
Direct link between metabolic regulation and the heat ... · Direct link between metabolic...
10
Direct link between metabolic regulation and the heat-shock response through the transcriptional regulator PGC-1α Neri Minsky and Robert G. Roeder 1 Laboratory of Biochemistry and Molecular Biology, The Rockefeller University, New York, NY 10065 Contributed by Robert G. Roeder, September 9, 2015 (sent for review June 29, 2015; reviewed by Jorge L. Ruas and Patrick Seale) In recent years an extensive effort has been made to elucidate the molecular pathways involved in metabolic signaling in health and disease. Here we show, surprisingly, that metabolic regulation and the heat-shock/stress response are directly linked. Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), a critical transcriptional coactivator of metabolic genes, acts as a direct tran- scriptional repressor of heat-shock factor 1 (HSF1), a key regulator of the heat-shock/stress response. Our findings reveal that heat- shock protein (HSP) gene expression is suppressed during fasting in mouse liver and in primary hepatocytes dependent on PGC-1α. HSF1 and PGC-1α associate physically and are colocalized on several HSP promoters. These observations are extended to several cancer cell lines in which PGC-1α is shown to repress the ability of HSF1 to activate gene-expression programs necessary for cancer survival. Our study reveals a surprising direct link between two major cellular transcriptional networks, highlighting a previously unrecognized facet of the activity of the central metabolic regulator PGC-1α be- yond its well-established ability to boost metabolic genes via its interactions with nuclear hormone receptors and nuclear respiratory factors. Our data point to PGC-1α as a critical repressor of HSF1- mediated transcriptional programs, a finding with possible implica- tions both for our understanding of the full scope of metabolically regulated target genes in vivo and, conceivably, for therapeutics. metabolism | transcription | stress response T he cellular response to metabolic demands and stimuli involves a complex network of transcription factors and coactivators that regulate the timely expression of a diverse array of target genes. Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) is a key metabolic transcriptional coactivator protein that associates with numerous transcription factors and increases their ability to induce expression of their cognate target genes (1, 2). A key attribute of PGC-1α is its ability to enhance oxi- dative metabolism and mitochondrial biogenesis (3). PGC-1α also can induce tissue-specific programs such as hepatic gluconeogen- esis (4), thermogenesis in brown adipose tissue (BAT) (5), and fiber-type switching in skeletal muscle (6). PGC-1α is induced by a variety of physiological stimuli in the tissues where it acts, in- cluding exercise in muscle, cold in BAT, and fasting or diabetes in the liver (1, 2). Mechanistically, PGC-1α induces gene expression via a strong transcriptional activation domain at its N terminus. This domain interacts with several lysine acetyltransferase com- plexes that include p300, CBP, and SRC-1 (7). The ability of PGC- 1α to coactivate nuclear hormone receptors depends on two N-terminal LXXLL motifs, designated L2 and L3, that mediate the interaction of PGC-1α with these transcription factors (8, 9). However, PGC-1α also was described as a coactivator of several nonnuclear hormone receptor transcription factors that include NRF1 and NRF2 (3). Although these interactions were demon- strated to regulate the expression of numerous transcriptional programs, the full scope of PGC-1α binding partners and regu- lated targets is not fully elucidated. A critical aspect of cellular protein homeostasis is the utilization of chaperone proteins responsible for correctly folding proteins and for stabilizing protein structure (10, 11). Chaperone proteins include heat-shock proteins (HSPs) within the HSP27, HSP40, HSP70, and HSP90 families as well as α-crystallin and class I and class II chaperonins. These proteins are part of the cellular heat- shock response (HSR), the cellular response to proteotoxic stimuli such as elevated temperatures and oxidative stress (10, 11). This response is controlled by the heat-shock factor (HSF) family of transcription factors (12) and in particular by HSF1, which is conserved from yeast to humans (13). Although HSF1 has long been known to bind to and activate promoters of various HSP genes as part of the HSR, it has been described more recently as a regulator of many additional transcriptional programs that include those controlling cell survival, ion transport, signal transduction, and other cellular processes (12, 14, 15). Consistent with its ver- satile functions in the regulation of numerous transcriptional programs, HSF1 also has been implicated in certain pathological conditions that include neurodegeneration and cancer (16, 17). Although HSF1 may not be strictly required for normal cellular or organismal survival in mammals (18), the role of HSF1 in regu- lating non-HSR targets, including those related to DNA damage repair and the cell cycle, recently has emerged as critical for cancer cell survival (19–21). Thus, inhibiting the activity of HSF1 has been suggested as a potential anticancer therapeutic approach (22). Although the HSPs are the products of genes activated by HSF1, they also have been reported to inhibit HSF1 in a negative feedback loop (23). For example, HSP70 and HSP40 interactions Significance In recent years an extensive effort has been made to elucidate the molecular pathways involved in metabolic signaling. Our study shows, surprisingly, a direct link between metabolic reg- ulation and the heat-shock response, a highly conserved tran- scriptional program that is activated in the presence of various environmental stresses. Specifically, we demonstrate that per- oxisome proliferator-activated receptor γ coactivator 1α, a criti- cal and well-established inducible transcriptional coactivator of metabolic genes, acts as a direct transcriptional repressor of heat- shock factor 1, a key regulator of the heat-shock/stress response and a factor more recently demonstrated to be necessary for cancer initiation and survival. Thus, our findings have possible implications both for our understanding of the full scope of metabolically regulated target genes in vivo and, conceivably, for therapeutics. Author contributions: N.M. and R.G.R. designed research; N.M. performed research; N.M. contributed new reagents/analytic tools; N.M. and R.G.R. analyzed data; and N.M. and R.G.R. wrote the paper. Reviewers: J.L.R., Karolinska Institute; and P.S., University of Pennsylvania. The authors declare no conflict of interest. Data deposition: Microarray data associated with this article have been deposited in the Gene Expression Omnibus database (accession no. GSE51498). 1 To whom correspondence should be addressed. Email: [email protected]. This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10. 1073/pnas.1516219112/-/DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1516219112 PNAS | Published online October 5, 2015 | E5669–E5678 CELL BIOLOGY PNAS PLUS
-
Upload
duongnguyet -
Category
Documents
-
view
224 -
download
1
Transcript of Direct link between metabolic regulation and the heat ... · Direct link between metabolic...

Direct link between metabolic regulation and theheat-shock response through the transcriptionalregulator PGC-1αNeri Minsky and Robert G. Roeder1
Laboratory of Biochemistry and Molecular Biology, The Rockefeller University, New York, NY 10065
Contributed by Robert G. Roeder, September 9, 2015 (sent for review June 29, 2015; reviewed by Jorge L. Ruas and Patrick Seale)
In recent years an extensive effort has been made to elucidate themolecular pathways involved in metabolic signaling in health anddisease. Here we show, surprisingly, that metabolic regulation andthe heat-shock/stress response are directly linked. Peroxisomeproliferator-activated receptor γ coactivator 1α (PGC-1α), a criticaltranscriptional coactivator of metabolic genes, acts as a direct tran-scriptional repressor of heat-shock factor 1 (HSF1), a key regulatorof the heat-shock/stress response. Our findings reveal that heat-shock protein (HSP) gene expression is suppressed during fastingin mouse liver and in primary hepatocytes dependent on PGC-1α.HSF1 and PGC-1α associate physically and are colocalized on severalHSP promoters. These observations are extended to several cancercell lines in which PGC-1α is shown to repress the ability of HSF1to activate gene-expression programs necessary for cancer survival.Our study reveals a surprising direct link between twomajor cellulartranscriptional networks, highlighting a previously unrecognizedfacet of the activity of the central metabolic regulator PGC-1α be-yond its well-established ability to boost metabolic genes via itsinteractions with nuclear hormone receptors and nuclear respiratoryfactors. Our data point to PGC-1α as a critical repressor of HSF1-mediated transcriptional programs, a finding with possible implica-tions both for our understanding of the full scope of metabolicallyregulated target genes in vivo and, conceivably, for therapeutics.
metabolism | transcription | stress response
The cellular response to metabolic demands and stimuli involvesa complex network of transcription factors and coactivators
that regulate the timely expression of a diverse array of targetgenes. Peroxisome proliferator-activated receptor γ coactivator 1α(PGC-1α) is a key metabolic transcriptional coactivator proteinthat associates with numerous transcription factors and increasestheir ability to induce expression of their cognate target genes(1, 2). A key attribute of PGC-1α is its ability to enhance oxi-dative metabolism and mitochondrial biogenesis (3). PGC-1α alsocan induce tissue-specific programs such as hepatic gluconeogen-esis (4), thermogenesis in brown adipose tissue (BAT) (5), andfiber-type switching in skeletal muscle (6). PGC-1α is induced by avariety of physiological stimuli in the tissues where it acts, in-cluding exercise in muscle, cold in BAT, and fasting or diabetes inthe liver (1, 2). Mechanistically, PGC-1α induces gene expressionvia a strong transcriptional activation domain at its N terminus.This domain interacts with several lysine acetyltransferase com-plexes that include p300, CBP, and SRC-1 (7). The ability of PGC-1α to coactivate nuclear hormone receptors depends on twoN-terminal LXXLL motifs, designated L2 and L3, that mediatethe interaction of PGC-1α with these transcription factors (8, 9).However, PGC-1α also was described as a coactivator of severalnonnuclear hormone receptor transcription factors that includeNRF1 and NRF2 (3). Although these interactions were demon-strated to regulate the expression of numerous transcriptionalprograms, the full scope of PGC-1α binding partners and regu-lated targets is not fully elucidated.A critical aspect of cellular protein homeostasis is the utilization
of chaperone proteins responsible for correctly folding proteins
and for stabilizing protein structure (10, 11). Chaperone proteinsinclude heat-shock proteins (HSPs) within the HSP27, HSP40,HSP70, and HSP90 families as well as α-crystallin and class I andclass II chaperonins. These proteins are part of the cellular heat-shock response (HSR), the cellular response to proteotoxic stimulisuch as elevated temperatures and oxidative stress (10, 11). Thisresponse is controlled by the heat-shock factor (HSF) family oftranscription factors (12) and in particular by HSF1, which isconserved from yeast to humans (13). Although HSF1 has longbeen known to bind to and activate promoters of various HSPgenes as part of the HSR, it has been described more recently as aregulator of many additional transcriptional programs that includethose controlling cell survival, ion transport, signal transduction,and other cellular processes (12, 14, 15). Consistent with its ver-satile functions in the regulation of numerous transcriptionalprograms, HSF1 also has been implicated in certain pathologicalconditions that include neurodegeneration and cancer (16, 17).Although HSF1 may not be strictly required for normal cellular ororganismal survival in mammals (18), the role of HSF1 in regu-lating non-HSR targets, including those related to DNA damagerepair and the cell cycle, recently has emerged as critical for cancercell survival (19–21). Thus, inhibiting the activity of HSF1 has beensuggested as a potential anticancer therapeutic approach (22).Although the HSPs are the products of genes activated by
HSF1, they also have been reported to inhibit HSF1 in a negativefeedback loop (23). For example, HSP70 and HSP40 interactions
Significance
In recent years an extensive effort has been made to elucidatethe molecular pathways involved in metabolic signaling. Ourstudy shows, surprisingly, a direct link between metabolic reg-ulation and the heat-shock response, a highly conserved tran-scriptional program that is activated in the presence of variousenvironmental stresses. Specifically, we demonstrate that per-oxisome proliferator-activated receptor γ coactivator 1α, a criti-cal and well-established inducible transcriptional coactivator ofmetabolic genes, acts as a direct transcriptional repressor of heat-shock factor 1, a key regulator of the heat-shock/stress responseand a factor more recently demonstrated to be necessary forcancer initiation and survival. Thus, our findings have possibleimplications both for our understanding of the full scope ofmetabolically regulated target genes in vivo and, conceivably,for therapeutics.
