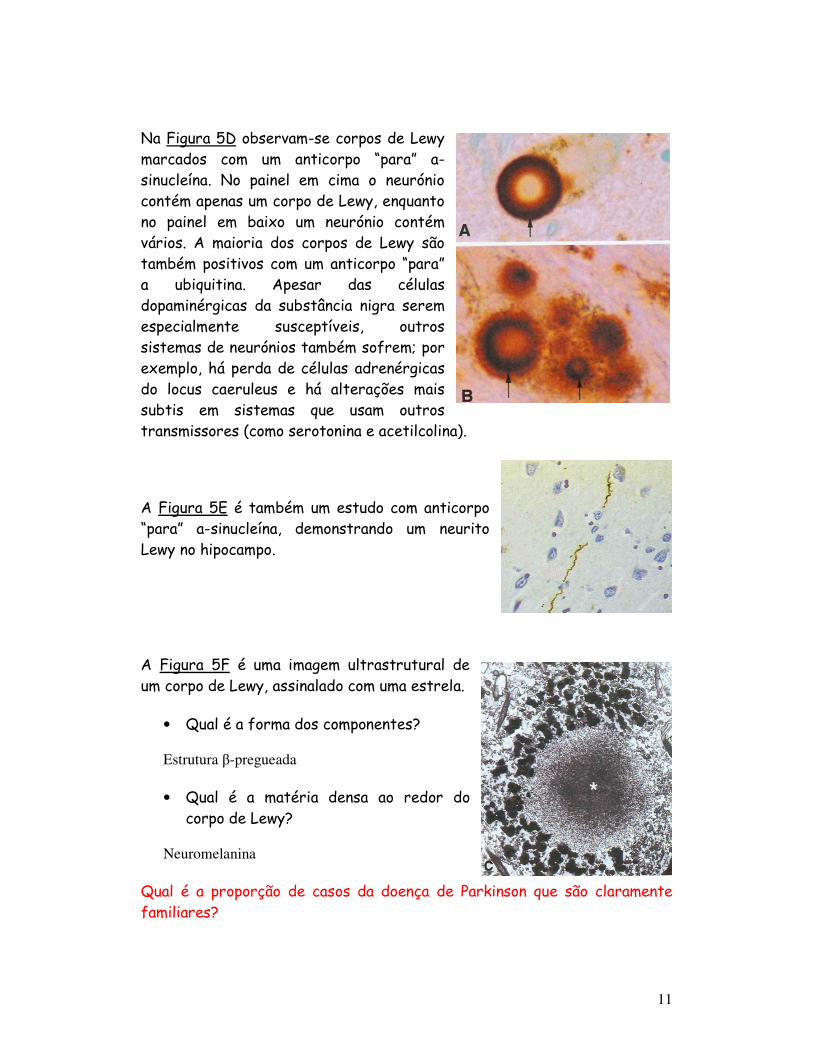
Na Figura 5D observam-se corpos de Lewy marcados com um anticorpo “para” a-sinucleína. No painel em cima o neurónio contém apenas um corpo de Lewy, enquanto no painel em baixo um neurónio contém vários. A maioria dos corpos de Lewy são também positivos com um anticorpo “para” a ubiquitina. Apesar das células dopaminérgicas da substância nigra serem especialmente susceptíveis, outros sistemas de neurónios também sofrem; por exemplo, há perda de células adrenérgicas do locus caeruleus e há alterações mais subtis em sistemas que usam outros transmissores (como serotonina e acetilcolina).
A Figura 5E é também um estudo com anticorpo “para” a-sinucleína, demonstrando um neurito Lewy no hipocampo.
A Figura 5F é uma imagem ultrastrutural de um corpo de Lewy, assinalado com uma estrela.
• Qual é a forma dos componentes?
Estrutura β-pregueada
• Qual é a matéria densa ao redor do corpo de Lewy?
Neuromelanina
Qual é a proporção de casos da doença de Parkinson que são claramente familiares?

12
Por que motivo é que as células da substância nigra são mais susceptíveis nesta doença?
Caso 6
Um homem de 72 anos começou a ter alucinações visuais pormenorizadas. A família notou variações marcadas na atenção e vigilância. No exame demonstrou dificuldade em iniciar movimentos, postura dobrada, cara inexpressiva. A dificuldade com a memória recente era mínima.
