
DEPARTAMENTO DE INGENIERÍA...
370
-
Upload
phungnguyet -
Category
Documents
-
view
220 -
download
0
Transcript of DEPARTAMENTO DE INGENIERÍA...



FACULTAD DE CIENCIA Y TECNOLOGÍA
DEPARTAMENTO DE INGENIERÍA QUÍMICA
CONDICIONES DE PROCESO Y MODELADO
CINÉTICO DEL REFORMADO CON VAPOR DE
ETANOL SOBRE CATALIZADOR Ni/La2O3-αAl2O3
TESIS DOCTORAL
CAROLINA MONTERO CALDERÓN
Enero, 2015


FACULTAD DE CIENCIA Y TECNOLOGÍA
DEPARTAMENTO DE INGENIERÍA QUÍMICA
CONDICIONES DE PROCESO Y MODELADO
CINÉTICO DEL REFORMADO CON VAPOR DE
ETANOL SOBRE CATALIZADOR Ni/La2O3-αAl2O3
MEMORIA
Que para optar al grado de Doctor en Ingeniería Química
presenta
Dña. Carolina Montero Calderón
Leioa, enero de 2015


AUTORIZACIÓN DEL/LA DIRECTOR/A DE
TESIS PARA SU PRESENTACIÓN
La Dra. ANA G. GAYUBO CAZORLA con N.I.F. 16040358C y el Dr.
JAVIER BILBAO ELORRIAGA con N.I.F. 14548967H como Directores de
la Tesis Doctoral: CONDICIONES DE PROCESO Y MODELADO
CINÉTICO DEL REFORMADO CON VAPOR DE ETANOL SOBRE
CATALIZADOR Ni/La2O3-αAl2O3
realizada en el Departamento de INGENIERÍA QUÍMICA por la Doctoranda
Dña. CAROLINA MONTERO CALDERÓN autorizamos la presentación de
la citada Tesis Doctoral, dado que reúne las condiciones necesarias para su
defensa.
En Leioa a 18 de diciembre de 2014
LOS DIRECTORES DE LA TESIS
Fdo.: ANA G. GAYUBO CAZORLA Fdo.: JAVIER BILBAO ELORRIAGA


CONFORMIDAD DEL DEPARTAMENTO
El Consejo del Departamento de INGENIERÍA QUÍMICA en reunión
celebrada el día 18 de diciembre de 2014, ha acordado dar la conformidad a la
admisión a trámite de presentación de la Tesis Doctoral titulada:
CONDICIONES DE PROCESO Y MODELADO CINÉTICO DEL
REFORMADO CON VAPOR DE ETANOL SOBRE CATALIZADOR
Ni/La2O3-αAl2O3
dirigida por la Dra. ANA G. GAYUBO CAZORLA y el Dr. JAVIER
BILBAO ELORRIAGA presentada por Doña CAROLINA MONTERO
CALDERÓN ante este Departamento.
En. Leioa, a 07 de enero de 2015
Vº Bº DIRECTOR DEL
DEPARTAMENTO
SECRETARIO
DEL DEPARTAMENTO
Fdo.: Fdo.:
Dr. Roberto Aguado Zarraga Dr. Javier Ereña Loaizaga


AUTORIZACIÓN DE LA COMISIÓN ACADÉMICA
DEL PROGRAMA DE DOCTORADO
La Comisión Académica del Programa de Doctorado en INGENIERIA
QUÍMICA
en reunión celebrada el día 18 de diciembre de 2014, ha acordado dar la
conformidad a la presentación de la Tesis Doctoral titulada: CONDICIONES
DE PROCESO Y MODELADO CINÉTICO DEL REFORMADO CON
VAPOR DE ETANOL SOBRE CATALIZADOR Ni/La2O3-αAl2O3
dirigida por la Dra. ANA G. GAYUBO CAZORLA con N.I.F. 16040358C y
el Dr. JAVIER BILBAO ELORRIAGA con N.I.F. 14548967H y presentada
por Don/ña. CAROLINA MONTERO CALDERÓN
e inscrita en el Departamento INGENIERIA QUÍMICA
En Leioa, 07 de enero de 2015
EL COORDINADOR DEL PROGRAMA DE DOCTORADO
Fdo.: ________________________________
Dr. José Ignacio Lombraña


ACTA DE GRADO DE DOCTOR O DOCTORA
ACTA DE DEFENSA DE TESIS DOCTORAL
DOCTORANDO DÑA. CAROLINA MONTERO CALDERÓN
TITULO DE LA TESIS: CONDICIONES DE PROCESO Y MODELADO
CINÉTICO DEL REFORMADO CON VAPOR DE ETANOL SOBRE
CATALIZADOR Ni/La2O3-αAl2O3
El Tribunal designado por la Subcomisión de Postgrado de la UPV/EHU para
calificar la Tesis Doctoral arriba indicada y reunido en el día de la fecha, una
vez efectuada la defensa por el doctorando y contestadas las objeciones y/o
sugerencias que se le han formulado, ha otorgado por___________________la
calificación de: unanimidad ó mayoría
SOBRESALIENTE/NOTABLE/APROBADO/NO APTO
Idioma/s de defensa (en caso de más de un idioma, especificar porcentaje
defendido en cada idioma): Español
En a de de
EL/LA PRESIDENTE/A, EL/LA SECRETARIO/A,
Fdo.: Fdo.:
Dr/a: ____________________ Dr/a: ______________________
VOCAL 1º, VOCAL 2º VOCAL 3º
Fdo.: Fdo.: Fdo.:
Dr/a:________________ Dr/a:________________ Dr/a:________________
EL/LA DOCTORANDO/A Fdo.:


“Dejar el terreno firme de la lengua materna.
Despegarse de la zona de confort que garantiza la frecuentación
de los compañeros de siempre y de los docentes que te palmean la
espalda desde niño.
Abandonar el refugio donde quizás tienes ganadas las batallas
antes de empezar.
Rebuscárselas para conseguir un lugar en un cupo mínimo a costa
de competir con los estudiantes más ambiciosos y con ganas de
esforzarse más.
Exponerse a la posibilidad del rechazo. Extrañar el hogar paterno.
Experimentar la humildad de ser nadie entre otros nadies de
distintos países del mundo que están dispuestos a hacerse valer sin
el peso de sus apellidos y a costa de su propio talento y
entusiasmo.
Verse obligado a escuchar a otros muy diferentes y a testear las
propias certezas confrontando con otras visiones.
Reconocer la existencia de un afuera más allá del propio territorio
nacional, un mundo donde el conocimiento se acelera.
Conducir las pasiones ideológicas hacia la construcción de
consensos con argumentos bien informados.
Todo eso da un doctorado que se hace afuera”
“El único presidente latinoamericano con doctorado”
Diario La Nación (Argentina), 20 de marzo 2014.


Han pasado ya varios años desde que tomé la decisión de realizar mi
doctorado y ahora cuando está muy próxima a concluir esta etapa académica
de mi vida, me lleno de emoción al recordar a todas las personas que han
hecho posible que este trabajo salga adelante, por lo cual con estas letras
quiero manifestar mis más sinceros agradecimientos.
En primer lugar a mis directores: Dra. Ana Gayubo y Dr. Javier Bilbao
para quienes las palabras se quedan cortas para expresar toda mi gratitud por
permitirme realizar este trabajo bajo su acertada dirección. Ana, gracias por
todas las enseñanzas que me ha brindado en este proceso, por las múltiples
dudas resueltas, las explicaciones detalladas, la paciencia enorme que me ha
tenido y por esas palabras justas en los momentos precisos. Javi, gracias por
haberme permitido ser parte del grupo de investigación, por la factibilidad
para realizar mi tesis sin ninguna limitación, y el apoyo final para que este
trabajo se culmine en el tiempo estipulado. Les deseo lo mejor en sus vidas
profesionales y personales, espero a la distancia poder trabajar conjuntamente
ya que sin duda aun me queda mucho por aprender de Ustedes.
A quienes conforman el Departamento de Ingeniería Química de la
Universidad del País Vasco UPV-EHU y al grupo ProCatVaRes, de manera
especial al Dr. Andrés Aguayo por su ayuda en el manejo de equipos y en el
desarrollo del programa de cálculo, y al Dr. Pedro Castaño por su guía en el
estudio de los catalizadores desactivados. Quiero agradecer también al
personal de SGiker por su ayuda en la caracterización de los catalizadores.
A mis compañeros del Laboratorio por estos años de vivencias, por los
momentos de training y debates científicos, así como por las actividades
extra-laborales realizadas que sin duda serán inolvidables. Con cariño quiero
agradecer a los Reforming Team por su apoyo directo en esta Tesis: Koke y
Aingeru por haberme apoyado en mi PFM, Bea por su guía en el área
bibliográfica, Lide por compartir nuestro “querido reactor”, Borja por el
adiestramiento en rácores, y Aitor en quien queda la responsabilidad de
encontrar la mejor ruta para el OSR.
Este trabajo ha sido posible a la beca doctoral concedida por la Secretaría
Nacional de Educación Superior, Ciencia, Tecnología e Innovación de
Ecuador-SENESCYT y al auspicio de la Universidad Central del Ecuador;
instituciones a las que quiero expresar mi más profunda gratitud y que espero
haber representado de la mejor manera. En la Facultad de Ingeniería Química-
UCE quiero agradecer a los Ingenieros Andrés De La Rosa y Luis Calle por las

cartas de recomendación para acceder a la beca, y al Ingeniero Jorge Medina
por incentivarme a conocer el mundo de la catálisis.
Gracias a todos quienes han hecho que mi estancia en Bilbao sea
inolvidable, les voy a extrañar y recordar muchísimo: Rober y su familia por el
cariño con que me han abierto las puertas de su casa, mis chicas del café en
especial a Elena por todas las toneladas de buena energía transmitidas a
diario, Eva por las inolvidables navidades en las que pude ser parte de tu
familia, y en especial a mis amigos Saira, Lumi y JuanFer por los muchos y
buenos recuerdos que me llevo, espero que nuestra amistad perdure siempre
y que la próxima vez que nos veamos sea en Guadalajara, Medellín o Quito.
A las familias Calderón-Yunda y Luna-Calderón que han estado muy
pendientes de mí en esta etapa, me van a hacer mucha falta y espero pronto
tenerles de vuelta en casa. A mis familiares y amigos que desde Ecuador me
brindaron su apoyo, valoré mucho cada saludo, mensaje, llamada, e-mail que
me enviaron, pronto nos veremos.
Para concluir quiero dedicar este trabajo a mi querida familia, ya que sin
ellos nada de esto hubiese sido posible. Abigail y Xavier, gracias por ser los
mejores hermanos que alguien puede tener, por su cariño, su complicidad, por
aguantarme en mis buenos y sobretodo en los malos ratos. Deseo que Ustedes
también cumplan todas las metas que se propongan. Sarita y Silvio, mis
queridos padres, mis ojos se llenan de emoción solo en escribir sus nombres,
ninguna palabra puede expresar todo el amor, gratitud y admiración que
siento por Ustedes, gracias por nunca poner límites a mis sueños, por su
ejemplo de rectitud y perseverancia, por enseñarme a ser constante y no
decaer ante nada, por apoyarme con la mejor herencia que es la educación, por
darme las alas pero sobretodo por enseñarme a volar.
A todos quienes de manera desinteresada me han apoyado en este
trabajo y en esta etapa de mi vida, que sin duda será inolvidable.
Carolina.
Bilbao, diciembre 2014

Índice i
Carolina Montero Calderón
OBJETIVOS 1
1. INTRODUCCIÓN 5
1.1. EL H2 COMO VECTOR ENERGÉTICO 10
1.2. PRODUCCIÓN DE HIDRÓGENO DESDE BIOMASA 11
1.2.1. Producción de H2 por reformado de oxigenados
derivados de la biomasa 12
1.2.1.1. Reformado de dimetil éter (DME) 13
1.2.1.2. Reformado con vapor de bio-oil 15
1.3. EL ETANOL COMO FUENTE DE HIDRÓGENO 17
1.3.1. Etanol lignocelulósico o de segunda generación 18
1.3.1.1. Etapas en la producción 19
1.3.1.2. Implantación industrial 21
1.3.2. Obtención de hidrógeno a partir de etanol 23
1.4. REFORMADO CON VAPOR DE ETANOL 25
1.4.1. Etapas de reacción y esquema cinético 26
1.4.2. Catalizadores 29
1.4.2.1. Catalizadores de metales nobles 30
1.4.2.2. Catalizadores de metales no nobles 32
1.4.3. Diseño del reactor de reformado con vapor de etanol 38
1.4.3.1. Estudios teóricos 38
1.4.3.2. Resultados en planta piloto 41
1.4.4. Economía del reformado con vapor de etanol 42
1.4.5. Otras tecnologías de reformado 44
1.4.5.1. Oxidación parcial 44
1.4.5.2. Reformado oxidativo y autotérmico 46
1.4.5.3. Reformado con CO2 (Reformado seco) 48
2. EXPERIMENTAL 51
2.1. SÍNTESIS DE LOS CATALIZADORES 53
2.2. CARACTERIZACIÓN DE LOS CATALIZADORES 55
2.2.1. Estructura porosa 55

ii
2.2.2. Composición química 56
2.2.3. Morfología del catalizador 56
2.2.4. Propiedades metálicas 57
2.2.4.1. Difracción de rayos X (XRD) (Estructura cristalina) 57
2.2.4.2. Espectroscopía fotoelectrónica de rayos X (XPS)
(Interacción metal-soporte) 58
2.2.4.3. Reducción a temperatura programada (TPR)
Reducibilidad de especies metálicas 60
2.2.4.4. Quimisorción de H2 (Superficie metálica, dispersión
y tamaño de cristal) 60
2.2.5. Análisis de la naturaleza y el contenido de coque 62
2.2.5.1. Oxidación a temperatura programada (TPO) 62
2.2.5.2. Espectroscopía infrarroja por transformada
de Fourier (FTIR) 64
2.2.5.3. Espectroscopía Raman 64
2.3. EQUIPO DE REACCIÓN Y ANÁLISIS 66
2.3.1. Equipo de reacción 66
2.3.2. Análisis de los productos de reacción 71
2.3.3. Condiciones fluidodinámicas 77
2.3.4. Condiciones de operación 78
2.3.5.Índices de reacción 78
3. OPTIMACIÓN DE LAS CONDICIONES DE PREPARACIÓN
DEL CATALIZADOR 81
3.1. EFECTO DE LA TEMPERATURA DE CALCINACIÓN
EN EL COMPORTAMIENTO CINÉTICO DEL CATALIZADOR 84
3.1.1.Índices de reacción a tiempo cero 84
3.1.2. Desactivación 87
3.2. EQUILIBRADO DEL CATALIZADOR 92
3.2.1. Comportamiento cinético en ciclos de reacción-regeneración 93
3.2.2. Propiedades físicas, composición y morfología
de los catalizadores 95
3.2.3. Propiedades metálicas de los catalizadores 98
3.2.3.1. Reducibilidad de las especies metálicas 98
3.2.3.2. Superficie metálica, dispersión y tamaño de cristal 100