Author contributions: N.M. and R.G.R. designed research; N.M. performed research; N.M.contributed new reagents/analytic tools; N.M. and R.G.R. analyzed data; and N.M. and R.G.R.wrote the paper.
Reviewers: J.L.R., Karolinska Institute; and P.S., University of Pennsylvania.
The authors declare no conflict of interest.
Data deposition: Microarray data associated with this article have been deposited in theGene Expression Omnibus database (accession no. GSE51498).1To whom correspondence should be addressed. Email: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1516219112/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1516219112 PNAS | Published online October 5, 2015 | E5669–E5678
CELL
BIOLO
GY
PNASPL
US
with HSF1 have been shown to inhibit its transactivation capacity,although the inhibitory mechanism is still unknown (24). Thus, theexact mechanisms and physiological stimuli that regulate HSF1remain incompletely understood.Here we show that PGC-1α is a direct binding partner of HSF1
and acts to repress its transcriptional activity. Using a variety ofcell types, as well as mice, we demonstrate the repressive effectsof PGC-1α on HSF1 in hepatocytes and in muscle. Although thereare a number of PGC-1α isoforms (25), our studies are focused onPGC-1α1, the coactivator variant with the best-established effectson cellular bioenergetics. Although initially focusing onHSP genesas targets of PGC-1α activity, we extend our observations to genesfrom additional transcriptional programs controlled by HSF1 incancer cells and show that PGC-1α can associate with the pro-moters of the target genes that it represses. In addition, by usingbiochemical and cellular approaches, we demonstrate a physicalassociation between HSF1 and PGC-1α and the effects of PGC-1αon HSF1-driven transcription. Thus, this work broadens our viewof PGC-1α as a transcriptional regulator and identifies the PGC-1α/HSF1 axis as a potential target for therapeutics.
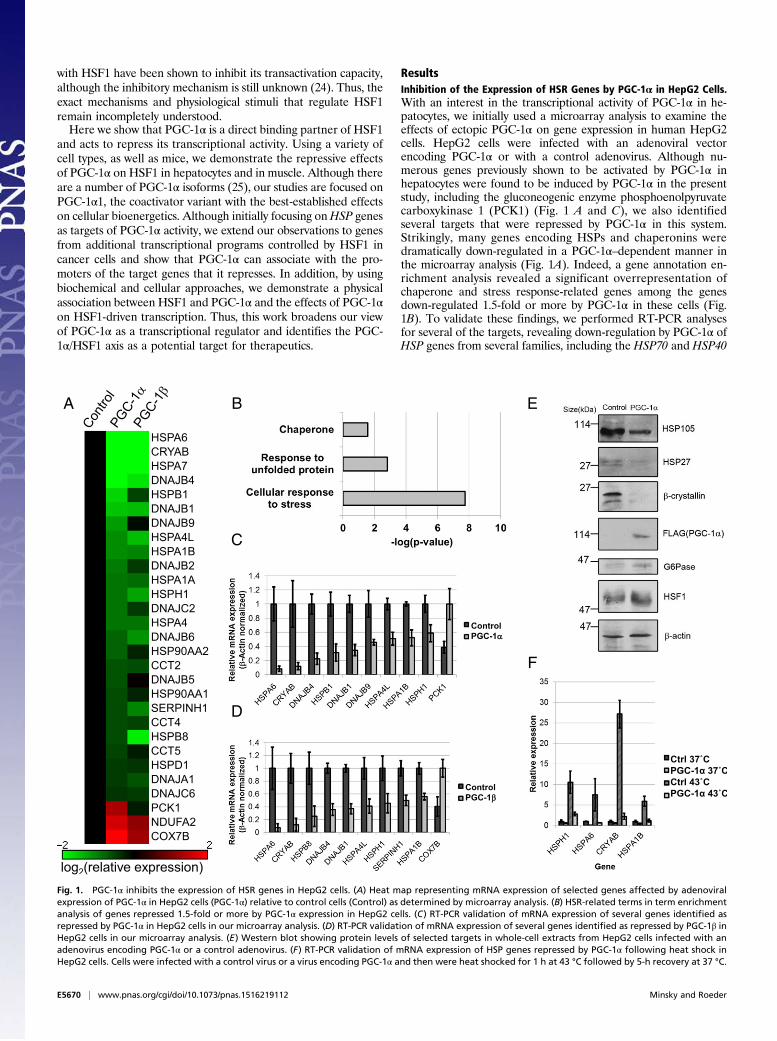
ResultsInhibition of the Expression of HSR Genes by PGC-1α in HepG2 Cells.With an interest in the transcriptional activity of PGC-1α in he-patocytes, we initially used a microarray analysis to examine theeffects of ectopic PGC-1α on gene expression in human HepG2cells. HepG2 cells were infected with an adenoviral vectorencoding PGC-1α or with a control adenovirus. Although nu-merous genes previously shown to be activated by PGC-1α inhepatocytes were found to be induced by PGC-1α in the presentstudy, including the gluconeogenic enzyme phosphoenolpyruvatecarboxykinase 1 (PCK1) (Fig. 1 A and C), we also identifiedseveral targets that were repressed by PGC-1α in this system.Strikingly, many genes encoding HSPs and chaperonins weredramatically down-regulated in a PGC-1α–dependent manner inthe microarray analysis (Fig. 1A). Indeed, a gene annotation en-richment analysis revealed a significant overrepresentation ofchaperone and stress response-related genes among the genesdown-regulated 1.5-fold or more by PGC-1α in these cells (Fig.1B). To validate these findings, we performed RT-PCR analysesfor several of the targets, revealing down-regulation by PGC-1α ofHSP genes from several families, including the HSP70 and HSP40
A B E
F
C
D
Fig. 1. PGC-1α inhibits the expression of HSR genes in HepG2 cells. (A) Heat map representing mRNA expression of selected genes affected by adenoviralexpression of PGC-1α in HepG2 cells (PGC-1α) relative to control cells (Control) as determined by microarray analysis. (B) HSR-related terms in term enrichmentanalysis of genes repressed 1.5-fold or more by PGC-1α expression in HepG2 cells. (C) RT-PCR validation of mRNA expression of several genes identified asrepressed by PGC-1α in HepG2 cells in our microarray analysis. (D) RT-PCR validation of mRNA expression of several genes identified as repressed by PGC-1β inHepG2 cells in our microarray analysis. (E) Western blot showing protein levels of selected targets in whole-cell extracts from HepG2 cells infected with anadenovirus encoding PGC-1α or a control adenovirus. (F) RT-PCR validation of mRNA expression of HSP genes repressed by PGC-1α following heat shock inHepG2 cells. Cells were infected with a control virus or a virus encoding PGC-1α and then were heat shocked for 1 h at 43 °C followed by 5-h recovery at 37 °C.
E5670 | www.pnas.org/cgi/doi/10.1073/pnas.1516219112 Minsky and Roeder
families (Fig. 1C). We also examined the expression of severaltargets by immunoblot, finding a consistent down-regulation of thelevels of HSP proteins, including HSP105 and HSP27 (Fig. 1E).PGC-1β, a closely related homolog of PGC-1α, has both over-
lapping and divergent transcriptional programs with PGC-1α inliver (26). Therefore, we also analyzed the effects of PGC-1β ongene expression in HepG2 cells. Although, as expected (26), PGC-1β failed to induce the gluconeogenic gene PCK1 (Fig. 1A), it in-duced the expression of other known targets of PGC-1 familymembers (Fig. 1 A and D). Importantly, it was able to inhibit theexpression of numerousHSP genes (Fig. 1 A andD), many of whichalso were repressed by PGC-1α in this system. Finally, PGC-1α wasable to repress the induced mRNA expression of HSP during heatstress in these cells (Fig. 1F), broadening the physiological contextin which PGC-1α may act to suppress HSP gene expression.