Na Figura 6A observam-se neurónios do córtex cerebral. O neurónio no centro tem uma inclusão eosinófila citoplasmática mal definida. Como se chama este tipo de inclusão?
Corpo de Lewy (α1-sinucleína)
Na Figura 6B observa-se uma inclusão do mesmo tipo com anticorpo “para” a-sinucleína.
A Figura 6C demonstra neuritos de Lewy no hipocampo com o mesmo anticorpo. Quando numerosas, estas inclusões indicam o diagnóstico de demência de corpos de Lewy. Esta é considerada o segundo tipo mais comum de demência nos idosos. Os critérios de diagnóstico patológico ainda não estão bem definidos. É comum encontrar placas do tipo Alzheimer em vários doentes com demência de corpos de Lewy, mas ambas as doenças parecem clinicamente distintas. Sinucleína e b-amilóide podem servir para iniciar deposição de uma e da outra, respectivamente (nas placas da doença de Alzheimer é vulgar encontrar sinucleína e a b-amilóide é um componente de corpos de Lewy). Uma síndroma de Parkinsonismo pode aparecer noutras doenças com envolvimento da substância nigra.

13
Esclerose Múltipla
A esclerose múltipla é a doença desmielinizante mais comum. As doenças desmielinizantes resultam de uma lesão preferencial na mielina, com relativa preservação dos axónios. A história natural das doenças desmielinizantes é determinada, em parte, pela capacidade limitada do SNC para regenerar mielina normal e pelo grau de lesão secundária aos axónios que ocorre à medida que a doença segue o seu curso.
A esclerose múltipla é uma doença auto-imune. As lesão observadas são causadas por uma resposta imune celular inapropriada contra os componentes da bainha de mielina. A doença é iniciada pelas células T CD4+ TH1 que reagem contra antigénios da própria mielina e secretam citocinas, como o IFN-γ, que activam os macrófagos. A desmielinização é causada por estes macrófagos e pelos seus produtos.
Caso 7
Uma mulher de 29 anos começou a sentir entorpecimento da mão direita associado a um grau ligeiro de fraqueza, que se desenvolveu durante um dia, durou um mês, e depois desapareceu. Nove meses depois, experimentou uma perda súbita de visão no olho esquerdo que melhorou durante uma semana. Esteve bem durante dois anos, mas então começou a sentir fraqueza progressiva do braço direito e da perna direita. No exame tinha sinais de Babinski bilaterais. A flexão passiva do pescoço produzia uma sensação eléctrica nas costas. Três meses depois desenvolveu diplopia.
Na Figura 7A observa-se um corte coronal do encéfalo de um doente que morreu depois de ter sofrido de esclerose múltipla durante muitos anos. Este corte é ao nível do tálamo posterior e da pineal.
• Pode identificar placas de esclerose múltipla? Quantas? Há uma localização preferencial?
Uma vez que a esclerose múltipla é uma doença da mielina, as suas evidências macroscópicas encontram-se na substância branca. As placas ocorrem normalmente ao lado dos ventrículos laterais, elas são bem circunscritas, algo deprimidas, vítreas e cinzentas.