Índice iii
Carolina Montero Calderón
3.2.3.3. Difracción de rayos X (XRD) 102
3.2.3.4. Espectroscopía Fotoelectrónica de Rayos X (XPS) 103
3.2.4. Discusión de los resultados 107
4. EFECTO DE LAS CONDICIONES DE OPERACIÓN 109
4.1. CONTRIBUCIÓN DE LAS RUTAS TÉRMICAS EN EL SRE 112
4.2. EFECTO DE LAS CONDICIONES DE REACCIÓN EN EL
COMPORTAMIENTO DEL CATALIZADOR ESTABILIZADO 118
4.2.1. Comportamiento cinético a tiempo cero 118
4.2.1.1. Efecto de la temperatura 118
4.2.1.2. Efecto del tiempo espacial 124
4.2.1.3. Efecto de la relación molar vapor/etanol (S/E) 128
4.2.2. Estabilidad del catalizador 132
4.2.2.1. Efecto de la temperatura 132
4.2.2.2. Efecto de la relación molar S/E 139
4.2.2.3. Efecto del tiempo espacial 141
4.2.2.4. Ensayos de muy larga duración 147
5. ESTUDIO DEL CATALIZADOR 10Ni/LaAl-E DESACTIVADO 151
5.1. EFECTO DE LA VARIABLES DE OPERACIÓN SOBRE LA
DEPOSICIÓN DE COQUE 155
5.1.1. Temperatura 155
5.1.2. Relación molar S/E 160
5.1.3. Tiempo espacial 163
5.2. EVOLUCIÓN DEL Ni y DEL COQUE CON EL TIEMPO DE
REACCIÓN 171
5.2.1. Difracción de rayos X (XRD) 173
5.2.2. Adsorción-desorción de N2 176
5.2.3. Microscopía electrónica de barrido (SEM) 178
5.2.4. Microscopía electrónica de trasmisión (TEM) 180
5.2.5. Oxidación a temperatura programada (TPO) 182
5.2.6. Espectroscopía FTIR 185
5.2.7. Espectroscopía Raman 187

iv
5.2.8. Espectroscopía fotoeléctrica de rayos X (XPS) 189
5.2.9. Interpretación de los resultados 192
5.3. REGENERABILIDAD DEL CATALIZADOR 10Ni/LaAl-E
MEDIANTE GASIFICACIÓN DEL COQUE 195
6. MODELADO CINÉTICO DEL SRE SOBRE CATALIZADOR
10Ni/LaAl-E 199
6.1. MODELADO CINÉTICO A TIEMPO CERO 203
6.1.1. Antecedentes bibliográficos 203
6.1.1.1. Modelos cinéticos para catalizadores de Ni 203
6.1.1.2. Modelos cinéticos para catalizadores de Co y de
metales nobles 214
6.1.2. Metodología del modelado cinético a tiempo cero 217
6.1.2.1. Cálculo de los parámetros cinéticos de mejor ajuste 217
6.1.2.2. Ecuaciones de velocidad de reacción
y balance de materia 219
6.1.3. Programa de cálculo 220
6.1.4. Significación y discriminación de los modelos cinéticos 223
6.1.5. Modelos cinéticos propuestos 225
6.1.6. Discriminación de modelos cinéticos 230
6.1.7. Incorporación del craqueo térmico en el modelado cinético 236
6.2. MODELADO CINÉTICO DE LA DESACTIVACIÓN POR COQUE 245
6.2.1. Metodología de cálculo de los parámetros cinéticos 246
6.2.2. Modelos cinéticos de desactivación no selectiva 249
6.2.3. Modelo cinético de desactivación selectiva 255
6.2.3.1. Análisis de la necesidad de un modelo de
desactivación selectiva 255
6.2.3.2. Propuesta de un modelo de desactivación selectiva 258
6.3. MODELO CINÉTICO PROPUESTO 262
7. RESUMEN 269
8. CONCLUSIONES 275

Índice v
Carolina Montero Calderón
9. NOMENCLATURA 291
10. BIBLIOGRAFIA 301
ANEXOS 319
ANEXO A. BALANCE DE MATERIA EN EL REACTOR 321
ANEXO B. ESTUDIO TERMODINAMICO DEL PROCESO SRE 325
B.1. FUNDAMENTOS 324
327 B.2. PROCEDIMIENTO DE SIMULACIÓN
B.3. RENDIMIENTO DE PRODUCTOS EN EL EQUILIBRIO 329
B.3.1. Composición de la corriente gaseosa en el SRE 329
B.3.2. Composición de la corriente gaseosa en el OSRE 330
B.3.3. Rendimientos de coque en el SRE y OSRE 333
B.3.4. Comparación con resultados de la bibliografía 334
B.4. REQUERIMIENTOS ENERGÉTICOS EN LOS
PROCESOS DE REFORMADO DE ETANOL 335
ANEXO C. SELECCIÓN DE LA EXPRESIÓN PARA LA
FUNCION (ATENUACIÓN POR ADSORCIÓN
DE COMPONENTES DEL MEDIO DE REACCIÓN) 339
ANEXO D. SELECCIÓN DE UN MODELO CINÉTICO
PARA CUANTIFICAR LA CONTRIBUCIÓN DE LAS
RUTAS TÉRMICAS
342


1
Carolina Montero Calderón
OBJETIVOS
El objetivo de esta Tesis Doctoral ha sido el estudio del reformado con vapor de
etanol. Se enmarca dentro de una línea de investigación del Grupo ProCatVares
(Procesos Catalíticos y Valorización de Residuos) para la producción de H2 por
reformado de compuestos oxigenados derivados de biomasa, como vía de valorización
de estos compuestos alternativa a la obtención de hidrocarburos o de dimetil éter
(DME). Esta línea de investigación se inició en el grupo ProCatVaRes hace 8 años,
ante el creciente interés del H2 como combustible y como materia prima, y ha sido
financiada a través 3 convocatorias sucesivas del Plan Nacional de I+D+i (Proyectos
CTQ2006-12005/PPQ, CTQ2009-13428/PPQ, y CTQ2012-35263/PPQ, aún en vigor).
Ambas líneas de investigación sobre la valorización de oxigenados derivados de
biomasa (producción de H2 o bien de hidrocarburos y de DME) se enmarcan dentro
del más amplio concepto de Bio-Refinería.
Entre los compuestos oxigenados cuyo reformado con vapor (SR) ha sido
estudiado por el grupo ProCatVaRes se incluyen: i) dimetil éter (obtenido vía gas de
síntesis a partir de biomasa); ii) bio-oil (obtenido mediante pirólisis) y iii) bio-etanol
(que se obtendría mediante hidrólisis-fermentación). La investigación llevada a cabo
en esta temática ha dado lugar a la defensa de 3 Tesis Doctorales (Remiro, 2012;
Vicente, 2012; Oar-Arteta, 2014).
La resultados de la Tesis Doctoral de Vicente (2012), centrada en la selección
de catalizadores y condiciones de proceso para el reformado con vapor de DME
(SRD) y de etanol (SRE), pusieron de manifiesto el buen comportamiento para el
proceso de SRE de catalizadores de Ni soportados en SiO2 y en La2O3-αAl2O3. Se
profundizó en dicha Tesis en el estudio del catalizador soportado en SiO2, que da lugar
a una muy elevada deposición de coque que, si bien no causa una elevada
desactivación debido a su naturaleza fibrilar, sí podría originar problemas
operacionales por excesiva acumulación de coque en el lecho. También se puso de
manifiesto el excelente comportamiento del catalizador 10Ni/La2O3-αAl2O3, el cual
requiere un tratamiento previo de equilibrado para alcanzar un comportamiento
reproducible en sucesivos ciclos de reacción-regeneración.

2
Por otro lado, Remiro (2012), desarrolló un proceso continuo con 2 etapas
(térmica y catalítica) para el SR del bio-oil completo, con intensificación en la
producción de H2 mediante captura in situ de CO2. En esta Tesis se comprobó el buen
comportamiento del mismo catalizador 10Ni/La2O3-αAl2O3 para el reformado con
vapor de bio-oil, o de mezclas etanol/bio-oil. Posteriormente, se comprobó que el
comportamiento de este catalizador está muy influenciado por las condiciones de su
preparación (temperaturas de calcinación y de reducción) (Valle y cols., 2014a).
Estas primeras Tesis permitieron progresar en la selección de catalizadores y de
condiciones de proceso apropiados para llevar a cabo el SR de DME, etanol y bio-oil,
así como delimitar los aspectos susceptibles de mejora en la preparación y propuesta
de nuevos catalizadores. Sin embargo, el progreso hacia el desarrollo a nivel industrial
de estos procesos requiere, además, el desarrollo de herramientas matemáticas para el
diseño y simulación del reactor. Es clave para ello el desarrollo de modelos cinéticos
que describan la evolución de los componentes del medio de reacción con las
condiciones de operación, y que cuantifiquen la desactivación del catalizador. Este ha
sido uno de los objetivos de una reciente Tesis Doctoral (Oar-Arteta, 2014) sobre el
SRD, utilizando un catalizador bifuncional novedoso, de espinela CuF2O4/ boehmita,
así como de otra Tesis en realización sobre SR de bio-oil, con fecha próxima de
lectura prevista (Aramburu, 2015).
Los antecedentes propios del grupo y de la bibliografía respecto al reformado
con vapor de etanol, ponen de manifiesto que, a pesar del interés potencial del proceso
para la producción sostenible de H2 a gran escala, el desarrollo tecnológico está
limitado por las notables lagunas en el conocimiento del reactor, esquema y modelado
cinético, y en particular de la desactivación, aspecto abordado cualitativamente y sin
que existan antecedentes de modelos cinéticos que cuantifiquen la desactivación, que
es fundamental para la simulación y escalado del proceso.
A la vista de estos antecedentes, los objetivos planteados en esta Tesis, centrada
en el proceso SRE, han sido:
1. Selección de las condiciones de preparación del catalizador 10Ni/La2O3-αAl2O3
que proporcionen elevada actividad, selectividad de H2 y estabilidad en el proceso

Objetivos 3
Carolina Montero Calderón
SRE, así como comportamiento reproducible para la operación en ciclos sucesivos
de reacción-regeneración. Se estudiarán dos vías de actuación:
i) Optimización de la temperatura de calcinación del catalizador
ii) Equilibrado (estabilización) del catalizador.
2. Estudio paramétrico en un amplio intervalo de las variables de operación
(temperatura, relación molar vapor de agua/etanol, tiempo espacial, tiempo de
reacción) para:
i) Determinar el efecto de dichas variables sobre el comportamiento cinético en
el proceso SRE del catalizador 10Ni/La2O3-αAl2O3 equilibrado.
ii) Obtener suficientes datos cinéticos para abordar con rigor el modelado
cinético del proceso.
3. Delimitación de las causas de desactivación del catalizador 10Ni/La2O3-αAl2O3
equilibrado (deposición de coque y/o sinterización), prestando especial atención al
análisis del efecto de las condiciones de reformado (temperatura, tiempo espacial,
relación molar S/E, tiempo de reacción) sobre el contenido y naturaleza del coque
depositado y las propiedades del catalizador desactivado. Igualmente, se
identificarán los componentes del medio de reacción que son precursores de la
formación de coque. Para este objetivo, se utilizarán técnicas variadas de análisis
de las propiedades y estructura del catalizador y del coque.
4. Estudio de la gasificación del coque, como posible vía de regeneración del
catalizador, alternativa a la combustión con aire.
5. Desarrollo de un modelo cinético capaz de predecir el efecto de las condiciones de
reacción (temperatura, tiempo espacial, relación molar agua y etanol, tiempo)
sobre la distribución de todos los productos de reacción en el proceso SRE sobre
el catalizador 10Ni/La2O3-αAl2O3 equilibrado. Se abordará el modelado en 2
etapas:
i) Modelado cinético a tiempo cero, mediante un modelo basado en el esquema
cinético que describa los resultados experimentales de composición de los
productos de reacción para diferentes valores de las variables de operación
(temperatura, relación molar S/E y tiempo espacial)

4
ii) Modelado cinético de la desactivación, mediante una ecuación cinética que
incluya el efecto de la temperatura y concentración de componentes en el
medio de reacción, y considerando la posible necesidad de un modelo de
desactivación selectiva (con diferentes actividades para las diferentes etapas
del esquema cinético). La propuesta de este modelo se basará en el
conocimiento adquirido sobre los orígenes de la desactivación y la
incidencia de ésta en la evolución con el tiempo, de la concentración de
productos.