PGC-1α Suppresses HSR Gene Expression in Human Primary Hepatocytesand in Mouse Liver.Because HepG2 cells are cancer cells, we wishedto examine whether PGC-1α can exert its inhibitory effects on HSP
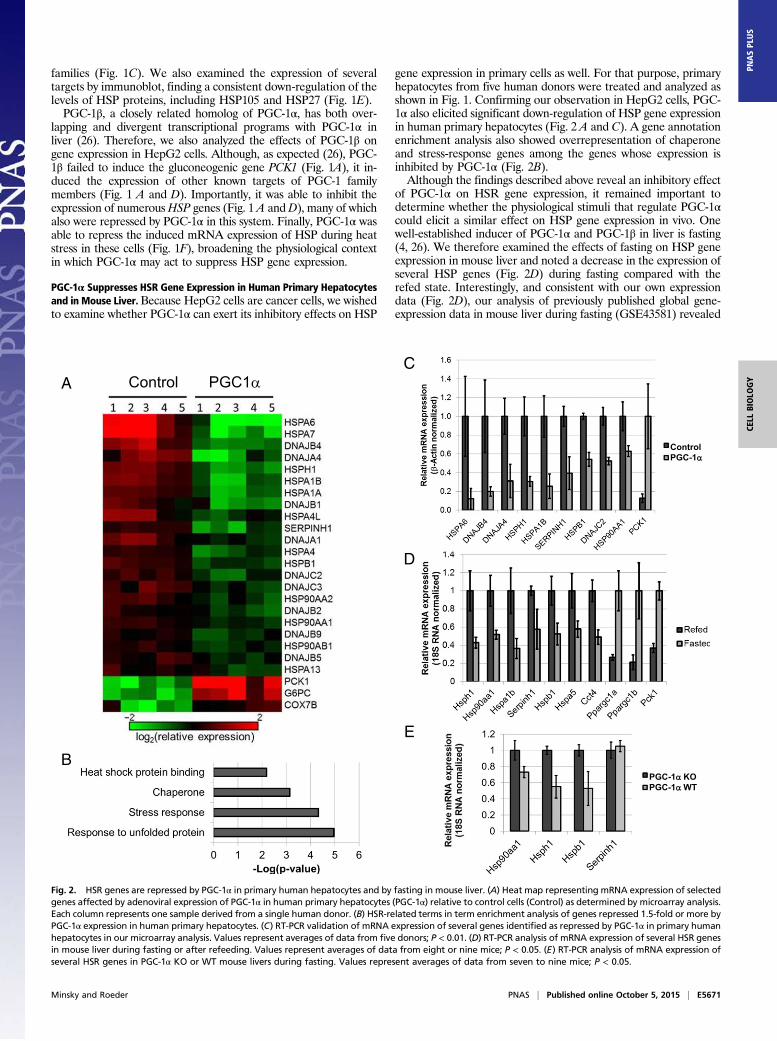
gene expression in primary cells as well. For that purpose, primaryhepatocytes from five human donors were treated and analyzed asshown in Fig. 1. Confirming our observation in HepG2 cells, PGC-1α also elicited significant down-regulation of HSP gene expressionin human primary hepatocytes (Fig. 2 A and C). A gene annotationenrichment analysis also showed overrepresentation of chaperoneand stress-response genes among the genes whose expression isinhibited by PGC-1α (Fig. 2B).Although the findings described above reveal an inhibitory effect
of PGC-1α on HSR gene expression, it remained important todetermine whether the physiological stimuli that regulate PGC-1αcould elicit a similar effect on HSP gene expression in vivo. Onewell-established inducer of PGC-1α and PGC-1β in liver is fasting(4, 26). We therefore examined the effects of fasting on HSP geneexpression in mouse liver and noted a decrease in the expression ofseveral HSP genes (Fig. 2D) during fasting compared with therefed state. Interestingly, and consistent with our own expressiondata (Fig. 2D), our analysis of previously published global gene-expression data in mouse liver during fasting (GSE43581) revealed
AC
D
B
E
Fig. 2. HSR genes are repressed by PGC-1α in primary human hepatocytes and by fasting in mouse liver. (A) Heat map representing mRNA expression of selectedgenes affected by adenoviral expression of PGC-1α in human primary hepatocytes (PGC-1α) relative to control cells (Control) as determined by microarray analysis.Each column represents one sample derived from a single human donor. (B) HSR-related terms in term enrichment analysis of genes repressed 1.5-fold or more byPGC-1α expression in human primary hepatocytes. (C) RT-PCR validation of mRNA expression of several genes identified as repressed by PGC-1α in primary humanhepatocytes in our microarray analysis. Values represent averages of data from five donors; P < 0.01. (D) RT-PCR analysis of mRNA expression of several HSR genesin mouse liver during fasting or after refeeding. Values represent averages of data from eight or nine mice; P < 0.05. (E) RT-PCR analysis of mRNA expression ofseveral HSR genes in PGC-1α KO or WT mouse livers during fasting. Values represent averages of data from seven to nine mice; P < 0.05.
Minsky and Roeder PNAS | Published online October 5, 2015 | E5671
CELL
BIOLO
GY
PNASPL
US
a significant overrepresentation of genes related to chaperonesand protein-folding among the genes repressed during fasting(Fig. S1).Although PGC-1α and PGC-1β appear to have somewhat
overlapping functions with respect to HSR regulation (Fig. 1 A,C, and D), we wondered whether ablating the activity of en-dogenous PGC-1α alone would be sufficient to affect HSR geneexpression. For this purpose we used a previously describedmouse strain harboring a floxed allele of the gene encodingPGC-1α (27) crossed with a strain harboring CRE recombinaseunder the control of the liver-specific albumin promoter. Whenexamining gene expression in Pgc1α WT or KO livers underfasting conditions, we noted that several HSP genes were re-pressed in the WT livers compared with the KO livers (Fig. 2E).
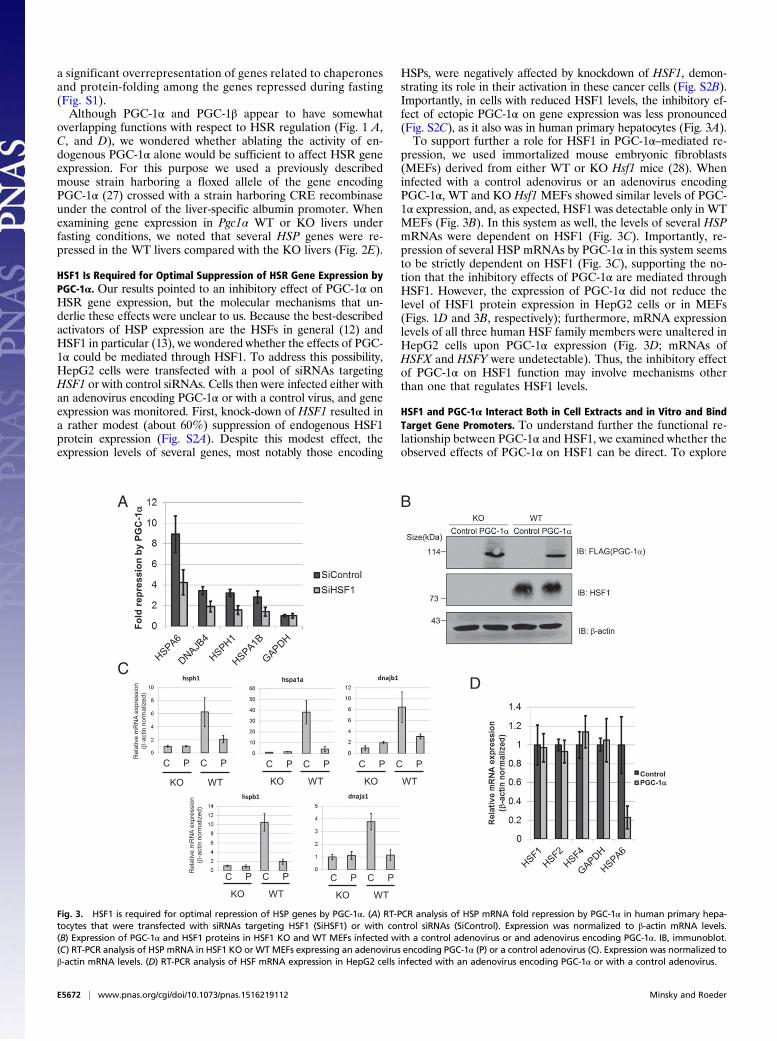
HSF1 Is Required for Optimal Suppression of HSR Gene Expression byPGC-1α. Our results pointed to an inhibitory effect of PGC-1α onHSR gene expression, but the molecular mechanisms that un-derlie these effects were unclear to us. Because the best-describedactivators of HSP expression are the HSFs in general (12) andHSF1 in particular (13), we wondered whether the effects of PGC-1α could be mediated through HSF1. To address this possibility,HepG2 cells were transfected with a pool of siRNAs targetingHSF1 or with control siRNAs. Cells then were infected either withan adenovirus encoding PGC-1α or with a control virus, and geneexpression was monitored. First, knock-down of HSF1 resulted ina rather modest (about 60%) suppression of endogenous HSF1protein expression (Fig. S2A). Despite this modest effect, theexpression levels of several genes, most notably those encoding
HSPs, were negatively affected by knockdown of HSF1, demon-strating its role in their activation in these cancer cells (Fig. S2B).Importantly, in cells with reduced HSF1 levels, the inhibitory ef-fect of ectopic PGC-1α on gene expression was less pronounced(Fig. S2C), as it also was in human primary hepatocytes (Fig. 3A).To support further a role for HSF1 in PGC-1α–mediated re-
pression, we used immortalized mouse embryonic fibroblasts(MEFs) derived from either WT or KO Hsf1 mice (28). Wheninfected with a control adenovirus or an adenovirus encodingPGC-1α, WT and KO Hsf1 MEFs showed similar levels of PGC-1α expression, and, as expected, HSF1 was detectable only in WTMEFs (Fig. 3B). In this system as well, the levels of several HSPmRNAs were dependent on HSF1 (Fig. 3C). Importantly, re-pression of several HSP mRNAs by PGC-1α in this system seemsto be strictly dependent on HSF1 (Fig. 3C), supporting the no-tion that the inhibitory effects of PGC-1α are mediated throughHSF1. However, the expression of PGC-1α did not reduce thelevel of HSF1 protein expression in HepG2 cells or in MEFs(Figs. 1D and 3B, respectively); furthermore, mRNA expressionlevels of all three human HSF family members were unaltered inHepG2 cells upon PGC-1α expression (Fig. 3D; mRNAs ofHSFX and HSFY were undetectable). Thus, the inhibitory effectof PGC-1α on HSF1 function may involve mechanisms otherthan one that regulates HSF1 levels.