14
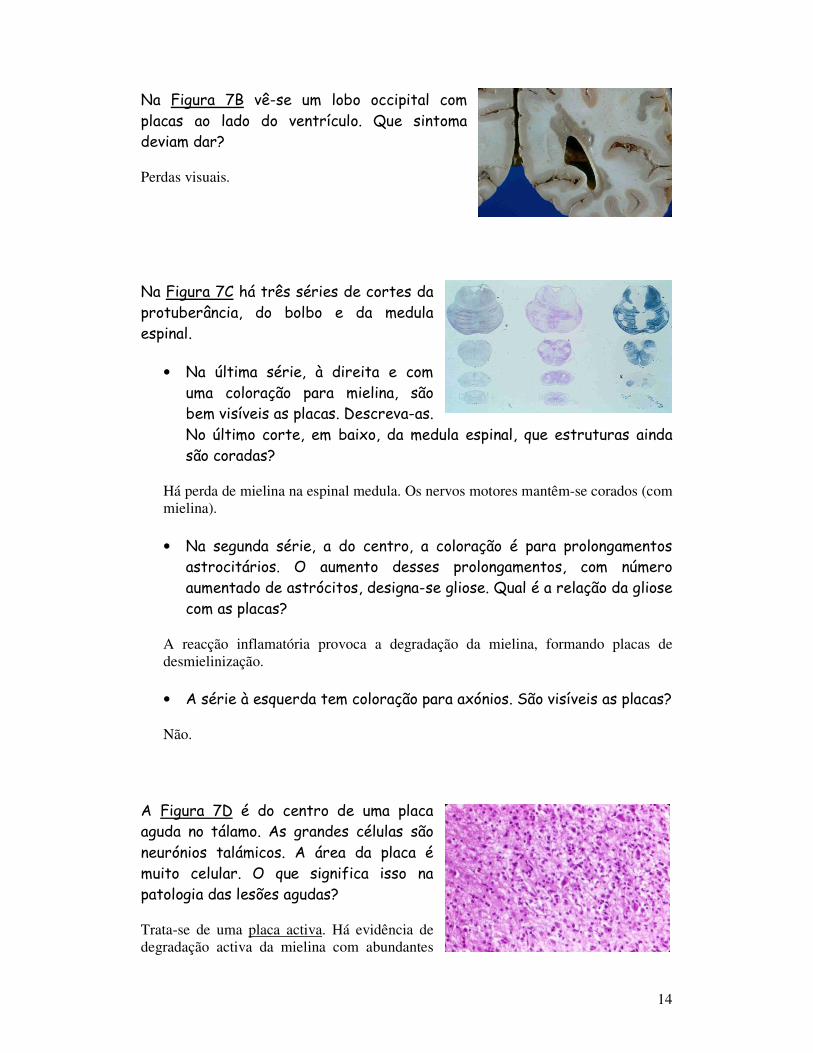
Na Figura 7B vê-se um lobo occipital com placas ao lado do ventrículo. Que sintoma deviam dar?
Perdas visuais.
Na Figura 7C há três séries de cortes da protuberância, do bolbo e da medula espinal.
• Na última série, à direita e com uma coloração para mielina, são bem visíveis as placas. Descreva-as. No último corte, em baixo, da medula espinal, que estruturas ainda são coradas?
Há perda de mielina na espinal medula. Os nervos motores mantêm-se corados (com mielina).
• Na segunda série, a do centro, a coloração é para prolongamentos astrocitários. O aumento desses prolongamentos, com número aumentado de astrócitos, designa-se gliose. Qual é a relação da gliose com as placas?
A reacção inflamatória provoca a degradação da mielina, formando placas de desmielinização.
• A série à esquerda tem coloração para axónios. São visíveis as placas?
Não.
A Figura 7D é do centro de uma placa aguda no tálamo. As grandes células são neurónios talámicos. A área da placa é muito celular. O que significa isso na patologia das lesões agudas?
Trata-se de uma placa activa. Há evidência de degradação activa da mielina com abundantes

15
macrófagos contendo resíduos ricos em lipídeos PAS positivos. Células inflamatórias (linfócitos e monócitos) estão presentes na maior parte dos aglomerados perivasculares, principalmente na margem externa da lesão. No interior da placa há uma preservação relativa dos axónios e redução no número de oligodendrócitos.
A Figura 7E mostra o centro de uma placa antiga. A densidade celular está normal? Que tipo de células em particular está diminuída?
Há uma diminuição do infiltrado celular inflamatório e dos macrófagos. No interior da placa inactiva há pouca ou nenhuma mielina, e há uma redução do número dos oligodentrócitos; ao contrário são proeminentes a proliferação astrocítica e a gliose. Os axónios nas placas glióticas antigas mostram severa depleção da mielina e estão também diminuídos em número.
A etiologia da esclerose múltipla ainda não é conhecida apesar de muitos anos de experimentação. Um modelo que influenciou muito as teorias desta doença foi o da encefalite auto-imune experimental. Um extracto de tecido cerebral misturado com um adjuvante é injectado no pé de um animal de uma estirpe sensível, e este animal 10 dias depois desenvolve doença desmielinizante. Várias proteínas da mielina podem causar esta resposta. O problema é a ausência de um marcador imunitário específico “para” a esclerose múltipla. Os tratamentos mais eficazes implicam um tipo de imunossupressão.
FIM
Pedimos desculpa pela demora...As perguntas que estiverem a vermelho não têm resposta ou porque não foram respondidas
na aula ou porque não estavam no livro...Desculpas por esta falha...e qualquer outra que encontrem...


