1. INTRODUCCIÓN


7
Carolina Montero Calderón
1. INTRODUCCIÓN
El aumento de la demanda energética, asociada al mantenimiento del bienestar
social de los países desarrollados y a la consecución del bienestar social de los países
en desarrollo de Asia y Latinoamérica, ha llevado a una sobreexplotación del crudo
petrolífero. En este contexto, el último informe de la Agencia Internacional de Energía
(World Energy Outlook 2013) destaca como puntos relevantes:
� Se requiere un análisis del efecto de las abultadas diferencias del precio de la
energía entre regiones en la promoción o frustración del crecimiento económico,
porque estas diferencias afectan a la competitividad industrial, incidiendo en las
decisiones de inversión y en las estrategias empresariales.
� Los países pueden amortiguar el impacto de los elevados precios promoviendo
mercados de la energía más eficientes, competitivos e interconectados.
� Mejorar la competitividad energética no significa disminuir los esfuerzos por
luchar contra el cambio climático.
� La evolución de la demanda de productos petroquímicos requiere una utilización
del petróleo en ascenso hasta 2035.
� El petróleo ligero, de fácil extracción y valorización, se agotará en los próximos 10
años, dejando una seria incertidumbre sobre el abastecimiento a largo plazo.
Esta evaluación señala que la demanda mundial de energía continuará creciendo
al menos hasta 2050, sin que se alcance el punto máximo de la producción del petróleo
en el futuro previsible y, con el contrasentido que 880 millones de personas en el
mundo continúen sin acceso a la energía. Además, que no se logrará el objetivo de que
las emisiones de gases de efecto invernadero disminuyan en un 50 % para el año 2050.
Estas tendencias han de condicionar la transición energética en las próximas
décadas, siempre sujeta a la viabilidad económica e influenciada por la presión social
derivada del impacto medioambiental de las alternativas energéticas que en cada
momento se planteen.
8
Capítulo 1
En este escenario, se estima que con el desarrollo de tecnologías que maximicen
el aprovechamiento de la energía derivada de fuentes renovables se podría suplir hasta
3000 veces la demanda energética actual (Ellabban y cols., 2014). Entre estas
tecnologías tienen un papel relevante las de biorefinería y biotransformación, para
convertir diferentes tipos de biomasa en combustibles.
La biomasa puede ser transformada en energía, directa o indirectamente, a
través de procesos termoquímicos y bioquímicos, entre los que se incluyen la
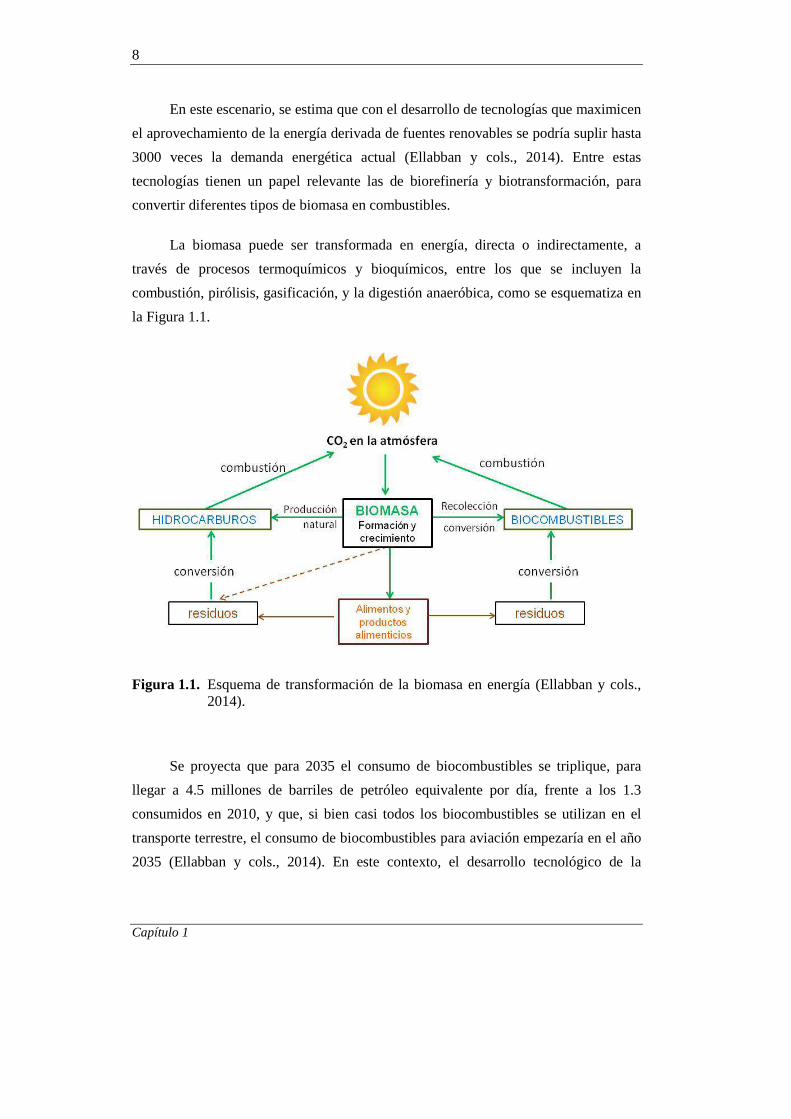
combustión, pirólisis, gasificación, y la digestión anaeróbica, como se esquematiza en
la Figura 1.1.
Figura 1.1. Esquema de transformación de la biomasa en energía (Ellabban y cols., 2014).
Se proyecta que para 2035 el consumo de biocombustibles se triplique, para
llegar a 4.5 millones de barriles de petróleo equivalente por día, frente a los 1.3
consumidos en 2010, y que, si bien casi todos los biocombustibles se utilizan en el
transporte terrestre, el consumo de biocombustibles para aviación empezaría en el año
2035 (Ellabban y cols., 2014). En este contexto, el desarrollo tecnológico de la

1. Introducción 9
Carolina Montero Calderón
obtención de H2 mediante reformado de oxigenados derivados de la biomasa ha
adquirido un interés estratégico prioritario, ya que, además de ser un potencial
combustible, es una materia prima de creciente demanda en la industria petroquímica
y agroquímica.

10
Capítulo 1
1.1. EL H2 COMO VECTOR ENERGÉTICO
Desde el punto de vista energético, el H2 es el combustible con mayor potencia
calorífica neta, con 120 MJ/kg de H2, frente a los 47 MJ/kg del gas natural, 43 MJ/kg
de la gasolina, 29 MJ/kg del DME, 26 MJ/kg del etanol y 15 MJ/kg del bagazo de
caña de azúcar. Además, tiene la ventaja de que el único producto de su combustión es
el vapor de agua, mientras que los otros combustibles generan CO2 y NOx, gases a los
que se atribuye principalmente el efecto invernadero. En consecuencia, se considera
idóneo y una alternativa viable para ser utilizado en el sector del transporte, que
consume ~ 18 % de la energía primaria mundial.
Con estas perspectivas surge el concepto de la “economía del H2”, en la cual el
H2 sería fundamental para generar electricidad (turbinas accionadas con la energía
desprendida de la hidrolisis del agua) y para el transporte, por combustión directa y
por medio de celdas de combustible (Figura 1.2).
Figura 1.2. Rutas para la obtención y valorización del H2 como combustible (Andrews y Shabani, 2012).

1. Introducción 11
Carolina Montero Calderón
1.2. PRODUCCIÓN DE HIDROGENO DESDE BIOMASA
El H2, que es el elemento químico más abundante en el universo, no se
encuentra en estado puro, sino únicamente en forma molecular o iónica, por lo cual es
necesario separarlo de los compuestos a los que esté vinculado, que principalmente
son agua e hidrocarburos.
La producción de H2 en Europa fue en 2009 de 8 mil millones de m3.
Actualmente es la clave en la industria del petróleo para los procesos de hidrocraqueo,
con objeto de producir combustibles líquidos a partir de fracciones pesadas de
hidrocarburos y con los requerimientos de composición acordes con la normativa de
protección ambiental. Además, tiene también un uso a gran escala para la fabricación
de NH3, materia prima de los fertilizantes. Entre otros usos que se dan al H2 podemos
señalar: como el combustible que alimenta las lanzaderas de los cohete; en la industria
química para fabricar fibras textiles, tales como espumas de nylon, poliuretano o
plásticos; en la industria del vidrio, para fabricar vidrio plano; en electrónica, como
gas vector; y en metalurgia, para el tratamiento térmico del acero. Además, el interés
en utilizarlo como vector energético ha tomado impulso en los últimos años, debido a
que el producto de su combustión es únicamente agua y se reducirían las emisiones de
gases de efecto invernadero.
Los métodos de obtención de H2 son variados y en función de la materia prima
pueden clasificarse en: i) reformado de fuentes fósiles; ii) rutas desde la biomasa, y;
iii) separación electroquímica del agua (Peláez-Samaniego y cols., 2014). Entre éstas,
la obtención de H2 desde biomasa lignocelulósica (no adecuada para la cadena
alimenticia), tiene un indiscutible interés para su implantación a gran escala a corto-
medio plazo, por su pequeña contribución a la emisión de CO2, disponibilidad
universal, integración con la explotación de recursos naturales y contribución a una
economía social y sostenible (Trane-Restrup y cols., 2013).
Las rutas de obtención de H2 a partir de biomasa pueden clasificarse en: a)
obtención directa (gasificación, pirólisis a alta temperatura, pirólisis catalítica y
procesos biológicos), y; b) rutas con etapas intermedias de obtención de oxigenados

12
Capítulo 1
(etanol, metanol, dimetil éter, bio-oil, etc.) para su posterior reformado (Nahar y
Dupont, 2012).
Cabe destacar que dentro del grupo de investigación se han realizado
previamente tesis doctorales relacionadas con la obtención de H2 por medio de
reformado de oxigenados derivados de biomasa, como el bio-oil (Remiro, 2012) y
dimetil éter (Oar-Arteta, 2014), así como un trabajo previo de discriminación de
catalizadores para el reformado con vapor de etanol (Vicente, 2012). También se ha
estudiado la obtención de H2 por gasificación directa de biomasa (Erkiaga, 2014).
1.2.1. Producción de H2 por reformado de oxigenados derivados de la biomasa
Actualmente, cerca del 90 % del H2 es obtenido por reformado de gas natural o
de nafta (Bshish y cols., 2011), por lo que el desarrollo tecnológico de la obtención de
H2 mediante reformado de oxigenados derivados de la biomasa ha merecido una
notable atención en la bibliografía, debido a que al interés desde la perspectiva de la
sostenibilidad hay que añadir la alta densidad energética y relativa seguridad en el
transporte y almacenaje de estos compuestos. En la Figura 1.3 se observa un resumen
de las rutas completas para la obtención de H2 desde biomasa vía reformado de
oxigenados intermedios.
El proceso de reformado de los oxigenados puede efectuarse con diferentes
condiciones:
Reformado con vapor (SR), alimentando una corriente de vapor de agua. Es un
proceso altamente endotérmico, pero con mayor rendimiento de H2.
Oxidación Parcial (PO), con la alimentación de O2 (aire). Proceso exotérmico, que
evita los requerimientos energéticos del reformado, pero resta eficiencia al proceso,
disminuyendo el rendimiento de H2.
Reformado oxidativo con vapor (OSR), con la alimentación de H2O y O2 y que es
una combinación del SR y PO, de modo que se puede mitigar el requerimiento
energético sin una disminución drástica del rendimiento de H2. Con un control

1. Introducción 13
Carolina Montero Calderón
adecuado de los caudales de de H2O y O2 se anula el requerimiento energético
(reformado autotérmico, ATR).
Figura 1.3. Rutas para la obtención de H2 por reformado de oxigenados derivados de diferentes procesos de valorización de la biomasa.
1.2.1.1. Reformado de dimetil éter (DME)
El DME es un producto relativamente inerte, degradable, no-corrosivo, no
mutagénico y no-cancerígeno, que puede almacenarse y manejarse como los LPGs,
tiene número de cetano similar al del combustible diesel (entre 55-60) y quema sin
producir cenizas, por lo que es considerado un combustible alternativo atractivo para
motores de compresión-ignición (Arcoumanis y cols., 2008). Además, la síntesis del
DME está termodinámicamente favorecida respecto a la de metanol y permite la
coalimentación de CO2 junto con el gas de síntesis (Ereña y cols., 2013).
El reformado con vapor de DME (SRD) puede hacerse a temperaturas
relativamente bajas (250-400 ºC), solo ligeramente superiores a las necesarias para el
reformado de metanol (SR-MeOH), lo que unido a las ventajas comentadas respecto a
facilidad de manejo y de síntesis, hacen que el SRD sea una ruta prometedora,
alternativa al SR-MeOH, para la producción de H2 a bordo para ser utilizada en pilas

14
Capítulo 1
de combustible, especialmente de baja temperatura PEMFC (proton exchange
membrane fuel cell) (Vicente y cols., 2013).
El SRD es un proceso endotérmico que puede tener lugar a presión atmosférica
y temperaturas moderadas sobre un catalizador bifuncional, y consiste en 2 etapas en
serie: la hidrólisis del DME sobre la función ácida (Ec. 1.1), seguida del reformado
con vapor del metanol (MeOH) sobre la función metálica (Ec. 1.2):
Hidrólisis de DME: (CH3)2O + H2O ↔ 2CH3OH (1.1)
Reformado de MeOH: CH3OH + H2O ↔ 3H2+CO2 (1.2)
Proceso global (SRD): (CH3)2O + 3H2O ↔ 6H2 + 2CO2 (1.3)
Además de la hidrólisis de DME y del reformado del metanol, pueden ocurrir
reacciones indeseadas como la reacción inversa de gas de agua (rWGS), (Ec. 1.4)
sobre la función metálica del catalizador y la descomposición del DME (Ec. 1.5), que
produce CH4, cuando se emplea un catalizador fuertemente ácido o temperaturas de
reformado elevadas (Faungnawakij y cols., 2007).
Reacción inversa de gas de agua (rWGS): H2 + CO2 ↔ CO + H2O (1.4)
Descomposición del DME: (CH3)2O ↔ H2 + CH4 + CO (1.5)
Por tanto, es necesario elegir funciones metálica y ácida adecuadas para
alcanzar una elevada conversión de DME y una alta selectividad de H2, evitando la
formación de CO (para evitar el envenenamiento del catalizador anódico en las
PEMFC) y de CH4 como subproductos. Entre los catalizadores bifuncionales con buen
comportamiento, se encuentran los basados en funciones metálicas de Cu (función
convencional CZA y del tipo espinela CuFe2O4) y en una zeolita HZSM-5 como
función ácida (pura o con tratamiento alcalino para disminuir su acidez) (Oar-Arteta y
cols., 2014).

1. Introducción 15
Carolina Montero Calderón
1.2.1.2. Reformado con vapor de bio-oil
El bio-oil, producto de la pirólisis rápida de biomasa vegetal, es un líquido
marrón, polar e hidrofílico, constituido por productos de despolimerización y
fragmentación de celulosa, hemicelulosa y lignina. La reactividad de estos compuestos
dan al bio-oil un carácter inestable. Está compuesto por agua y oxigenados, de
concentración dependiente de la biomasa de partida y de las condiciones de pirólisis.
Se han identificado más de 300 componentes individuales por GC-MS, que pueden
agruparse en cinco familias: 1) hidroxiacetaldehídos; 2) hidroxicetonas; 3) azúcares;
4) ácidos carboxílicos, y; 5) compuestos fenólicos (Valle y cols., 2014B).
El interés de la valorización del bio-oil se basa en una estrategia de
deslocalización de los puntos geográficos de pirólisis de la biomasa lignocelulósica y
la posterior valorización del bio-oil, centralizada y a gran escala. La reacción del
reformado con vapor del bio-oil tiene la estequiometría:
CnHmOk + (n-k)H2O ↔ nCO + (n + m/2 - k)H2 (1.6)
Considerando la reacción de gas de agua (WGS), reacción inversa a la Ec. 1.4,
la reacción global es:
CnHmOk + (2n-k)H2O ↔ nCO2 + (2n + m/2-k)H2 (1.7)
Ahora bien, la presencia en el bio-oil de los derivados de la pirólisis de la
lignina constituyente de la biomasa lignocelulósica, genera problemas por la re-
polimerización de estos componentes cuando el bio-oil es calentado durante la
vaporización y posteriormente en el reactor de reformado (Trane-Restrup y cols.,
2013). La utilización de un reactor de lecho fluidizado burbujeante o aún mejor de
lecho circulante, puede evitar la aglomeración de las partículas recubiertas de coque y
el bloqueo del lecho (que se da inevitablemente en lecho fijo y en lecho móvil). Por
otro lado, la desactivación del catalizador es atenuada por un elevado contenido de
vapor de agua en el medio, y por la operación a elevada temperatura, que favorece la
gasificación del coque. Contribuye a la atenuación de la desactivación el movimiento
e isotermicidad en el reactor fluidizado.