HSF1 and PGC-1α Interact Both in Cell Extracts and in Vitro and BindTarget Gene Promoters. To understand further the functional re-lationship between PGC-1α and HSF1, we examined whether theobserved effects of PGC-1α on HSF1 can be direct. To explore
A
CD
B
Fig. 3. HSF1 is required for optimal repression of HSP genes by PGC-1α. (A) RT-PCR analysis of HSP mRNA fold repression by PGC-1α in human primary hepa-tocytes that were transfected with siRNAs targeting HSF1 (SiHSF1) or with control siRNAs (SiControl). Expression was normalized to β-actin mRNA levels.(B) Expression of PGC-1α and HSF1 proteins in HSF1 KO and WT MEFs infected with a control adenovirus or and adenovirus encoding PGC-1α. IB, immunoblot.(C) RT-PCR analysis of HSP mRNA in HSF1 KO or WTMEFs expressing an adenovirus encoding PGC-1α (P) or a control adenovirus (C). Expression was normalized toβ-actin mRNA levels. (D) RT-PCR analysis of HSF mRNA expression in HepG2 cells infected with an adenovirus encoding PGC-1α or with a control adenovirus.
E5672 | www.pnas.org/cgi/doi/10.1073/pnas.1516219112 Minsky and Roeder
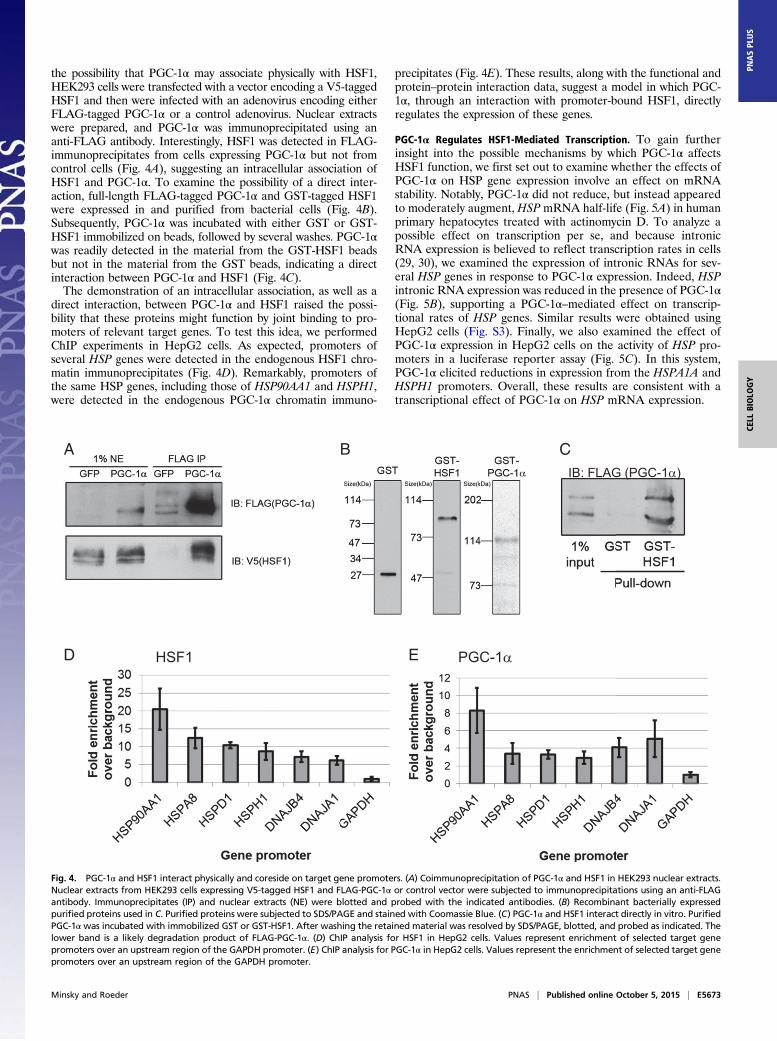
the possibility that PGC-1α may associate physically with HSF1,HEK293 cells were transfected with a vector encoding a V5-taggedHSF1 and then were infected with an adenovirus encoding eitherFLAG-tagged PGC-1α or a control adenovirus. Nuclear extractswere prepared, and PGC-1α was immunoprecipitated using ananti-FLAG antibody. Interestingly, HSF1 was detected in FLAG-immunoprecipitates from cells expressing PGC-1α but not fromcontrol cells (Fig. 4A), suggesting an intracellular association ofHSF1 and PGC-1α. To examine the possibility of a direct inter-action, full-length FLAG-tagged PGC-1α and GST-tagged HSF1were expressed in and purified from bacterial cells (Fig. 4B).Subsequently, PGC-1α was incubated with either GST or GST-HSF1 immobilized on beads, followed by several washes. PGC-1αwas readily detected in the material from the GST-HSF1 beadsbut not in the material from the GST beads, indicating a directinteraction between PGC-1α and HSF1 (Fig. 4C).The demonstration of an intracellular association, as well as a
direct interaction, between PGC-1α and HSF1 raised the possi-bility that these proteins might function by joint binding to pro-moters of relevant target genes. To test this idea, we performedChIP experiments in HepG2 cells. As expected, promoters ofseveral HSP genes were detected in the endogenous HSF1 chro-matin immunoprecipitates (Fig. 4D). Remarkably, promoters ofthe same HSP genes, including those of HSP90AA1 and HSPH1,were detected in the endogenous PGC-1α chromatin immuno-
precipitates (Fig. 4E). These results, along with the functional andprotein–protein interaction data, suggest a model in which PGC-1α, through an interaction with promoter-bound HSF1, directlyregulates the expression of these genes.
PGC-1α Regulates HSF1-Mediated Transcription. To gain furtherinsight into the possible mechanisms by which PGC-1α affectsHSF1 function, we first set out to examine whether the effects ofPGC-1α on HSP gene expression involve an effect on mRNAstability. Notably, PGC-1α did not reduce, but instead appearedto moderately augment, HSP mRNA half-life (Fig. 5A) in humanprimary hepatocytes treated with actinomycin D. To analyze apossible effect on transcription per se, and because intronicRNA expression is believed to reflect transcription rates in cells(29, 30), we examined the expression of intronic RNAs for sev-eral HSP genes in response to PGC-1α expression. Indeed, HSPintronic RNA expression was reduced in the presence of PGC-1α(Fig. 5B), supporting a PGC-1α–mediated effect on transcrip-tional rates of HSP genes. Similar results were obtained usingHepG2 cells (Fig. S3). Finally, we also examined the effect ofPGC-1α expression in HepG2 cells on the activity of HSP pro-moters in a luciferase reporter assay (Fig. 5C). In this system,PGC-1α elicited reductions in expression from the HSPA1A andHSPH1 promoters. Overall, these results are consistent with atranscriptional effect of PGC-1α on HSP mRNA expression.
A
D E
B C
Fig. 4. PGC-1α and HSF1 interact physically and coreside on target gene promoters. (A) Coimmunoprecipitation of PGC-1α and HSF1 in HEK293 nuclear extracts.Nuclear extracts from HEK293 cells expressing V5-tagged HSF1 and FLAG-PGC-1α or control vector were subjected to immunoprecipitations using an anti-FLAGantibody. Immunoprecipitates (IP) and nuclear extracts (NE) were blotted and probed with the indicated antibodies. (B) Recombinant bacterially expressedpurified proteins used in C. Purified proteins were subjected to SDS/PAGE and stained with Coomassie Blue. (C) PGC-1α and HSF1 interact directly in vitro. PurifiedPGC-1α was incubated with immobilized GST or GST-HSF1. After washing the retained material was resolved by SDS/PAGE, blotted, and probed as indicated. Thelower band is a likely degradation product of FLAG-PGC-1α. (D) ChIP analysis for HSF1 in HepG2 cells. Values represent enrichment of selected target genepromoters over an upstream region of the GAPDH promoter. (E) ChIP analysis for PGC-1α in HepG2 cells. Values represent the enrichment of selected target genepromoters over an upstream region of the GAPDH promoter.