16
Capítulo 1
Para la resolución de los problemas inherentes a la valorización del bio-oil
completo se han propuesto diferentes estrategias, si bien la solución completamente
satisfactoria y escalable es un reto pendiente, lo que ha obligado a que la mayor parte
de los estudios fundamentales sobre la comparación de catalizadores y sobre la
reactividad del bio-oil se hayan realizado con compuestos modelo contenidos en el
bio-oil, principalmente ácido acético, acetona y etilenglicol. Los progresos en el
tratamiento del bio-oil completo han estado precedidos de la valorización de la
fracción acuosa, cuyo contenido de componentes polimerizables es notablemente
menor que en el bio-oil completo.
Remiro y cols. (2013a) han llevado a cabo el estudio del reformado de la
fracción acuosa del bio-oil (obtenida mediante la separación de fases al añadir agua al
bio-oil completo), con y sin captura in situ de CO2, utilizando un catalizador de Ni
sobre La2O3-Al 2O3 y con una selectividad de H2 de casi el 100 %, lo cual facilita las
posteriores etapas de purificación.
El reformado del bio-oil completo en una etapa catalítica ha sido poco
estudiado. Remiro y cols. (2013a) hacen un análisis de las diferentes estrategias
utilizadas en la bibliografía para este objetivo y proponen un proceso con dos
reactores en serie: uno de tratamiento térmico, donde se deposita la lignina pirolítica y
otro de reformado catalítico de los volátiles de salida. En un estudio reciente, Valle y
cols. (2014b) proponen la utilización de un catalizador de bajo coste (dolomita).

1. Introducción 17
Carolina Montero Calderón
1.3. EL ETANOL COMO FUENTE DE HIDRÓGENO
Es bien conocido el uso de etanol como combustible de automoción mezclado
con gasolina (en proporciones de hasta el 15 % en la denominada gasolina E-85), y
despierta gran interés como materia prima para la producción de H2 mediante
reformado.
El etanol es obtenido principalmente por dos vías: una química, la hidratación
del etileno, y mediante tecnologías bien desarrolladas de hidrólisis-fermentación de
azúcares. El impulso que se ha dado al desarrollo de tecnologías basadas en la
hidrólisis enzimática para obtenerlo a partir de biomasa lignocelulósica (bioetanol de
segunda generación (Menon y Rao, 2012)), junto con su baja toxicidad (frente a otros
alcoholes, como el metanol), y su facilidad para el manejo y almacenamiento (respecto
a otros oxigenados), hacen del etanol uno de los biocombustibles con mejor
perspectiva de valorización. Además, cabe recalcar que la producción de 1000 kg de
etanol derivado de caña de azúcar, desde la preparación del suelo hasta la llegada al
consumidor final, genera 309 kg de CO2, mientras que en el ciclo de producción de
gasolina se generan 3368 kg de CO2 (Silveira y cols., 2009), lo cual evidencia también
una gran ventaja medioambiental del etanol sobre otros combustibles fósiles.
La producción mundial de bioetanol en 2013 superó los 104 mil millones de
litros, y según las proyecciones presentadas por la FAO en su informe “OECD-FAO
Agricultural Outlook 2014-2023”, este valor se incrementará en un 50 % para el año
2023.
Entre los principales productores de bioetanol se encuentran Estados Unidos y
Brasil, que de manera conjunta sumaron el 85 % de la producción mundial en 2013
(Figura 1.4), principalmente a partir de la fermentación de la sacarosa obtenida de la
caña de azúcar en Brasil, del almidón de maíz en Estados Unidos, de trigo en Europa,
de yuca en China y en menor medida de otras fuentes como la remolacha, cebada y el
centeno. Sin embargo, estos materiales son de consumo alimentario, por lo cual se
deben plantear materias primas alternativas.

18
Capítulo 1
Estados Unidos 56%
Brasil 29%
Europa 9%
China 2% Otros 4%
Figura 1.4. Distribución de la producción mundial de bio-etanol en 2013 (Renewables Global Status Report 2014).
1.3.1. Etanol lignocelulósico o de segunda generación
El uso generalizado del etanol como combustible, tiene limitaciones debido a
que su producción depende de productos agrícolas con gran potencial alimenticio, por
lo cual se ha impulsado la valorización de residuos y cultivos lignocelulósicos para la
obtención del denominado etanol de segunda generación. Se estima que cada año
puede obtenerse del orden de 150 billones t de biomasa lignocelulósica derivada de
residuos agroindustriales, como el bagazo de caña de azúcar y maíz, la cascarilla de
arroz y trigo, los residuos de palma aceitera, banana y flores, así como de las algas, y
la obtención de etanol es una alternativa idónea para la valorización de estos residuos
(Menon y Rao, 2012). En el caso particular de la caña de azúcar, se estima que con la
integración de los procesos de obtención de etanol de primera y segunda generación se
pueden lograr rendimientos de hasta 16 mil L de etanol por ha de cultivo (Raele y
cols., 2014).

1. Introducción 19
Carolina Montero Calderón
1.3.1.1. Etapas en la producción
La producción de bioetanol de segunda generación consta principalmente de
tres etapas: pretratamiento, hidrolisis enzimática y fermentación. De manera
esquemática el proceso se muestra en la Figura 1.5.
Figura 1.5. Esquema de producción de etanol de segunda generación (IFP Energies Nouvelles, 2010).
Pretratamiento de la biomasa
El pretratamiento es considerado como la etapa más exigente del proceso de
obtención de biocombustibles a partir de agro-residuos (Gupta y Verma, 2015). En
esta etapa se solubilizan o separan la celulosa, hemicelulosa y lignina, que son los
componentes mayoritarios de la biomasa, logrando de esta manera que la celulosa sea
susceptible de ser posteriormente hidrolizada. Para que este tratamiento sea viable,
además de tener un coste asumible, debe ser efectivo para promover la formación de
azúcares hidrolizables, evitar la pérdida o degradación de estos azúcares, evitar la
formación de productos inhibidores y facilitar la retirada de la lignina. Los métodos se
clasifican en:

20
Capítulo 1
� Pretratamiento físico; consiste en la reducción de la cristalinidad de la celulosa y
del tamaño de partícula, lo que a su vez aumenta el área superficial para favorecer
el ataque de las enzimas en la hidrólisis. Entre las operaciones se incluyen la
extrusión, molienda y triturado en seco o en húmedo de la biomasa, así como
tratamientos térmicos con vapor y agua caliente. Dependiendo del tipo de biomasa,
los requerimientos energéticos de estos pretratamientos pueden ser excesivos y no
aplicables a gran escala (Menon y Rao, 2012).
� Pretratamiento Químico; bien desarrollado para la retirada de lignina de materiales
celulósicos en la industria papelera, y que se aplica en primera instancia para
verificar la biodegradabilidad de la celulosa. Se utilizan ácidos (sulfúrico,
perclórico, fórmico), bases (como el hidróxido de sodio y de amonio); y otros
disolventes orgánicos como la etilendiamina (Gupta y Verma, 2015).
� Pretratamiento Físico-Químico; es una combinación de los pretratamientos físico y
químico, siendo los más aplicados el tratamiento con vapor combinado con CO2 o
SO2 y el tratamiento de explosión fibrilar con amoniaco (AFEX).
� Pretratamiento Biológico; consiste en la utilización de microorganismos (hongos y
bacterias) para degradar la lignocelulosa y que tiene como desventaja que es más
lento que los anteriores.
Hidrólisis Enzimática
La hidrólisis enzimática es una etapa clave en la producción del etanol de
segunda generación. Consiste en la conversión de la celulosa y hemicelulosa en
azucares solubles fermentables mediante la acción de enzimas. Estas enzimas son
principalmente celulasas (endoglucanasas, exoglucanasas y B-glucosidasas) y
hemicelulasas (endoxilanasa y endomannasa), que son sintetizadas por diferentes
microorganismos, principalmente hongos, bacterias y microplantas. Se logra que la
celulosa sea hidrolizada a glucosa, mientras que la hemicelulosa es hidrolizada a
pentosas y hexosas. La principal fuente de producción de celulasa son los hongos del
tipo Tricoderma reesei, mientras que el Aspergillus niger es utilizado en la fabricación
industrial de hemicelulasas como las xilanasas.

1. Introducción 21
Carolina Montero Calderón
Para que el proceso de hidrólisis sea efectivo, debe garantizarse que en el
pretratamiento sea separada la lignina, ya que puede bloquear el acceso de las enzimas
a la celulosa, disminuyendo la velocidad de hidrólisis.
La hidrólisis enzimática permite alcanzar altos rendimientos con condiciones
medias de temperatura (40-50 ºC) y pH (4-5). Además, tiene como ventajas respecto a
la hidrólisis ácida o básica, el menor coste de mantenimiento de equipos, ya que evita
los problemas de corrosión y es más respetuosa con el medio ambiente.
Fermentación
Para la producción de etanol de segunda generación se requieren
microorganismos capaces de fermentar las pentosas y las hexosas. La metodología
tradicional es realizar la fermentación tras la hidrólisis (SHF); como alternativa se
utilizan métodos integrados como, la sacarificación simultánea y fermentación (SSF),
sacarificación simultánea y co-fermentación (SSCF), y el denominado proceso
consolidado de transformación de la biomasa (CBP), teniendo los procesos integrados
la ventaja de reducir los costes.
A escala industrial el método SHF es el más usado, ya que permite lograr los
mayores rendimientos. El método consiste en fermentar los azucares obtenidos de la
hidrólisis, utilizando levaduras del tipo Saccharomices, Kluvriomices, Debariomices,
Zimomomas y combinaciones de estas. Se estima que aplicando esta metodología con
Saccharomices cerevisiae, y con un pretratamiento de tipo explosión fibrilar AFEX, se
puede obtener un rendimiento de 178 g de etanol/ kg de biomasa (pasto), con una
concentración de etanol de 34.6 g/l (Jin y cols., 2010).
1.3.1.2. Implantación industrial
El relevante desarrollo a escala de laboratorio ha permitido el aumento de escala
y recientemente han sido implantadas varias plantas para la producción de etanol de
segunda generación. Entre las iniciativas se pueden citar (Menon y Rao, 2012;
Kudakasseril Kurian y cols., 2013; Biotechnology Industrial Organization, 2014):

22
Capítulo 1
� Abengoa España: con un pretratamiento con vapor y por medio de hidrolisis
enzimática produce etanol a partir de paja de cebada y trigo. La planta, ubicada en
Salamanca, comenzó a operar en 2009 y tiene capacidad para producir 1.3 millones
de galones de etanol al año. En octubre de 2014, han inaugurado una nueva planta
en Hugoton, Kansas, en la cual pueden producir hasta 26.5 millones de galones de
etanol/año.
� Iogen Corporation: planta ubicada en Ottawa (Canadá) desde 2011 y que utiliza
residuos agrícolas que puede producir 70000 t de etanol/año, aplicando a la
biomasa un pretratamiento de explosión con vapor seguido de una etapa de
hidrolisis enzimática.
� En Estados Unidos se han implantado recientemente varias plantas, como: la de
Fiberight en Iowa, que empezó a producir en 2013 y tiene una capacidad de 6
millones de galones de etanol/año; INEOS New Planet Energy en Florida,
operativa desde 2012, que tiene capacidad para producir 8 millones de galones de
etanol/año; POET-DSM Advanced Biofuels también en el estado de Iowa, con
capacidad de 25 millones de galones de etanol/ año y operativa desde 2013.
� ST1 Biofuel, en Finlandia que opera con diferentes tipos de residuos
lignocelulósicos y tiene una capacidad de 11 millones de litros de etanol/año.
� SEKAB, en Suecia, que empezó a operar en 2011 y utiliza como materia prima
residuos de madera, a los que se aplica un pretratamiento térmico-mecánico. Esta
planta podría llegar a producir hasta 4500 t de etanol/año.
� Planta RESETA de la Pontificia Universidad Católica del Ecuador que tiene un
rendimiento de 45 mil L/año de etanol derivado de residuos de palma aceitera
(Carvajal y cols., 2013).
Por otro lado, a partir de la fracción orgánica de los residuos municipales
podrían llegar a producirse hasta 593 mil millones de litros de etanol de segunda
generación a nivel mundial (Uckun Kiran y cols., 2014). Entre las plantas piloto
desarrolladas para este objetivo, podemos citar la de la Universidad de Kukamoto
(Japón) que tiene un rendimiento de 60 L etanol/ t de residuos.
1. Introducción 23
Carolina Montero Calderón
1.3.2. Obtención de hidrógeno a partir de etanol
Las buenas perspectivas sobre la disponibilidad de etanol de segunda
generación aumentan el interés de las rutas de su valorización como combustible y
materia prima, las cuales pueden contribuir en gran medida a desarrollar a gran escala
el concepto de biorefinería.
Las propiedades físicas y la composición hacen que el etanol sea adecuado
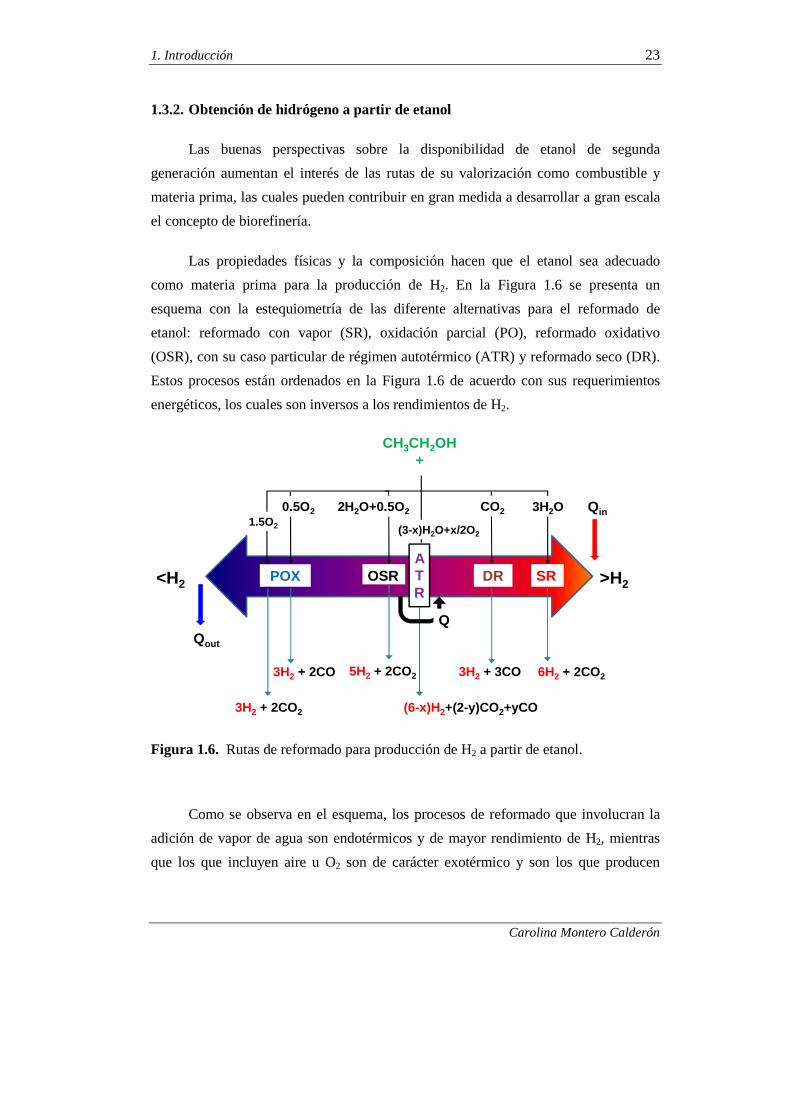
como materia prima para la producción de H2. En la Figura 1.6 se presenta un
esquema con la estequiometría de las diferente alternativas para el reformado de
etanol: reformado con vapor (SR), oxidación parcial (PO), reformado oxidativo
(OSR), con su caso particular de régimen autotérmico (ATR) y reformado seco (DR).
Estos procesos están ordenados en la Figura 1.6 de acuerdo con sus requerimientos
energéticos, los cuales son inversos a los rendimientos de H2.
POXATR
OSR SR >H2<H2
Qin
Qout
CH3CH2OH +
Q
6H2 + 2CO2
(6-x)H2+(2-y)CO2+yCO3H2 + 2CO2
3H2O
(3-x)H2O+x/2O21.5O2
2H2O+0.5O2
5H2 + 2CO23H2 + 2CO
0.5O2
DR
3H2 + 3CO
CO2
Figura 1.6. Rutas de reformado para producción de H2 a partir de etanol.
Como se observa en el esquema, los procesos de reformado que involucran la
adición de vapor de agua son endotérmicos y de mayor rendimiento de H2, mientras
que los que incluyen aire u O2 son de carácter exotérmico y son los que producen