Minsky and Roeder PNAS | Published online October 5, 2015 | E5673
CELL
BIOLO
GY
PNASPL
US
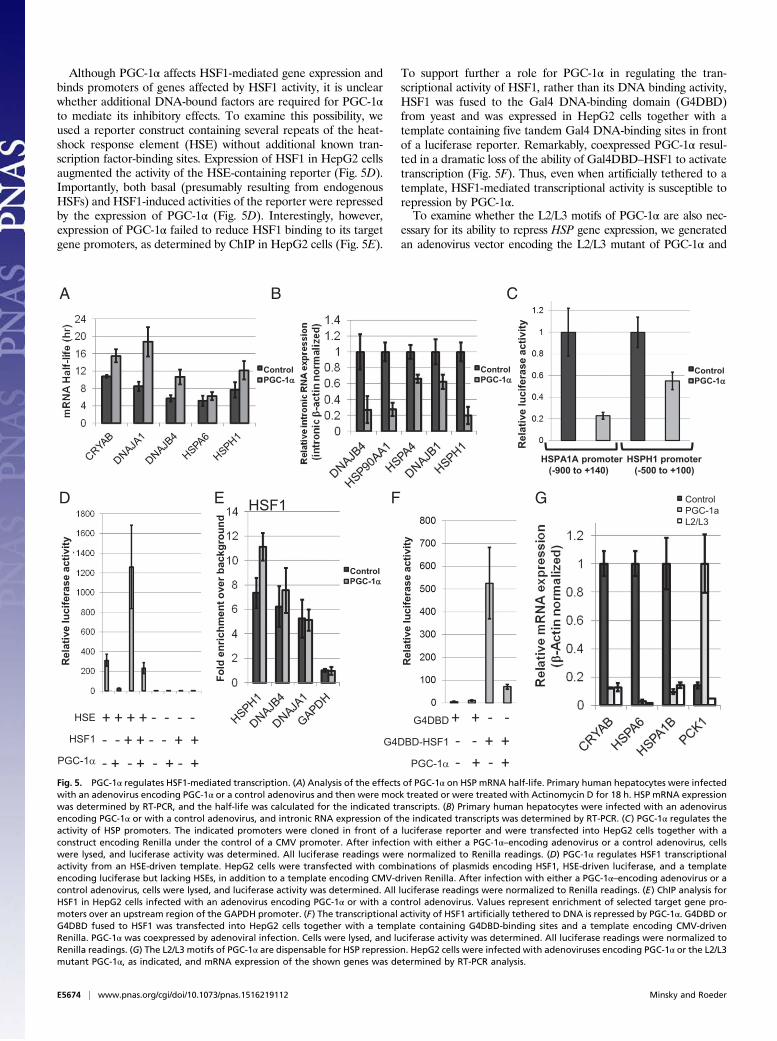
Although PGC-1α affects HSF1-mediated gene expression andbinds promoters of genes affected by HSF1 activity, it is unclearwhether additional DNA-bound factors are required for PGC-1αto mediate its inhibitory effects. To examine this possibility, weused a reporter construct containing several repeats of the heat-shock response element (HSE) without additional known tran-scription factor-binding sites. Expression of HSF1 in HepG2 cellsaugmented the activity of the HSE-containing reporter (Fig. 5D).Importantly, both basal (presumably resulting from endogenousHSFs) and HSF1-induced activities of the reporter were repressedby the expression of PGC-1α (Fig. 5D). Interestingly, however,expression of PGC-1α failed to reduce HSF1 binding to its targetgene promoters, as determined by ChIP in HepG2 cells (Fig. 5E).
To support further a role for PGC-1α in regulating the tran-scriptional activity of HSF1, rather than its DNA binding activity,HSF1 was fused to the Gal4 DNA-binding domain (G4DBD)from yeast and was expressed in HepG2 cells together with atemplate containing five tandem Gal4 DNA-binding sites in frontof a luciferase reporter. Remarkably, coexpressed PGC-1α resul-ted in a dramatic loss of the ability of Gal4DBD–HSF1 to activatetranscription (Fig. 5F). Thus, even when artificially tethered to atemplate, HSF1-mediated transcriptional activity is susceptible torepression by PGC-1α.To examine whether the L2/L3 motifs of PGC-1α are also nec-
essary for its ability to repress HSP gene expression, we generatedan adenovirus vector encoding the L2/L3 mutant of PGC-1α and
A
D E F G
B C
Fig. 5. PGC-1α regulates HSF1-mediated transcription. (A) Analysis of the effects of PGC-1α on HSP mRNA half-life. Primary human hepatocytes were infectedwith an adenovirus encoding PGC-1α or a control adenovirus and then were mock treated or were treated with Actinomycin D for 18 h. HSP mRNA expressionwas determined by RT-PCR, and the half-life was calculated for the indicated transcripts. (B) Primary human hepatocytes were infected with an adenovirusencoding PGC-1α or with a control adenovirus, and intronic RNA expression of the indicated transcripts was determined by RT-PCR. (C) PGC-1α regulates theactivity of HSP promoters. The indicated promoters were cloned in front of a luciferase reporter and were transfected into HepG2 cells together with aconstruct encoding Renilla under the control of a CMV promoter. After infection with either a PGC-1α–encoding adenovirus or a control adenovirus, cellswere lysed, and luciferase activity was determined. All luciferase readings were normalized to Renilla readings. (D) PGC-1α regulates HSF1 transcriptionalactivity from an HSE-driven template. HepG2 cells were transfected with combinations of plasmids encoding HSF1, HSE-driven luciferase, and a templateencoding luciferase but lacking HSEs, in addition to a template encoding CMV-driven Renilla. After infection with either a PGC-1α–encoding adenovirus or acontrol adenovirus, cells were lysed, and luciferase activity was determined. All luciferase readings were normalized to Renilla readings. (E) ChIP analysis forHSF1 in HepG2 cells infected with an adenovirus encoding PGC-1α or with a control adenovirus. Values represent enrichment of selected target gene pro-moters over an upstream region of the GAPDH promoter. (F) The transcriptional activity of HSF1 artificially tethered to DNA is repressed by PGC-1α. G4DBD orG4DBD fused to HSF1 was transfected into HepG2 cells together with a template containing G4DBD-binding sites and a template encoding CMV-drivenRenilla. PGC-1α was coexpressed by adenoviral infection. Cells were lysed, and luciferase activity was determined. All luciferase readings were normalized toRenilla readings. (G) The L2/L3 motifs of PGC-1α are dispensable for HSP repression. HepG2 cells were infected with adenoviruses encoding PGC-1α or the L2/L3mutant PGC-1α, as indicated, and mRNA expression of the shown genes was determined by RT-PCR analysis.
E5674 | www.pnas.org/cgi/doi/10.1073/pnas.1516219112 Minsky and Roeder
used it to infect HepG2 cells. The PGC-1α WT and L2/L3 mutantproteins were equally efficient in repressing several HSF1 targetgenes (Fig. 5G), suggesting that these motifs and nuclear hormonereceptor binding via these motifs (8, 9) are dispensable for theinhibitory effects of PGC-1α on HSF1.
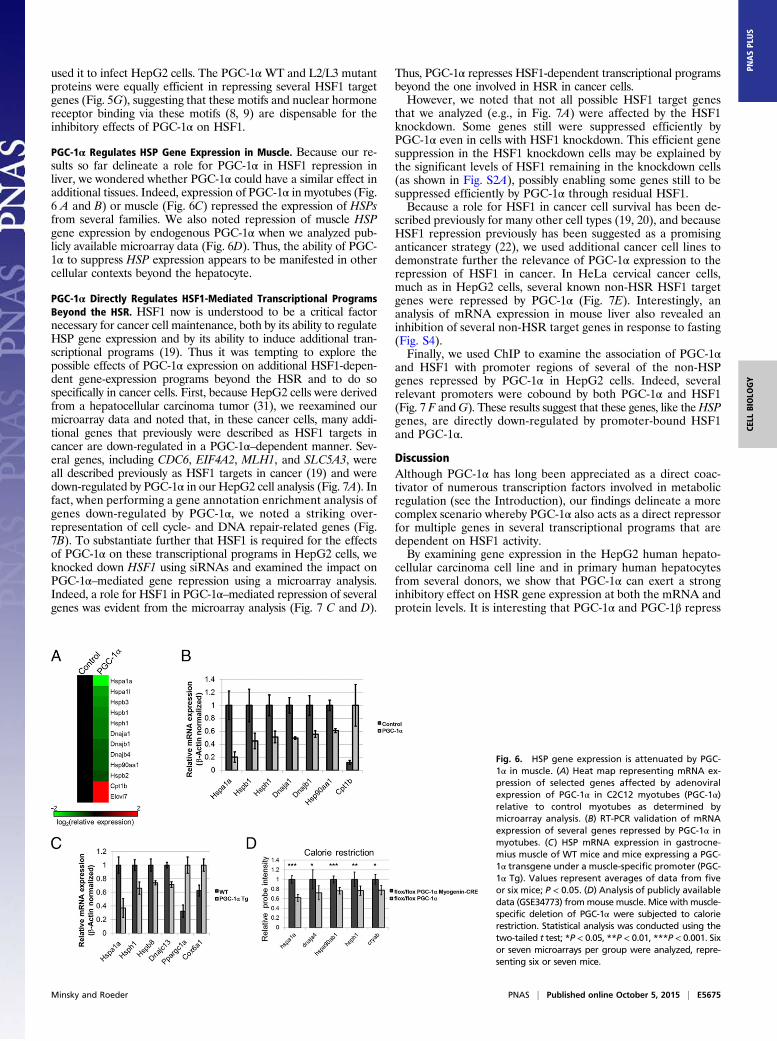
PGC-1α Regulates HSP Gene Expression in Muscle. Because our re-sults so far delineate a role for PGC-1α in HSF1 repression inliver, we wondered whether PGC-1α could have a similar effect inadditional tissues. Indeed, expression of PGC-1α in myotubes (Fig.6 A and B) or muscle (Fig. 6C) repressed the expression of HSPsfrom several families. We also noted repression of muscle HSPgene expression by endogenous PGC-1α when we analyzed pub-licly available microarray data (Fig. 6D). Thus, the ability of PGC-1α to suppress HSP expression appears to be manifested in othercellular contexts beyond the hepatocyte.