24
Capítulo 1
menor cantidad de H2 aunque pueden ser una alternativa interesante ya que
disminuirían la cantidad de coque depositado.
Si bien el reformado con vapor de etanol es la alternativa con un mayor
rendimiento de H2, tiene el mayor requerimiento energético. Sin embargo, debe
señalarse que el reformado con vapor permite la valorización directa del bio-etanol
(~86 % de H2O), evitando el elevado coste de su deshidratación para ser utilizado
como combustible (etanol anhidro). El coste de la destilación se estima que es el 50%
del coste total del producto (Subramammi y cols., 2007).

1. Introducción 25
Carolina Montero Calderón
1.4. REFORMADO CON VAPOR DE ETANOL
Los primeros estudios de reformado con vapor de etanol datan de comienzos de
los 90 con el estudio termodinámico de García y Laborde (1991), en el que se
determinó que un exceso en la alimentación de agua y temperaturas superiores a 350
ºC minimizan la formación de CH4 e inhiben la formación de coque. Los estudios
experimentales empezaron a presentarse a partir de 1996. Cavallaro y Freni (1996)
ensayaron catalizadores metálicos con diferentes soportes, con la relación S/E=3,
observando una mayor actividad en el CuO/ZnO/Al2O3 y NiO/CuO/SiO2 que la
correspondiente a los catalizadores de metales nobles. Los primeros intentos por
determinar un modelo cinético fueron realizados por Therdthianwong y cols. (2001),
que establecieron un modelo empírico para un catalizador comercial de Ni/Al2O3, en
el intervalo entre 400-650 ºC.
En una búsqueda realizada en la base de datos Scopus ( ~850 publicaciones con
las palabras clave “ethanol steam reforming”) se puede observa un repunte de las
publicaciones a partir del año 2008, impulsado por la búsqueda de alternativas para: i)
reducir las emisiones de gases de efecto invernadero, tras la entrada en vigor del
Protocolo de Kioto el 16 de febrero del 2005; ii) la búsqueda de fuentes alternativas de
energía tras la crisis en el precio del petróleo del periodo 2004-2008; iii) el
resurgimiento de la producción de etanol que supuso en 2003 el programa Proalcool
de Brasil, y; iv) por la propuesta de la economía basada en el H2, impulsada en 2003
por el gobierno de Estados Unidos.
Los resultados de la revisión bibliográfica permiten observar que China es el
país que cuenta con mayor número de publicaciones (150), seguida por Italia y España
(101 y 97). Entre los principales productores de etanol, Estados Unidos cuenta con 94
publicaciones, seguida por Brasil con 85 y la India con 23. Cabe resaltar que la
mayoría de las publicaciones se centran en el desarrollo de los catalizadores, siendo
los más estudiados los formulados con Ni (164 publicaciones), Co (108), Pt (70) y Ru
(58). Entre los soportes, el más utilizado es el de CeO2 (84 publicaciones) y la Al2O3
(54). Sin embargo, son pocos los estudios que han abordado la determinación del
modelo cinético.

26
Capítulo 1
Como antecedente a esta tesis doctoral, en el grupo de investigación se ha
realizado una tesis (Vicente, 2012) en la cual se estudió el comportamiento de los
catalizadores de Ni y Co en diferentes soportes como SiO2, ZnO y α-Al 2O3, sin
modificar y modificada con La2O3. Se realizó una comparación de la estabilidad de los
catalizadores entre 500 ºC (donde hay mínima formación de CO) y 700 ºC (formación
insignificante de coque). Se observó que entre 500 y 600 ºC los catalizadores de Co
presentan un ligero mayor rendimiento que los de Ni, aunque estos últimospresentan
una mayor estabilidad a alta temperatura (700 ºC), especialmente los soportados en α-
Al 2O3. (Gayubo y cols., 2014). También fue abordado el efecto de la deposición de
coque en la desactivación de los catalizadores de Ni y Co (Vicente y cols., 2014a),
aspecto poco estudiado en la bibliografía. Entre los resultados destaca la relación entre
la naturaleza del coque y la estabilidad del catalizador, así como la dependencia de
esta naturaleza del coque con las condiciones de reacción.
1.4.1. Etapas de reacción y esquema cinético
El reformado con vapor de etanol (SRE), es un proceso endotérmico,
termodinámicamente posible a temperaturas relativamente bajas (entre 300 y 800 ºC),
y cuya reacción global se rige por la siguiente estequiometria:
C2H5OH + 3H2O → 6H2 + 2CO2 =∆ 0298H 173.3 kJ/mol (1.8)
Si bien la estequiometría exige una relación molar de vapor de agua/etanol
(S/E) = 3, la alimentación de bioetanol acuoso corresponde a una mayor relación, lo
que tiene ventajas, al aumentar la velocidad de reacción, favorecer la selectividad y
atenuar la formación de coque.
El estudio termodinámico predice un elevado rendimiento de H2, 85 %, para la
relación S/E = 6 a 700 ºC (Sun y cols., 2012). Sin embargo, el mecanismo de reacción
es complejo, y está fuertemente influenciado tanto por las condiciones de reacción
(temperatura, relación molar vapor/etanol, tiempo espacial) como por la composición
del catalizador, que inciden en la presencia de reacciones secundarias que generan
subproductos y productos intermedios, los cuales reducen el rendimiento de H2 y son

1. Introducción 27
Carolina Montero Calderón
precursores de la formación de coque, principal causa de la desactivación de los
catalizadores. Entre las reacciones que se pueden presentar:
Deshidrogenación del etanol: C2H5OH ↔ C2H4O + H2 (1.9)
Deshidratación del etanol: C2H5OH → C2H4 + H2O (1.10)
Descomposición del etanol: C2H5OH → H2 + CO + CH4 (1.11)
Formación de ácido acético: C2H5OH + H2O → CH3COOH + 2H2 (1.12)
Formación de acetona: 2 C2H5OH → CH3COCH3 + CO + H2 (1.13)
Reformado con vapor de acetona: CH3COCH3 + 2H2O → 3CO + 5H2 (1.14)
Reformado incompleto de etanol: C2H5OH + H2O → CH4 + 2H2 + CO2 (1.15)
Descomposición del acetaldehído: C2H4O → CH4 + CO (1.16)
Reformado con vapor del acetaldehído:
C2H4O+H2O→2CO+3H2 (1.17)
Reformado con vapor de acetaldehído hacia CO2:
C2H4O + 3H2O → 2CO2 + 5H2 (1.18)
Reacción Water Gas Shift: CO + H2O ↔ H2 + CO2 (1.19)
Metanación de CO2: CO2 + 4H2 → CH4 + 2H2O (1.20)
Reformado con vapor del CH4: CH4 + H2O ↔ 3H2 + CO (1.21)
Reformado de etileno: C2H4 + 2H2O → 4H2 + 2CO (1.22)
Polimerización del etileno: C2H4 → polímeros → C (1.23)
Reacción de Boudouard: 2CO ↔ C + CO2 (1.24)
Descomposición de CH4: CH4 → 2H2 + C (1.25)
Gasificación del coque: C + H2O → CO + H2 (1.26)
Cabe resaltar que en el caso del reformado con vapor de bio-etanol crudo
estarían involucradas además las reacciones relacionadas con las impurezas presentes
en el medio, principalmente el ácido láctico y el glicerol.

28
Capítulo 1
En la Figura 1.7 se muestra un es quema de las etapas del mecanismo de
reacción del reformado de etanol sobre un catalizador de Ni/SiO2 (Vicente y cols.,
2014b). Este esquema se ha obtenido a partir del conocimiento de la distribución de
los productos para un amplio intervalo de las condiciones de operación (temperatura,
relación molar S/E, presión parcial y temperatura de reacción).
En el esquema cinético se han identificado como los precursores del coque
encapsulante a los compuestos intermedios: etileno, acetaldehído, acetona y etanol
(adsorbido como iones etoxi). Los precursores del coque fibrilar son el CO y el CH4,
por medio de la reacción de Boudouard y la descomposición de CH4, respectivamente.
Esta diferenciación de dos tipos de coque tiene una importancia relevante por la
diferente incidencia en la desactivación del catalizador, siendo el coque encapsulante
el principal responsable de esta.
C2H5OH
descomposición
CH3CHO
CO + CH4H2 +
± H2
metanación/SRM± H2O
CO2 + H2
+ H2O
H2 +
descomposición
reformado
desh
idra
taci
ón
deshidrogenación
cond
ensa
ició
n
C3H6O + H2 + CO
C2H4
-H2O
WGS
+ H2O
± H2
metanación/SRM
Coque fibrilar
Coque encapsulante
desc
ompo
sici
ón
+ H2
Bou
duar
d
+ CO2
Tª
TE
MP
ER
AT
UR
A
Figura 1.7. Esquema cinético para el reformado con vapor de etanol, para un catalizador Ni/SiO2 (Vicente y cols., 2014b).

1. Introducción 29
Carolina Montero Calderón
1.4.2. Catalizadores
La viabilidad tecnológica y económica del proceso SRE requiere el desarrollo
de catalizadores que sean muy activos y selectivos para la formación de H2,
minimizando las reacciones secundarias, y que además sean estables y poco afectados
por la formación de coque. Varias revisiones bibliográficas han analizado el uso de
catalizadores tanto de metales nobles como no nobles, soportados en diferentes óxidos
(Haryanto y cols., 2005; Ni y cols., 2007; Bshish y cols., 2011; Nahar y Dupont 2012;
Contreras y cols., 2014; Kumar y cols, 2014). Se ha establecido la necesidad de que
los soportes sean inertes, porque los soportes ácidos favorecen las reacciones de
deshidratación a etileno, seguido de una polimerización del mismo para formar coque,
mientras que los soportes básicos favorecen la deshidrogenación del etanol, así como
reacciones de condensación para producir acetaldehído y acetona.
Los catalizadores de Ni y Co son muy activos para la producción de coque, si
bien el aumento de la relación vapor/etanol en la alimentación resulta eficaz para la
atenuación de la deposición (Xu y cols., 2013). Ahora bien, no hay una relación
directa entre el nivel de desactivación del catalizador y el contenido de coque, sino
que la disminución de la actividad depende principalmente de la naturaleza del coque
y de las propiedades físico-químicas de los catalizadores (Vicente y cols., 2014a).
Además, la naturaleza del coque es a su vez dependiente de las propiedades del
catalizador y de las condiciones de operación.
La principal causa de la pérdida de actividad es el bloqueo de los centros
metálicos por coque encapsulante, el cual evoluciona hacia un coque de estructura
filamentosa (coque fibrilar), que tiene una notable porosidad y no impide la adsorción
del etanol. La formación del coque fibrilar es por tanto menos perjudicial que la del
coque encapsulante, especialmente en catalizadores de elevada superficie específica, y
la desactivación solo es importante cuando el crecimiento de los filamentos es muy
elevado y llega a bloquear el acceso del etanol y agua a los poros del catalizador
(Wang y cols., 2009a; Karim y cols., 2010; Djinovic y cols., 2012). Una baja
dispersión metálica favorece el bloqueo de los centros metálicos, lo que explica
porqué los catalizadores de Co son menos estables que los de Ni (Vicente y cols.,
2014a).

30
Capítulo 1
1.4.2.1. Catalizadores de metales nobles
Los catalizadores de metales nobles, como Rh, Ru, Pt, Pd, Ir, Au, Ag sobre
diferentes soportes, son atractivos para el SRE debido a las altas selectividades de H2 y
las relativas bajas temperaturas de operación necesarias (en comparación con otros
catalizadores). El método de preparación más utilizado es el de impregnación en base
húmeda y el orden de actividad es: Rh >> Pt > Pd > Ru. Sin embargo, su uso está
limitado por el alto coste.
Catalizadores de Rodio
El Rh es el metal noble más estudiado en esta reacción. Moura y cols. (2012)
observaron que catalizadores de Rh soportados en Al2O3 dan conversiones de etanol
altas, pero por la deshidratación de etanol y no por el reformado, dado el bajo
rendimiento de H2 (8.4 % a 500 ºC y S/E=3). Sin embargo, el soporte de MgO
favorece la selectividad de H2, por lo que sugirieron como adecuado el soporte mixto
Al 2O3-MgO. Según Wu y Kawi (2010), en el intervalo 650-800 ºC la actividad de los
catalizadores de Rh corresponde al orden de los soportes Y2O3 > CeO2 > La2O3 >
Al 2O3.
La superficie específica del soporte tiene gran importancia. Así, Da Silva y cols.
(2011) compararon el comportamiento de catalizadores con soporte de CeO2 de baja
superficie (por calcinación directa de sales de Ce) y de alta superficie (por co-
precipitación), observando que el de alta superficie es más estable, lo que es atribuible
a la mayor capacidad de adsorción de O2 en el soporte, que atenúa el crecimiento de
coque. Por otro lado, comparando los resultados de Da Silva y cols. (2011), con los de
Wu y Kawi (2010), para el catalizador 1 %Rh/CeO2, se observa que el aumento de la
temperatura de operación mejora la conversión de etanol (55 % a 500 ºC y completa a
700 ºC), si bien no existen grandes diferencias en la selectividad (aprox.70 %) de H2.
En un estudio comparativo de catalizadores de Rh y Pt sobre Al2O3 (Bilal y
cols., 2013) se ha determinado que el Rh es más activo a 500 y 600 ºC, y solo
ligeramente más selectivo a H2, debido a la actividad de estos metales para la
descomposición y deshidratación del etanol, mayor que para el reformado.

1. Introducción 31
Carolina Montero Calderón
Una alternativa a los soportes convencionales son las espinelas. Graschinsky y
cols. (2010) señalan que con el catalizador de Rh soportado sobre espinela
MgAl 2O4/Al 2O3 el proceso tiene lugar con las etapas de: descomposición y reformado
de etanol, reformado de CH4 y reacción WGS.
Catalizadores de Platino
Los catalizadores de Pt soportados con cargas metálicas bajas entre 0.5 y 5 %
son activos en el intervalo 200 - 500 ºC (Ito y Tomishige, 2010; Panagiotopoulou y
Verikios, 2012), con selectividades de H2 entre 29 y 70 % dependiendo del soporte.
Los soportes de Al2O3 favorecen la reacción de deshidratación, mientras que el ZrO2,
CeO2 y su combinación favorecen el reformado. La SiO2 tiene peor comportamiento,
si bien es mejorado con la adición de Nb, V y Mo. Una fuerte interacción entre el Pt y
el soporte junto con una débil acidez superficial, favorece la producción de H2 y
atenúa la formación de coque y el orden de actividad de los catalizadores es: Pt/CeO2
> Pt/ZrO2 > Pt//TiO2-Pt/C (He y cols., 2012).
También se han estudiado catalizadores bimetálicos. Palma y cols. (2013, 2014)
han establecido que para catalizadores de Pt-Co, y Pt-Ni soportados en CeO2 la
deshidrogenación de etanol y la descomposición de acetaldehído en CH4 y CO, junto
con la reacción WGS son las etapas principales del reformado. Simson y cols., (2011)
han obtenido con catalizadores comerciales de Rh/Pt y velocidades espaciales bajas
(22000 h-1) conversiones del 100 % de la mezcla de etanol y gasolina (85/15), con
concentraciones de equilibrio de H2, CO, CO2 y CH4 y sin desactivación durante 110
h. Estos autores plantearon el problema de la desactivación irreversible, aconsejando
un equilibrado previo del catalizador para que se reproduzca su comportamiento
cinético en ciclos de reacción-regeneración.
Catalizadores de otros metales nobles
El Ru, Pd y Ag soportados sobre CeO2/YSZ han sido estudiados por Ramos y
cols. (2012). El Ru/CeO2/YSZ es activo y selectivo en las condiciones de reacción
estudiadas (86 % de H2 a 550 ºC y S/E=5), mientras que el Pd, con buena actividad
inicial (80 % H2), sufre una desactivación significativa (~ 80 % de disminución de la

32
Capítulo 1
conversión de etanol tras 70 h). Los resultados de caracterización mediante XRD
demostraron que los dos metales sufren sinterización parcial entre 550 y 600 ºC. El
contenido de coque de los catalizadores desactivados, cuantificado por
termogravimetría, es muy bajo (0.5 % para Ru y 0.7 % para Pd), siendo en el de Pd un
coque amorfo, lo que justifica la desactivación drástica del catalizador. Cabe señalar
que el catalizador de Ag es menos activo (rendimiento de H2 del 13 %).
Entre los estudios sobre el uso de Ir, Wang y cols. (2011) han propuesto un
catalizador de Ir/CeO2 que mejora la selectividad (60 %) respecto a los catalizadores
de Pt, pero requiere temperaturas de operación por encima de 500 ºC. También
proponen la modificación del soporte por la adición de PrOx (Ir/Ce0.9Pr0.1O2), para
mejorar la capacidad de almacenamiento de oxígeno (mejorando la capacidad rédox
del soporte) y brindar estabilidad térmica al catalizador, con lo cual a 650 ºC se
obtiene conversión completa y el catalizador es estable durante 300 h. Chiou y cols.
(2012) han realizado un estudio comparativo de catalizadores con soporte de CeO2,
concluyendo que, si bien el catalizador Ir/CeO2 requiere mayor temperatura para la
conversión completa (425 ºC) que los catalizadores de Co (400 ºC ) y Pt (300 ºC), sin
embargo el Ir da lugar a un mayor rendimiento de H2 (88 % a 475 ºC), lo que es
atribuido a que favorece la formación de acetona, que es reformada, aumentando el
rendimiento de H2.
1.4.2.2. Catalizadores de metales no nobles
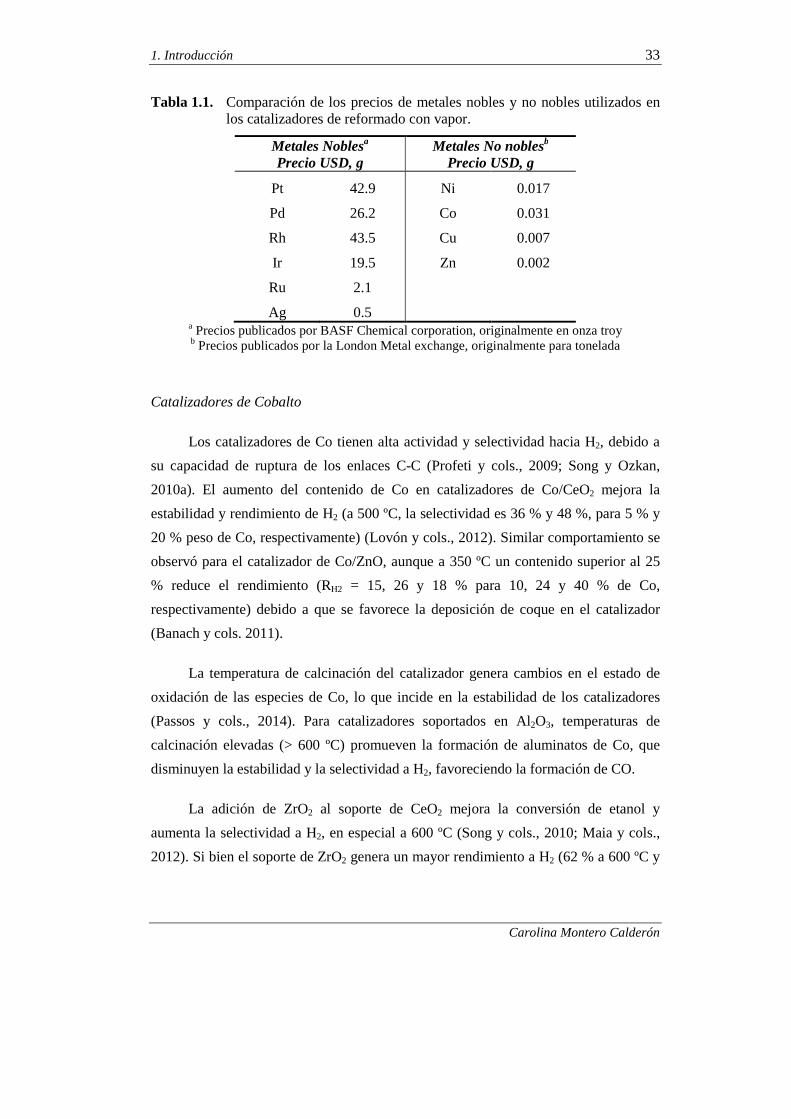
El interés de los catalizadores de metales no nobles se justifica con la Tabla 1.1,
donde se muestran los precios aproximados (actualizados a septiembre 2014) de varios
de los metales utilizados en los catalizadores para reformado, y donde resulta evidente
la diferencia del precio de las dos familias de metales.
Si bien se han publicado resultados para catalizadores de Cu, Zn, Fe y Sn, el
mayor número de estudios corresponde a catalizadores de Ni y Co, que son los usados
comúnmente en procesos comerciales de reformado de metano y naftas, y por su
reconocida capacidad para activar reacciones de hidrogenación/deshidrogenación
(Chiou y cols., 2014). En consecuencia, a continuación centramos la atención en los
resultados con estos catalizadores
1. Introducción 33
Carolina Montero Calderón
Tabla 1.1. Comparación de los precios de metales nobles y no nobles utilizados en los catalizadores de reformado con vapor.
a Precios publicados por BASF Chemical corporation, originalmente en onza troy b Precios publicados por la London Metal exchange, originalmente para tonelada
Catalizadores de Cobalto
Los catalizadores de Co tienen alta actividad y selectividad hacia H2, debido a
su capacidad de ruptura de los enlaces C-C (Profeti y cols., 2009; Song y Ozkan,
2010a). El aumento del contenido de Co en catalizadores de Co/CeO2 mejora la
estabilidad y rendimiento de H2 (a 500 ºC, la selectividad es 36 % y 48 %, para 5 % y
20 % peso de Co, respectivamente) (Lovón y cols., 2012). Similar comportamiento se
observó para el catalizador de Co/ZnO, aunque a 350 ºC un contenido superior al 25
% reduce el rendimiento (RH2 = 15, 26 y 18 % para 10, 24 y 40 % de Co,
respectivamente) debido a que se favorece la deposición de coque en el catalizador
(Banach y cols. 2011).
La temperatura de calcinación del catalizador genera cambios en el estado de
oxidación de las especies de Co, lo que incide en la estabilidad de los catalizadores
(Passos y cols., 2014). Para catalizadores soportados en Al2O3, temperaturas de
calcinación elevadas (> 600 ºC) promueven la formación de aluminatos de Co, que
disminuyen la estabilidad y la selectividad a H2, favoreciendo la formación de CO.
La adición de ZrO2 al soporte de CeO2 mejora la conversión de etanol y
aumenta la selectividad a H2, en especial a 600 ºC (Song y cols., 2010; Maia y cols.,
2012). Si bien el soporte de ZrO2 genera un mayor rendimiento a H2 (62 % a 600 ºC y
Metales Noblesa Precio USD, g
Metales No noblesb
Precio USD, g
Pt 42.9 Ni 0.017
Pd 26.2 Co 0.031
Rh 43.5 Cu 0.007
Ir 19.5 Zn 0.002
Ru 2.1
Ag 0.5

34
Capítulo 1
S/E=3) que el de CeO2 (55 %), la mezcla de los óxidos promueve la movilidad de
oxígeno, lo cual reduce la formación de CO.
La utilización de metanol como disolvente en la preparación por impregnación
húmeda mejora la selectividad a H2 en catalizadores de Co/SiO2 (68 % respecto al 45
% del catalizador preparado impregnando con agua). Por otro lado, en catalizadores de
Co/Al2O3 la naturaleza ácida del soporte influye en el rendimiento de la reacción, en
mayor medida que el disolvente utilizado en la preparación (Lucredio y cols., 2011).
En el reformado con vapor, al ser un proceso endotérmico, el aumento de la
temperatura de reacción mejora notablemente la conversión de etanol y la selectividad
a H2 (XEtOH = 68-100 %, SH2 = 36-62 %; en el intervalo 500-600 ºC, S/E = 3 y 5
%Co/CeO2 (Lovón y cols., 2012)).
Los catalizadores soportados en CeO2 han sido muy estudiados, por su
resistencia a la deposición de coque. Modificaciones del soporte con hasta un 5 % de
CaO atenúan esta deposición y favorecen la producción de H2 (90 %, a 550 °C, S/E =
6) (Pang y cols., 2012). Además, se ha comprobado que el efecto de la morfología del
CeO2 afecta a la capacidad de reducción del catalizador (Soykal y cols., 2012), de
forma que el Co soportado en nanocubos de CeO2 tiene una mayor capacidad de
reducción que el soportado en nanovarillas de tamaño de partícula similar,
aumentando el rendimiento de H2 a temperaturas intermedias (a 450 ºC, y S/E = 10,
RH2 = 40.5 % para nanocubos y 8.5 % para nanovarillas).
Catalizadores de Níquel
Los catalizadores de Ni han sido ampliamente usados en el reformado con
vapor de CH4 y gas natural (Koo y cols., 2014). La posibilidad de ser regenerados para
su reutilización los hacen atractivos para el reformado de oxigenados, particularmente
en el caso del etanol. El Ni tiene como ventaja frente a los otros metales su bajo coste
(Tabla 1.1) y alta actividad para la hidrogenación, lo cual facilita la combinación de
los átomos de H2 adsorbidos en la superficie del catalizador para formar moléculas de
H2 (Elias y cols., 2013; Trane-Restrup y cols., 2013). Sin embargo, los catalizadores
de Ni soportados son susceptibles de desactivación por sinterización (a temperaturas
1. Introducción 35
Carolina Montero Calderón
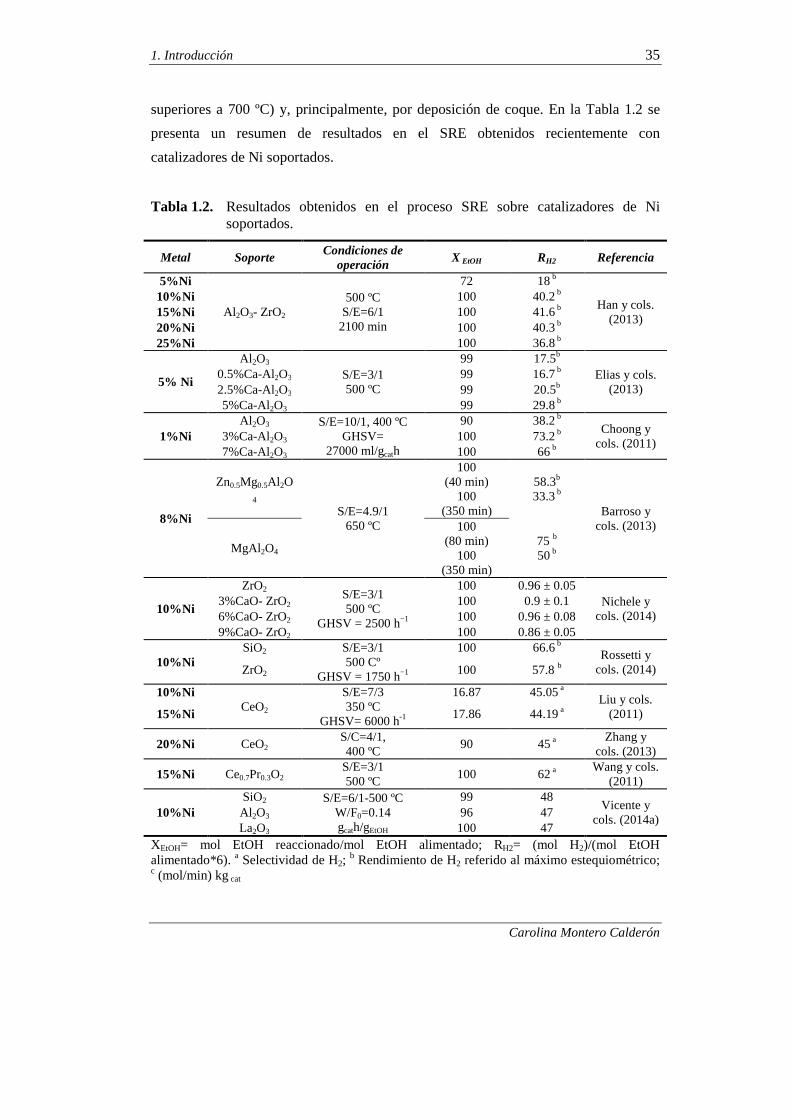
superiores a 700 ºC) y, principalmente, por deposición de coque. En la Tabla 1.2 se
presenta un resumen de resultados en el SRE obtenidos recientemente con
catalizadores de Ni soportados.
Tabla 1.2. Resultados obtenidos en el proceso SRE sobre catalizadores de Ni soportados.
Metal Soporte Condiciones de
operación X EtOH RH2 Referencia
5%Ni
Al 2O3- ZrO2 500 ºC
S/E=6/1 2100 min
72 18 b
Han y cols. (2013)
10%Ni 100 40.2 b 15%Ni 100 41.6 b 20%Ni 100 40.3 b 25%Ni 100 36.8 b
5% Ni
Al 2O3 S/E=3/1 500 ºC
99 17.5b Elias y cols.
(2013) 0.5%Ca-Al2O3 99 16.7 b 2.5%Ca-Al2O3 99 20.5b 5%Ca-Al2O3 99 29.8 b
1%Ni Al 2O3 S/E=10/1, 400 ºC
GHSV= 27000 ml/gcath
90 38.2 b Choong y
cols. (2011) 3%Ca-Al2O3 100 73.2 b 7%Ca-Al2O3 100 66 b
8%Ni
Zn0.5Mg0.5Al 2O4
S/E=4.9/1 650 ºC
100 (40 min)
100 (350 min)
58.3b 33.3 b
Barroso y cols. (2013)
MgAl 2O4
100 (80 min)
100 (350 min)
75 b 50 b
10%Ni
ZrO2 S/E=3/1 500 ºC
GHSV = 2500 h−1
100 0.96 ± 0.05 Nichele y
cols. (2014) 3%CaO- ZrO2 100 0.9 ± 0.1 6%CaO- ZrO2 100 0.96 ± 0.08 9%CaO- ZrO2 100 0.86 ± 0.05
10%Ni SiO2 S/E=3/1
500 Cº GHSV = 1750 h−1
100 66.6 b Rossetti y
cols. (2014) ZrO2 100 57.8 b
10%Ni CeO2
S/E=7/3 350 ºC
GHSV= 6000 h-1
16.87 45.05 a Liu y cols.
(2011) 15%Ni 17.86 44.19 a
20%Ni CeO2 S/C=4/1, 400 ºC
90 45 a Zhang y
cols. (2013)
15%Ni Ce0.7Pr0.3O2 S/E=3/1 500 ºC
100 62 a Wang y cols.
(2011)
10%Ni SiO2 S/E=6/1-500 ºC
W/F0=0.14 gcath/gEtOH
99 48 Vicente y
cols. (2014a) Al 2O3 96 47 La2O3 100 47
XEtOH= mol EtOH reaccionado/mol EtOH alimentado; RH2= (mol H2)/(mol EtOH alimentado*6). a Selectividad de H2;
b Rendimiento de H2 referido al máximo estequiométrico;
c (mol/min) kg cat