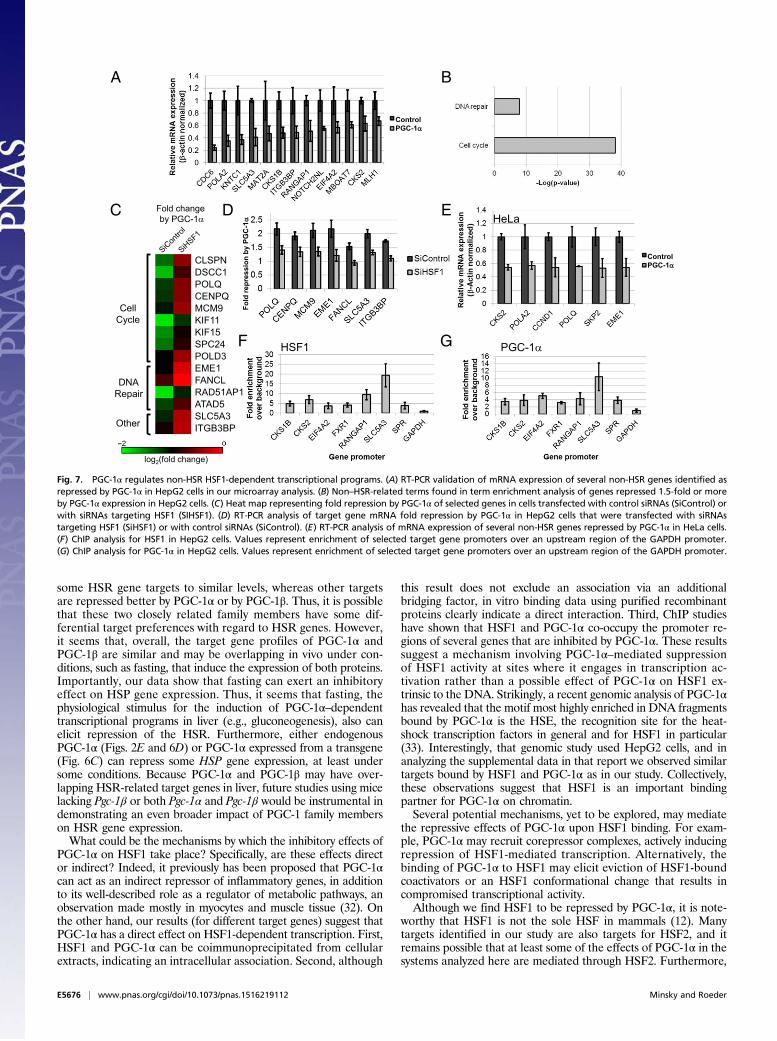
PGC-1α Directly Regulates HSF1-Mediated Transcriptional ProgramsBeyond the HSR. HSF1 now is understood to be a critical factornecessary for cancer cell maintenance, both by its ability to regulateHSP gene expression and by its ability to induce additional tran-scriptional programs (19). Thus it was tempting to explore thepossible effects of PGC-1α expression on additional HSF1-depen-dent gene-expression programs beyond the HSR and to do sospecifically in cancer cells. First, because HepG2 cells were derivedfrom a hepatocellular carcinoma tumor (31), we reexamined ourmicroarray data and noted that, in these cancer cells, many addi-tional genes that previously were described as HSF1 targets incancer are down-regulated in a PGC-1α–dependent manner. Sev-eral genes, including CDC6, EIF4A2, MLH1, and SLC5A3, wereall described previously as HSF1 targets in cancer (19) and weredown-regulated by PGC-1α in our HepG2 cell analysis (Fig. 7A). Infact, when performing a gene annotation enrichment analysis ofgenes down-regulated by PGC-1α, we noted a striking over-representation of cell cycle- and DNA repair-related genes (Fig.7B). To substantiate further that HSF1 is required for the effectsof PGC-1α on these transcriptional programs in HepG2 cells, weknocked down HSF1 using siRNAs and examined the impact onPGC-1α–mediated gene repression using a microarray analysis.Indeed, a role for HSF1 in PGC-1α–mediated repression of severalgenes was evident from the microarray analysis (Fig. 7 C and D).
Thus, PGC-1α represses HSF1-dependent transcriptional programsbeyond the one involved in HSR in cancer cells.However, we noted that not all possible HSF1 target genes
that we analyzed (e.g., in Fig. 7A) were affected by the HSF1knockdown. Some genes still were suppressed efficiently byPGC-1α even in cells with HSF1 knockdown. This efficient genesuppression in the HSF1 knockdown cells may be explained bythe significant levels of HSF1 remaining in the knockdown cells(as shown in Fig. S2A), possibly enabling some genes still to besuppressed efficiently by PGC-1α through residual HSF1.Because a role for HSF1 in cancer cell survival has been de-
scribed previously for many other cell types (19, 20), and becauseHSF1 repression previously has been suggested as a promisinganticancer strategy (22), we used additional cancer cell lines todemonstrate further the relevance of PGC-1α expression to therepression of HSF1 in cancer. In HeLa cervical cancer cells,much as in HepG2 cells, several known non-HSR HSF1 targetgenes were repressed by PGC-1α (Fig. 7E). Interestingly, ananalysis of mRNA expression in mouse liver also revealed aninhibition of several non-HSR target genes in response to fasting(Fig. S4).Finally, we used ChIP to examine the association of PGC-1α
and HSF1 with promoter regions of several of the non-HSPgenes repressed by PGC-1α in HepG2 cells. Indeed, severalrelevant promoters were cobound by both PGC-1α and HSF1(Fig. 7 F andG). These results suggest that these genes, like theHSPgenes, are directly down-regulated by promoter-bound HSF1and PGC-1α.
DiscussionAlthough PGC-1α has long been appreciated as a direct coac-tivator of numerous transcription factors involved in metabolicregulation (see the Introduction), our findings delineate a morecomplex scenario whereby PGC-1α also acts as a direct repressorfor multiple genes in several transcriptional programs that aredependent on HSF1 activity.By examining gene expression in the HepG2 human hepato-
cellular carcinoma cell line and in primary human hepatocytesfrom several donors, we show that PGC-1α can exert a stronginhibitory effect on HSR gene expression at both the mRNA andprotein levels. It is interesting that PGC-1α and PGC-1β repress
Fig. 6. HSP gene expression is attenuated by PGC-1α in muscle. (A) Heat map representing mRNA ex-pression of selected genes affected by adenoviralexpression of PGC-1α in C2C12 myotubes (PGC-1α)relative to control myotubes as determined bymicroarray analysis. (B) RT-PCR validation of mRNAexpression of several genes repressed by PGC-1α inmyotubes. (C) HSP mRNA expression in gastrocne-mius muscle of WT mice and mice expressing a PGC-1α transgene under a muscle-specific promoter (PGC-1α Tg). Values represent averages of data from fiveor six mice; P < 0.05. (D) Analysis of publicly availabledata (GSE34773) frommousemuscle. Mice with muscle-specific deletion of PGC-1α were subjected to calorierestriction. Statistical analysis was conducted using thetwo-tailed t test; *P < 0.05, **P < 0.01, ***P < 0.001. Sixor seven microarrays per group were analyzed, repre-senting six or seven mice.
Minsky and Roeder PNAS | Published online October 5, 2015 | E5675
CELL
BIOLO
GY
PNASPL
US
some HSR gene targets to similar levels, whereas other targetsare repressed better by PGC-1α or by PGC-1β. Thus, it is possiblethat these two closely related family members have some dif-ferential target preferences with regard to HSR genes. However,it seems that, overall, the target gene profiles of PGC-1α andPGC-1β are similar and may be overlapping in vivo under con-ditions, such as fasting, that induce the expression of both proteins.Importantly, our data show that fasting can exert an inhibitoryeffect on HSP gene expression. Thus, it seems that fasting, thephysiological stimulus for the induction of PGC-1α–dependenttranscriptional programs in liver (e.g., gluconeogenesis), also canelicit repression of the HSR. Furthermore, either endogenousPGC-1α (Figs. 2E and 6D) or PGC-1α expressed from a transgene(Fig. 6C) can repress some HSP gene expression, at least undersome conditions. Because PGC-1α and PGC-1β may have over-lapping HSR-related target genes in liver, future studies using micelacking Pgc-1β or both Pgc-1α and Pgc-1β would be instrumental indemonstrating an even broader impact of PGC-1 family memberson HSR gene expression.What could be the mechanisms by which the inhibitory effects of
PGC-1α on HSF1 take place? Specifically, are these effects director indirect? Indeed, it previously has been proposed that PGC-1αcan act as an indirect repressor of inflammatory genes, in additionto its well-described role as a regulator of metabolic pathways, anobservation made mostly in myocytes and muscle tissue (32). Onthe other hand, our results (for different target genes) suggest thatPGC-1α has a direct effect on HSF1-dependent transcription. First,HSF1 and PGC-1α can be coimmunoprecipitated from cellularextracts, indicating an intracellular association. Second, although
this result does not exclude an association via an additionalbridging factor, in vitro binding data using purified recombinantproteins clearly indicate a direct interaction. Third, ChIP studieshave shown that HSF1 and PGC-1α co-occupy the promoter re-gions of several genes that are inhibited by PGC-1α. These resultssuggest a mechanism involving PGC-1α–mediated suppressionof HSF1 activity at sites where it engages in transcription ac-tivation rather than a possible effect of PGC-1α on HSF1 ex-trinsic to the DNA. Strikingly, a recent genomic analysis of PGC-1αhas revealed that the motif most highly enriched in DNA fragmentsbound by PGC-1α is the HSE, the recognition site for the heat-shock transcription factors in general and for HSF1 in particular(33). Interestingly, that genomic study used HepG2 cells, and inanalyzing the supplemental data in that report we observed similartargets bound by HSF1 and PGC-1α as in our study. Collectively,these observations suggest that HSF1 is an important bindingpartner for PGC-1α on chromatin.Several potential mechanisms, yet to be explored, may mediate
the repressive effects of PGC-1α upon HSF1 binding. For exam-ple, PGC-1α may recruit corepressor complexes, actively inducingrepression of HSF1-mediated transcription. Alternatively, thebinding of PGC-1α to HSF1 may elicit eviction of HSF1-boundcoactivators or an HSF1 conformational change that results incompromised transcriptional activity.Although we find HSF1 to be repressed by PGC-1α, it is note-
worthy that HSF1 is not the sole HSF in mammals (12). Manytargets identified in our study are also targets for HSF2, and itremains possible that at least some of the effects of PGC-1α in thesystems analyzed here are mediated through HSF2. Furthermore,
A
C D E
F G
B
Fig. 7. PGC-1α regulates non-HSR HSF1-dependent transcriptional programs. (A) RT-PCR validation of mRNA expression of several non-HSR genes identified asrepressed by PGC-1α in HepG2 cells in our microarray analysis. (B) Non–HSR-related terms found in term enrichment analysis of genes repressed 1.5-fold or moreby PGC-1α expression in HepG2 cells. (C) Heat map representing fold repression by PGC-1α of selected genes in cells transfected with control siRNAs (SiControl) orwith siRNAs targeting HSF1 (SIHSF1). (D) RT-PCR analysis of target gene mRNA fold repression by PGC-1α in HepG2 cells that were transfected with siRNAstargeting HSF1 (SiHSF1) or with control siRNAs (SiControl). (E) RT-PCR analysis of mRNA expression of several non-HSR genes repressed by PGC-1α in HeLa cells.(F) ChIP analysis for HSF1 in HepG2 cells. Values represent enrichment of selected target gene promoters over an upstream region of the GAPDH promoter.(G) ChIP analysis for PGC-1α in HepG2 cells. Values represent enrichment of selected target gene promoters over an upstream region of the GAPDH promoter.
E5676 | www.pnas.org/cgi/doi/10.1073/pnas.1516219112 Minsky and Roeder

HSF2 and HSF4 also have some unique targets that might bemodulated by PGC-1α activity under some conditions. BecauseHSF family members are known to form heterotrimers (12), italso is possible that PGC-1α gains access to genes regulated byHSF2 and HSF4 indirectly, via its association with HSF1.Interestingly, we find that mutations in the L2/L3 motifs of
PGC-1α do not affect the ability of PGC-1α to repress HSP geneexpression (Fig. 5G). This finding is consistent with the notion thatthe PGC-1α effect on HSF1 is direct, because the binding ofnuclear hormone receptors via these motifs is dispensable forPGC-1α–mediated repression of HSPs. Future studies will helpdetermine the precise mechanisms that underlie the inhibitoryeffects of PGC-1α on HSF1-mediated transcription.Finally, our observations in liver and hepatocytes were extended
to muscle (Fig. 6). Thus, our findings are not restricted solely toliver, and future studies might reveal additional cell types andtissues in which PGC-1α can exert its inhibitory activity on HSPgene expression. Because adipocytes and neurons are also targetcells for PGC-1α, they constitute good candidates for future ex-ploration of the ability of PGC-1α to repress HSF1 functions.HSF1 is now widely recognized as a transcriptional regulator
not only of the HSR but also of numerous other transcriptionalprograms (19). It thus is notable that PGC-1α also can regulatethese aspects of HSF1 activity. Specifically, cancer cells are con-sidered to be highly dependent on HSF1 activity, which regulatesseveral transcriptional programs that include those controlling cellcycle, DNA damage repair, and protein translation (17, 19). Ouranalysis shows that at least some of these programs are regulateddirectly by PGC-1α; future studies will explore the possibility thatadditional non-HSR transcriptional programs are directly regu-lated by the action of PGC-1α on HSF1. Our studies used severalcancer cell lines, including HepG2, HeLa, and MCF7. AlthoughHepG2 cells naturally express PGC-1α, it is unclear whether othercancer cells such as HeLa and MCF7 express significant levels ofPGC-1α. However, regardless of the endogenous levels of PGC-1αin specific cancers, our studies constitute a proof of principle thattargeting the PGC-1α/HSF1 interface can be a valid approach torepress HSF1 activity in cancer.Why would PGC-1α both boost metabolic gene transcription
and at the same time repress HSF1? One possibility may involvethe ability of PGC-1α, under some conditions, to activate genesthat are implicated in the response to oxidative stress (34–36)and thus lead to a reduction of cellular oxidative stress levels.PGC-1α therefore may orchestrate the timely repression of HSPsin response to lower levels of oxidative stress.One other possibility may involve the previously described in-
hibition of cell proliferation by fasting (37–39). Because HSF1is required for several processes that involve normal cell pro-liferation, such as spleen cell proliferation following immunization(40), muscle regeneration (41), and postnatal growth in C57BL/6mice (42), it is possible that PGC-1α induction by fasting in liver(4) and muscle (43) represses HSF1 to mediate some of theantiproliferative effects of fasting. These scenarios also may holdtrue in cancer cells, although the potential effect of PGC-1α oncancer cell growth and survival is controversial, with reports sug-gesting both positive (44, 45) and negative (46–50) associationsbetween PGC-1α activity and cancer cell survival [including anegative association in HepG2 cells (51)], depending on the cel-lular context and on PGC-1α expression levels. It thus is possiblethat PGC-1α effects in cancer depend on a balance between itsability to induce metabolic genes and oxidative stress-responsegenes, a property that may promote cancer growth, and its abilityto repress HSF1 activity.Thus, our findings that PGC-1α acts as a critical repressor of
HSF1-mediated transcriptional programs link two major cellulartranscriptional networks and form a basis for future studies lookinginto the cross-talk between metabolic regulation and the cellularstress response in physiological and pathophysiological scenarios.
Materials and MethodsCell Culture and Treatments. HepG2 cells (ATCC HB-8065) and HeLa cells weregrown in Eagle’s minimum essential medium containing 10% (vol/vol) FBS andantibiotics. HEK293 cells were grown in DMEM containing 10% (vol/vol) FBSand antibiotics. Preplated fresh primary hepatocytes from different humandonors were obtained from Becton-Dickinson in collagen I-coated plates.C2C12 cells (ATCC CRL-1772) were grown in DMEM containing 20% (vol/vol)FBS and antibiotics and were differentiated into myotubes in DMEM supple-mented with 2% (vol/vol) donor equine serum and 1 μM insulin for 7 d. HSF1WT and KO immortalizedMEFs were a kind gift from Ivor J. Benjamin, MedicalCollege of Wisconsin, Milwaukee, and were maintained in DMEM containing10% FBS and antibiotics. Adenoviruses were produced in HEK293 cells aspreviously described (52) and were added directly to the culture mediumwhere indicated. Control and HSF1-targeting ON-TARGETplus SMARTpoolsiRNAs from Dharmacon were transfected into cells using the DharmaFECT 1reagent (Dharmacon) according to the manufacturer’s instructions.
In Vitro Binding and Immunoprecipitation Studies. GST-tagged proteins werepurified from bacteria as previously described (53). Recombinant FLAG-tagged PGC-1α was expressed from a pET vector in BL21 cells following in-duction with 1 mM isopropyl β-D-1-thiogalactopyranoside for 3 h. Followingsonication in BC500 [25% glycerol, 20 mM Hepes·KOH (pH 7.9), 500 mM KCl,0.1% Nonidet P-40, and protease inhibitors] and centrifugation, the super-natant was incubated with M2 anti-FLAG beads (Sigma). Beads were washedin BC500, and FLAG-PGC-1α was eluted with 3× FLAG peptide (Sigma) inBC100. For in vitro binding assays, immobilized proteins were incubatedwith purified PGC-1α in BC120/0.01% Nonidet P-40, were washed with thesame buffer, and then were boiled in SDS/PAGE protein-loading buffer forelution. For coimmunoprecipitation studies, nuclear extracts were preparedas previously described (54). Extracts then were adjusted to 100 mM KCl andwere supplemented with 0.01% Nonidet P-40 and M2 anti-FLAG beads(Sigma). After incubation and washes in BC100/0.01% Nonidet P-40, proteinswere eluted by boiling in SDS/PAGE protein-loading buffer. ChIP studieswere performed as previously described (32).
Luciferase Assays. Luciferase assays were performed using the dual luciferasekit from Promega. Cells were transfected with the Trans-IT reagent (Mirus)according to the manufacturer’s instructions. For HSE-driven luciferase assays,the Cignal Heat Shock Response Reporter kit from SA Biosciences was used.
Antibodies. Antibodies used were FLAG (F3165; Sigma), β-actin (sc-47778),HSF1 (sc-9144), PGC-1α (sc-13067), HSP27 (sc-1048), HSP105 (sc-6241), β-crys-tallin (sc-22744), and G6Pase (sc-25840) (all from Santa Cruz), and V5-tag(ab9116; Abcam).
Mice. All animal experiments were performed according to procedures ap-proved by the Institutional Animal Care and Use Committee of the Rock-efeller University. Mice were on a standard chow diet and were housed in apathogen-free facility under a standard 12-h/12-h light/dark cycle. Seven- tonine-week-old male mice were used for experiments. For fasting experi-ments, mice were placed in individual cages with access to water ad libitumfor 20 h and then were either given access to food or not for four additionalhours. Mice harboring a floxed allele of the gene encoding PGC-1α werepreviously described (27) and were purchased from Jackson Laboratories.
Gene-Expression Analysis. RNA was extracted using the RNeasy kit fromQiagen. RNA was reverse-transcribed using the SuperScript III First StrandSynthesis kit from Invitrogen. Real-time PCR was performed on an AppliedBiosystems 7300 Real Time PCR System using QuantiTect SYBR Green mix.Complete primer information is given in Table S1. Microarray analysis wasperformed using the Affymetrix GeneChip Gene 1.0 ST arrays (either humanor mouse) according to the manufacturer’s instructions. The data expressedas CEL files were normalized by the robust multiarray average method withthe Expression Console software (Affymetrix) and have been deposited inthe Gene Expression Omnibus database as GSE51498. Gene annotation en-richment analysis was performed using the Database for Annotation, Visu-alization, and Integrated Discovery (55) with the Benjamini procedure tocontrol for false-discovery rate.
ACKNOWLEDGMENTS. We thank Ivor J. Benjamin for the HSF1-KO mouseembryonic fibroblasts (MEFs). This work was supported by a Machiah Foun-dation Fellowship and a Merck Postdoctoral Fellowship (to N.M.) and by NIHGrant DK071900 (to R.G.R.).
Minsky and Roeder PNAS | Published online October 5, 2015 | E5677
CELL
BIOLO
GY
PNASPL
US

1. Handschin C, Spiegelman BM (2006) Peroxisome proliferator-activated receptorgamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev27(7):728–735.
2. Liu C, Lin JD (2011) PGC-1 coactivators in the control of energy metabolism. ActaBiochim Biophys Sin (Shanghai) 43(4):248–257.
3. Wu Z, et al. (1999) Mechanisms controlling mitochondrial biogenesis and respirationthrough the thermogenic coactivator PGC-1. Cell 98(1):115–124.
4. Yoon JC, et al. (2001) Control of hepatic gluconeogenesis through the transcriptionalcoactivator PGC-1. Nature 413(6852):131–138.
5. Puigserver P, et al. (1998) A cold-inducible coactivator of nuclear receptors linked toadaptive thermogenesis. Cell 92(6):829–839.