36
Capítulo 1
El aumento del contenido metálico de Ni mejora la conversión de etanol, pero
no garantiza una mayor selectividad a H2. Han y cols. (2013) han establecido una
carga metálica óptima de 15 % peso de Ni para el soporte de Al2O3-ZrO2, que
preparado por la técnica sol-gel presenta una alta dispersión de Ni y gran resistencia a
la deposición de coque.
El soporte de γ-Al 2O3 ha sido muy estudiado debido a su estabilidad térmica y
mecánica, asociada a su alta superficie específica, que a su vez mejora la dispersión de
la fase activa. Sin embargo, su acidez favorece la deshidratación de etanol, que es
precursor de la deposición de coque vía etileno, lo que conduce a una rápida
desactivación del catalizador, por lo que se ha buscado neutralizar su acidez por medio
de aditivos básicos. Con los soportes de SiO2 y ZrO2, el Ni presenta mejor
comportamiento que el Co o el Cu (Rossetti y cols., 2014). A 500 ºC el
comportamiento catalítico es similar y la conversión inicial es completa con ambos
soportes, aunque el rendimiento de H2 es ligeramente mayor para el de SiO2. Sin
embargo, a 300 ºC es mayor la formación de acetaldehído con el catalizador de ZrO2,
por lo que tiene lugar la formación de coque polimérico (18.8 mg/gcat).
La adición de CaO, además de disminuir la acidez del soporte, aumenta la
interacción entre el Ni y la Al2O3, lo que facilita la reducción de especies de Ni+2 a Ni0
y, en consecuencia disminuye la temperatura de reducción en los catalizadores
dopados (Choong y cols., 2011; Elias y cols., 2013). De esta forma, el aumento de la
carga de CaO mejora la conversión de etanol y el rendimiento de H2, pero contenidos
> 5 % en peso de Ca aumentan el tamaño de las partículas de Ni activo, con lo que
disminuye el rendimiento de H2 (Elias y cols., 2013), y se promueve la formación de
coque encapsulante, responsable de la rápida desactivación del catalizador (Choong y
cols., 2011). Al añadir MgO al soporte de Al2O3 (Barroso y cols., 2013; Szijjártó y
cols., 2013; Zeng y cols., 2013) se obtuvieron similares resultados al dopaje con CaO.
Sin embargo, el dopaje con CaO del soporte de ZrO2 no afecta la reducibilidad del Ni
(Nichele y cols., 2014).
Por su alta superficie específica y facilidad para la movilidad del oxígeno, los
catalizadores con soporte de CeO2 resisten mejor la sinterización y la deposición de
coque respecto a otros soportes. Los métodos de preparación inciden en el

1. Introducción 37
Carolina Montero Calderón
comportamiento del catalizador, proponiéndose que la adición del Ni (tipo Raney) por
medio de molienda en el soporte (nanoconfinamiento), mejora la interacción soporte-
metal, alcanzándose mejores rendimientos y mayor estabilidad que con catalizadores
preparados por impregnación a humedad incipiente (Zhang y cols., 2013a). Se ha
observado también que la adición de hasta un 3 % en peso de Cu, en lugar de
aumentar la carga de Ni, mejora la actividad del catalizador (Liu y cols., 2011). Al
igual que para catalizadores de metales nobles, en los catalizadores de Ni soportado la
adición de Pr al soporte de CeO2 forma centros vacantes de oxígeno, lo que inhibe la
deposición de coque, e intensifica la interacción entre el metal y el soporte, con lo cual
se minimiza la sinterización (Wang y cols., 2011).
Como antecedente al presente trabajo, dentro del grupo de investigación se ha
estudiado la desactivación por deposición de coque de catalizadores de Ni y Co
(Vicente y cols., 2014a) en el reformado con vapor de etanol a 500 y 700 ºC
(condiciones de reacción S/E = 6; W/F0 = 0.14 gcath/gEtOH, t = 20 h). Se observó que la
disminución de la actividad depende principalmente de la naturaleza del coque
depositado, así como de las propiedades físico-químicas de los catalizadores (área
superficial, volumen de poro, superficie metálica). A 500 °C (temperatura que
favorece la reacción WGS, disminuye los requerimientos energéticos y evita la
sinterización de Ni), la principal causa de desactivación es la fracción de coque de tipo
encapsulante (monoatómico y polimérico) que bloquea los centros metálicos, mientras
que la fracción de coque fibrilar (partículas filamentosas) recubre el catalizador y, si
bien aumenta en cantidad y tamaño con el tiempo de reacción, sin embargo presenta
un efecto menor sobre la desactivación, especialmente en los catalizadores con alta
superficie específica. El catalizador de 10 % Ni sobre SiO2 presentó alta actividad y
estabilidad tras 20 h de reacción (XEtOH = 95 % y RH2 = ~45 %), principalmente debido
a que la estructura porosa del soporte de SiO2 favorece la formación de coque
filamentoso que tiene poco efecto de desactivación. También hay que señalar el
interés de realizar el proceso en reactor fluidizado, que además de garantizar la
isotermicidad del lecho favorece la colisión de las partículas, provocando la
eliminación del coque filamentoso externo, y minimizando el bloqueo de los poros de
la SiO2. La deposición de coque se atenúa drásticamente a 700 ° C, porque: i) a esta
temperatura la gasificación de coque es rápida, y; ii) debido a su alta relación C/H, el

38
Capítulo 1
coque remanente es muy condensado e inerte. Sin embargo, a esta temperatura
empieza a ser notable la sinterización del Ni, con un notable aumento en el tamaño del
cristal del metal.
1.4.3. Diseño del reactor de reformado con vapor de etanol
1.4.3.1. Estudios teóricos
La mayor parte de estudios se ocupan de la optimización de sistemas
combinados con celdas de combustible, especialmente de tipo PEMFC (De Falco,
2011; Wu y cols., 2014a), y con la opción de integrar la captura de CO2 (Cormos,
2014). Las etapas contempladas en el diseño son el reformado con vapor de etanol,
una etapa de WGS a baja temperatura y una etapa final de purificación del H2,
obtenido generalmente por PSA (pressure swing adsorption).
Basándose en los resultados cinéticos obtenidos para el catalizador de
Co(Fe)/ZnO, García y cols. (2009) diseñaron un reformador para bajas temperaturas
(350-400 ºC) y que consta de tres etapas: 1) deshidrogenación de etanol a acetaldehído
e H2 con SnO2; 2) reformado del acetaldehído con catalizador de Co(Fe)/ZnO; y, 3)
reacción WGS utilizando Fe2O3-Cr2O3 como catalizador. El diseño tiene como base de
cálculo la obtención de un caudal de H2 suficiente para alimentar una celda de
combustible de 1 kW. El reformador está diseñado para operar con relación S/E = 6,
considerando un flujo total de entrada de 10 mmol/s que ingresa al sistema a 375 ºC, y
con un diámetro máximo de 2 cm en cada monolito, requeriría un suministro total de
227 W para la unidad completa, que representa solo el 13 % de la energía que genera
el H2 producido. Se especifica además que la corriente final tiene una fracción molar
de CO de ~0.012, por lo cual se contempla implementar una etapa adicional de
purificación. Los detalles de este diseño se encuentran en la Tabla 1.3:
1. Introducción 39
Carolina Montero Calderón
Tabla 1.3. Diseño del reformador de tres etapas propuesto por García y cols. (2009).
Etapa 1 Etapa 2 Etapa 3
Volumen de reactor, L 1.2 1.6 1.2
Temperatura de horno, ºC 375 400 360
Conversión, % 94 EtOH 91 Acetaldehído 92 CO
Flujo de calor, W 86 requerido 23 requerido 92 generado
Como parte de un sistema híbrido de generación de energía, Wu y cols. (2014a),
han diseñado un reformador con la cinética obtenida con un catalizador de Co3O4-
ZnO. Además de las etapas de reformado de etanol, WGS y purificador, este diseño
implementa un quemador para eliminar las trazas de etanol, CH4 y CO. El reformador
está diseñado para operar con 5 kg de catalizador y tendría un diámetro de 0.5 m y
longitud de 1.5 m. Con S/E = 4.18, a 330 ºC se lograría una conversión de etanol del
95 %, produciendo 55.62 kmol/h de H2 a la salida del sistema.
Con la finalidad de diseñar un dispositivo compacto para ser utilizado como
fuente de energía en vehículos pequeños (4 kW, 2 pasajeros, y máximo 45 km/h), el
diseño realizado por De Falco (2011) corresponde a un reformador con membranas de
Pd-Ag, y que utiliza la cinética de reformado propuesta por Mas y cols. (2008a). En la
Tabla 1.4 se comparan las emisiones de gases de efecto invernadero (GHG)
producidos por km recorrido con diferentes tipos de motor y con el dispositivo
propuesto, poniendo de manifiesto la ventaja de éste.
Tabla 1.4. Comparación de las emisiones de gases de efecto invernadero (GHG) de diferentes sistemas de motor (De Falco, 2011).
Modelo Combustible Emisiones GHG,
gCO2 /km
Audi A2 1.2 TDI Diesel 86
Toyota Yaris 1.4 Diesel 113
Fiat Stilo 1.216 V Gasolina 149
Ford Ka 1.3 Gasolina 150
Reformador de membrana + PEMFC
Etanol 30–80