6. Lin J, et al. (2002) Transcriptional co-activator PGC-1 alpha drives the formation ofslow-twitch muscle fibres. Nature 418(6899):797–801.
7. Puigserver P, et al. (1999) Activation of PPARgamma coactivator-1 through tran-scription factor docking. Science 286(5443):1368–1371.
8. Li Y, Kovach A, Suino-Powell K, Martynowski D, Xu HE (2008) Structural and bio-chemical basis for the binding selectivity of peroxisome proliferator-activated re-ceptor gamma to PGC-1alpha. J Biol Chem 283(27):19132–19139.
9. Wu Y, Chin WW, Wang Y, Burris TP (2003) Ligand and coactivator identity determinesthe requirement of the charge clamp for coactivation of the peroxisome proliferator-activated receptor gamma. J Biol Chem 278(10):8637–8644.
10. Morimoto RI (2011) The heat shock response: Systems biology of proteotoxic stress inaging and disease. Cold Spring Harb Symp Quant Biol 76:91–99.
11. Gidalevitz T, Prahlad V, Morimoto RI (2011) The stress of protein misfolding: Fromsingle cells to multicellular organisms. Cold Spring Harb Perspect Biol 3(6):a009704.
12. Akerfelt M, Morimoto RI, Sistonen L (2010) Heat shock factors: Integrators of cellstress, development and lifespan. Nat Rev Mol Cell Biol 11(8):545–555.
13. Anckar J, Sistonen L (2011) Regulation of HSF1 function in the heat stress response:Implications in aging and disease. Annu Rev Biochem 80:1089–1115.
14. Hahn JS, Hu Z, Thiele DJ, Iyer VR (2004) Genome-wide analysis of the biology of stressresponses through heat shock transcription factor. Mol Cell Biol 24(12):5249–5256.
15. Gonsalves SE, Moses AM, Razak Z, Robert F, Westwood JT (2011) Whole-genomeanalysis reveals that active heat shock factor binding sites are mostly associated withnon-heat shock genes in Drosophila melanogaster. PLoS One 6(1):e15934.
16. Neef DW, Jaeger AM, Thiele DJ (2011) Heat shock transcription factor 1 as a thera-peutic target in neurodegenerative diseases. Nat Rev Drug Discov 10(12):930–944.
17. Ciocca DR, Arrigo AP, Calderwood SK (2013) Heat shock proteins and heat shock factor 1in carcinogenesis and tumor development: An update. Arch Toxicol 87(1):19–48.
18. Zhang Y, Huang L, Zhang J, Moskophidis D, Mivechi NF (2002) Targeted disruption ofhsf1 leads to lack of thermotolerance and defines tissue-specific regulation for stress-inducible Hsp molecular chaperones. J Cell Biochem 86(2):376–393.
19. Mendillo ML, et al. (2012) HSF1 drives a transcriptional program distinct from heatshock to support highly malignant human cancers. Cell 150(3):549–562.
20. Santagata S, et al. (2013) Tight coordination of protein translation and HSF1 activa-tion supports the anabolic malignant state. Science 341(6143):1238303.
21. Dai C, Whitesell L, Rogers AB, Lindquist S (2007) Heat shock factor 1 is a powerfulmultifaceted modifier of carcinogenesis. Cell 130(6):1005–1018.
22. Whitesell L, Lindquist S (2009) Inhibiting the transcription factor HSF1 as an anti-cancer strategy. Expert Opin Ther Targets 13(4):469–478.
23. Baler R, Welch WJ, Voellmy R (1992) Heat shock gene regulation by nascent poly-peptides and denatured proteins: Hsp70 as a potential autoregulatory factor. J CellBiol 117(6):1151–1159.
24. Shi Y, Mosser DD, Morimoto RI (1998) Molecular chaperones as HSF1-specific tran-scriptional repressors. Genes Dev 12(5):654–666.
25. Ruas JL, et al. (2012) A PGC-1α isoform induced by resistance training regulatesskeletal muscle hypertrophy. Cell 151(6):1319–1331.
26. Lin J, et al. (2003) PGC-1beta in the regulation of hepatic glucose and energy me-tabolism. J Biol Chem 278(33):30843–30848.
27. Lin J, et al. (2004) Defects in adaptive energy metabolism with CNS-linked hyperac-tivity in PGC-1alpha null mice. Cell 119(1):121–135.
28. McMillan DR, Xiao X, Shao L, Graves K, Benjamin IJ (1998) Targeted disruption of heatshock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J Biol Chem 273(13):7523–7528.
29. Elferink CJ, Reiners JJ, Jr (1996) Quantitative RT-PCR on CYP1A1 heterogeneous nu-clear RNA: A surrogate for the in vitro transcription run-on assay. Biotechniques 20(3):470–477.
30. Shema E, Kim J, Roeder RG, Oren M (2011) RNF20 inhibits TFIIS-facilitated transcrip-tional elongation to suppress pro-oncogenic gene expression.Mol Cell 42(4):477–488.
31. Knowles BB, Howe CC, Aden DP (1980) Human hepatocellular carcinoma cell linessecrete the major plasma proteins and hepatitis B surface antigen. Science 209(4455):497–499.
32. Handschin C, Spiegelman BM (2008) The role of exercise and PGC1alpha in in-flammation and chronic disease. Nature 454(7203):463–469.
33. Charos AE, et al. (2012) A highly integrated and complex PPARGC1A transcriptionfactor binding network in HepG2 cells. Genome Res 22(9):1668–1679.
34. St-Pierre J, et al. (2006) Suppression of reactive oxygen species and neurodegenerationby the PGC-1 transcriptional coactivators. Cell 127(2):397–408.
35. Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M (2005) PGC-1alpha regu-lates the mitochondrial antioxidant defense system in vascular endothelial cells.Cardiovasc Res 66(3):562–573.
36. Lu Z, et al. (2010) PGC-1 alpha regulates expression of myocardial mitochondrialantioxidants and myocardial oxidative stress after chronic systolic overload. AntioxidRedox Signal 13(7):1011–1022.
37. Kouda K, et al. (2004) Dietary restriction: Effects of short-term fasting on proteinuptake and cell death/proliferation in the rat liver. Mech Ageing Dev 125(5):375–380.
38. Delvaux G, Caes F, Willems G (1984) Refeeding of fasting rats stimulates epithelial cellproliferation in the excluded colon. Gastroenterology 86(5 Pt 1):802–807.
39. Phillips TL, Wachtel LW (1968) Suppression of cellular proliferation in weanling ratkidneys by short-term fasting. Proc Soc Exp Biol Med 128(2):566–570.
40. Inouye S, et al. (2004) Impaired IgG production in mice deficient for heat shocktranscription factor 1. J Biol Chem 279(37):38701–38709.
41. Nishizawa S, et al. (2013) Regeneration of injured skeletal muscle in heat shocktranscription factor 1-null mice. Physiol Rep 1(3):e00071.
42. Xiao X, et al. (1999) HSF1 is required for extra-embryonic development, postnatalgrowth and protection during inflammatory responses in mice. EMBO J 18(21):5943–5952.
43. Gerhart-Hines Z, et al. (2007) Metabolic control of muscle mitochondrial function andfatty acid oxidation through SIRT1/PGC-1alpha. EMBO J 26(7):1913–1923.
44. Vazquez F, et al. (2013) PGC1α expression defines a subset of human melanoma tu-mors with increased mitochondrial capacity and resistance to oxidative stress. CancerCell 23(3):287–301.
45. Shiota M, et al. (2010) Peroxisome proliferator-activated receptor gamma coactivator-1alpha interacts with the androgen receptor (AR) and promotes prostate cancer cellgrowth by activating the AR. Mol Endocrinol 24(1):114–127.
46. D’Errico I, et al. (2011) Bax is necessary for PGC1α pro-apoptotic effect in colorectalcancer cells. Cell Cycle 10(17):2937–2945.
47. D’Errico I, et al. (2011) Peroxisome proliferator-activated receptor-gamma coactivator1-alpha (PGC1alpha) is a metabolic regulator of intestinal epithelial cell fate. Proc NatlAcad Sci USA 108(16):6603–6608.
48. Jiang WG, Douglas-Jones A, Mansel RE (2003) Expression of peroxisome-proliferatoractivated receptor-gamma (PPARgamma) and the PPARgamma co-activator, PGC-1, inhuman breast cancer correlates with clinical outcomes. International Journal ofCancer 106(5):752–757.
49. Feilchenfeldt J, Bründler MA, Soravia C, Tötsch M, Meier CA (2004) Peroxisome pro-liferator-activated receptors (PPARs) and associated transcription factors in coloncancer: Reduced expression of PPARgamma-coactivator 1 (PGC-1). Cancer Lett 203(1):25–33.
50. Wang X, Moraes CT (2011) Increases in mitochondrial biogenesis impair carcinogen-esis at multiple levels. Mol Oncol 5(5):399–409.
51. Martínez-Jiménez CP, Gómez-Lechón MJ, Castell JV, Jover R (2006) Underexpressedcoactivators PGC1alpha and SRC1 impair hepatocyte nuclear factor 4 alpha function andpromote dedifferentiation in human hepatoma cells. J Biol Chem 281(40):29840–29849.
52. Luo J, et al. (2007) A protocol for rapid generation of recombinant adenoviruses usingthe AdEasy system. Nat Protoc 2(5):1236–1247.
53. Wallberg AE, Yamamura S, Malik S, Spiegelman BM, Roeder RG (2003) Coordinationof p300-mediated chromatin remodeling and TRAP/mediator function through co-activator PGC-1alpha. Mol Cell 12(5):1137–1149.
54. Dignam JD, Lebovitz RM, Roeder RG (1983) Accurate transcription initiation by RNApolymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res11(5):1475–1489.
55. Huang W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis oflarge gene lists using DAVID bioinformatics resources. Nat Protoc 4(1):44–57.
E5678 | www.pnas.org/cgi/doi/10.1073/pnas.1516219112 Minsky and Roeder


