40
Capítulo 1
El trabajo de Cormos (2014) presenta el diseño de un reformador con captura de
CO2, para la producción de 1x105 m3/h de H2 (~300 MW de energía térmica) a partir
de bioetanol (12 % de etanol, 88 % de agua) con las opciones de reformado con vapor
y autotérmico. Son estudiadas tres estrategias de captura de CO2, de acuerdo con los
siguientes procesos:
i) El reformado de bioetanol (catalizador de Ni), es seguido de una etapa de WGS
(catalizador de Cu-Zn). El gas de síntesis es desplazado y tratado en un ciclo de
adsorción/desorción, usando metil-dietanol-amina (MDEA) para la captura de
CO2. La corriente de H2 se purifica por PSA, y el gas remanente de la
purificación se utiliza en un quemador para cubrir la demanda de calor del
reformado con vapor, o se aprovecha para generación de vapor y energía en el
reformado autotérmico.
ii) Se realiza un ciclo químico con Fe mediante un bucle para convertir el gas de
síntesis en H2 y posteriormente capturar el CO2 (Figura 1.8). Se incluye una etapa
de reacción endotérmica con ilmenita, en la cual el gas de síntesis se transforma
en CO2 y H2O, la corriente es separada con trietilen glicol y el CO2 es
comprimido a 120 bar.
iii) Se basa en la conversión directa del bioetanol que se vaporiza y se alimenta al
reactor junto con el portador de oxígeno (ilmenita), donde se consigue que el
bioetanol sea totalmente oxidado a CO2 + H2O y posteriormente el CO2 sea
separado como se ha explicado en la configuración ii).
De la comparación se concluyó que son mejores los resultados con la
integración de un ciclo químico (Figura 1.8), que con un ciclo de adsorción gas-
líquido en MDEA, principalmente en términos de mayor eficiencia energética (59-63
% frente al 53-58 %) y la descarbonización casi total del combustible (> 99 % frente
al 80-92 % ).
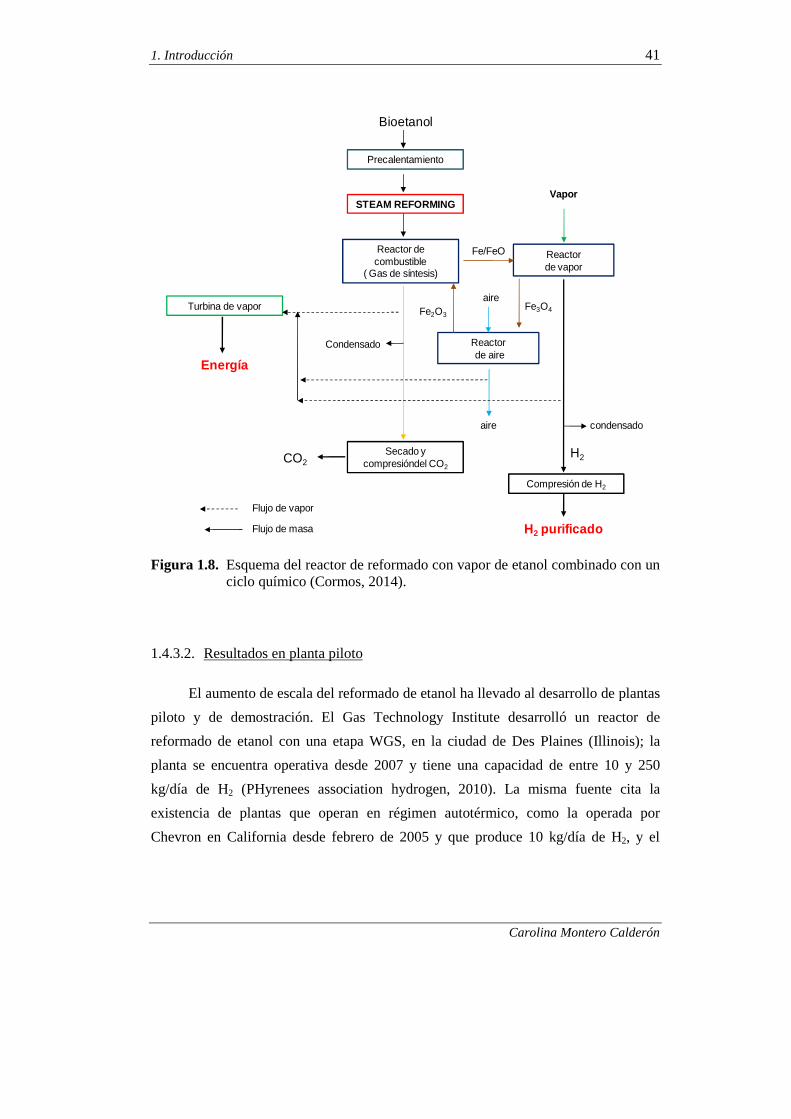
1. Introducción 41
Carolina Montero Calderón
Bioetanol
Precalentamiento
STEAM REFORMING
Reactor de combustible
( Gas de síntesis)
Reactor de vapor
Vapor
Fe/FeO
aireFe2O3
Fe3O4
Reactorde aire
Secado y compresióndel CO2
Turbina de vapor
Compresión de H2
H2 purificado
condensado
H2
Energía
Condensado
aire
CO2
Flujo de vapor
Flujo de masa
Figura 1.8. Esquema del reactor de reformado con vapor de etanol combinado con un ciclo químico (Cormos, 2014).
1.4.3.2. Resultados en planta piloto
El aumento de escala del reformado de etanol ha llevado al desarrollo de plantas
piloto y de demostración. El Gas Technology Institute desarrolló un reactor de
reformado de etanol con una etapa WGS, en la ciudad de Des Plaines (Illinois); la
planta se encuentra operativa desde 2007 y tiene una capacidad de entre 10 y 250
kg/día de H2 (PHyrenees association hydrogen, 2010). La misma fuente cita la
existencia de plantas que operan en régimen autotérmico, como la operada por
Chevron en California desde febrero de 2005 y que produce 10 kg/día de H2, y el
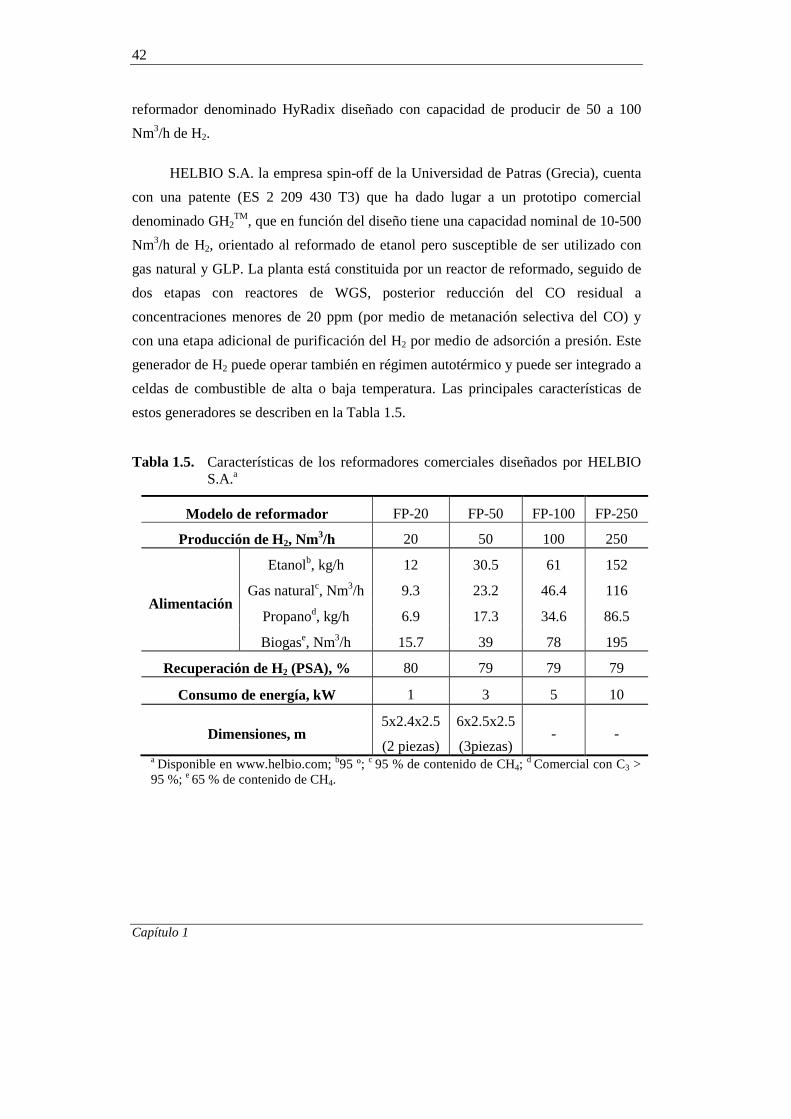
42
Capítulo 1
reformador denominado HyRadix diseñado con capacidad de producir de 50 a 100
Nm3/h de H2.
HELBIO S.A. la empresa spin-off de la Universidad de Patras (Grecia), cuenta
con una patente (ES 2 209 430 T3) que ha dado lugar a un prototipo comercial
denominado GH2TM, que en función del diseño tiene una capacidad nominal de 10-500
Nm3/h de H2, orientado al reformado de etanol pero susceptible de ser utilizado con
gas natural y GLP. La planta está constituida por un reactor de reformado, seguido de
dos etapas con reactores de WGS, posterior reducción del CO residual a
concentraciones menores de 20 ppm (por medio de metanación selectiva del CO) y
con una etapa adicional de purificación del H2 por medio de adsorción a presión. Este
generador de H2 puede operar también en régimen autotérmico y puede ser integrado a
celdas de combustible de alta o baja temperatura. Las principales características de
estos generadores se describen en la Tabla 1.5.
Tabla 1.5. Características de los reformadores comerciales diseñados por HELBIO S.A.a
Modelo de reformador FP-20 FP-50 FP-100 FP-250
Producción de H2, Nm3/h 20 50 100 250
Alimentación
Etanolb, kg/h 12 30.5 61 152
Gas naturalc, Nm3/h 9.3 23.2 46.4 116
Propanod, kg/h 6.9 17.3 34.6 86.5
Biogase, Nm3/h 15.7 39 78 195
Recuperación de H2 (PSA), % 80 79 79 79
Consumo de energía, kW 1 3 5 10
Dimensiones, m 5x2.4x2.5
(2 piezas)
6x2.5x2.5
(3piezas) - -
a Disponible en www.helbio.com; b95 º; c 95 % de contenido de CH4; d Comercial con C3 >
95 %; e 65 % de contenido de CH4.

1. Introducción 43
Carolina Montero Calderón
1.4.4. Economía del reformado con vapor de etanol
Se han realizado algunas predicciones de costes y precios, que incluyen el
análisis de sensibilidad de las variables que afectan el proceso, así como los costes de
emisión equivalente de CO2. Ciambelli y cols. (2009) consideraron un proceso
combinado en el que el calor requerido para el reformado se obtiene por la combustión
de los gases producto del reformado del CH4 remanente. Este esquema es una
alternativa a los reformadores actuales de gas natural, con una inversión inicial que se
reduciría en un 80 %, debido a la reducción del volumen del reformador y a que se
evitaría la etapa de desulfuración. Estos autores, calcularon que para una planta de
1500 m3/h de H2 y con un precio de etanol de 0.14 €/kg (~2.14 $/kgH2), el coste total
de producción es 0.113 €/m3H2 (~1.72 $/kgH2).
Con la finalidad de determinar el beneficio económico de utilizar H2 producido
por reformado de etanol (obtenido del bagazo de caña de azúcar) en las celdas de
combustibles de autobuses urbanos en Brasil, Silveira y cols. (2009) determinaron el
coste de producción de H2 para una estación de abastecimiento de 7200 m3/día. El
coste es 0.12 $/m3, menor que el correspondiente al reformado de gas natural (0.16
$/m3) y a la electrolisis con turbina eólica (0.29 $/m3). En el trabajo de Wu y cols.
(2014a) se establece que el coste de producción sería 0.71 $/m3 considerando un
reactor del tipo de adsorción selectiva.
El coste total del H2 ha sido estimado por Song y Ozkan (2010b) considerando
los costes de etanol (que puede ser un 70 % del total) y del catalizador. Este estudio
señala además la importancia del escalado para disminuir el coste de amortización. Se
obtuvo un coste total de 2.69 $/kgH2 para una central de 150 mil kg/día y 4.27 $/kgH2
para 1500 kg H2/día con un catalizador comercial de Co de ~10 $/kg. Al utilizar
catalizadores de metales nobles el coste total es de 22.34 $/kgH2. Sin embargo, en este
análisis no se ha contemplado el coste asociado a la desactivación del catalizador.
Con los datos de una planta de 11 kgH2/día, de la Universidad de Buenos Aires-
(Argentina), Gregorini y cols. (2010) han calculado un coste total de 31 $/kgH2, muy
superior al de Song y Ozkan (2010b), debido a la importancia de la escala de
producción.

44
Capítulo 1
En el estudio técnico-económico realizado por Mousavi y Chan (2014) se ha
analizado la viabilidad para obtener H2 por medio de reformado con vapor de metanol,
diesel y etanol, considerando además del coste de producción el coste de las materias
primas, la eficiencia energética del proceso y el coste social de la emisión de gases de
efecto invernadero (valor actual neto de los daños causados por las emisiones de CO2
considerando todos los sectores, regiones y periodos de tiempo). Este estudio concluye
que el reformado de metanol presenta mayor eficiencia energética, principalmente
porque requiere una menor temperatura de reformado que el de etanol y el diesel, por
lo cual señalan que sería la materia prima óptima cuando no existan diferencias
económicas significativas entre las tres alimentaciones. Sin embargo, los resultados
del coste social de la producción de CO2 ($/kg de H2 producido) son notablemente
menores para reformado de etanol (0.14) respecto al metanol (0.55) y el diesel (0.37),
por lo cual, desde el punto de vista ecológico, el reformado con vapor de etanol es el
proceso más sostenible.
1.4.5. Otras tecnologías de reformado
El inconveniente del elevado requerimiento energético del reformado con vapor
es atenuado o eliminado con otras tecnologías de reformado con generación de calor,
para la oxidación controlada del etanol. Estas tecnologías son: reformado seco, cuando
se lo realiza con CO2, oxidación parcial cuando se utiliza aire u oxígeno; y el
reformado oxidativo que conjuga el reformado con vapor y la oxidación parcial, con la
finalidad de mejorar el rendimiento de H2 respecto a la oxidación parcial y, a su vez,
eliminar el coque depositado, que es un condicionamiento del reformado con vapor.
1.4.5.1. Oxidación parcial
La oxidación parcial del etanol con O2 (o aire) es un proceso exotérmico,
evitando los requerimientos energéticos del reformado con vapor. Además, se realiza a
menor temperatura, con mayor velocidad de reacción, lo que, por tanto, requiere
menor tiempo espacial, reduciendo el tamaño de los reactores. Este proceso tiene la
siguiente estequiometria:

1. Introducción 45
Carolina Montero Calderón
C2H5OH + 0.5 O2 → 3H2 + 2CO =∆ 0298H -54.7 kJ/mol (1.27)
o bien, en condiciones de exceso de O2:
C2H5OH + 1.5 O2 → 3H2 + 2CO2 =∆ 0298H -620.7 kJ/mol (1.28)
Además, en función del catalizador utilizado se observa la presencia de
acetaldehído, acetona y acido acético, como subproductos no reformados.
El estudio termodinámico (Al-Hamamre y Hararah, 2010), señala condiciones
óptimas en torno a 750 °C y relación aire/etanol de 0.2, con un rendimiento de H2 de
~90 %. El mecanismo considerado en la simulación consta de tres etapas: 1) oxidación
de etanol, 2) reacción WGS y 3) reformado, siendo esta última la etapa limitante del
proceso. En consecuencia, en la primera etapa se promueven rápidamente las
reacciones de oxidación parcial y oxidación total, por lo que se consume etanol y
oxígeno, y se forman H2O, CO2, H2, además de CO, CH4 y C2H4, que son los
principales precursores del coque. En la segunda etapa se producen reacciones más
lentas, como la reacción WGS y la de reformado de CH4, lo cual explica el aumento
de CO2 en esta zona. En el caso del CO, éste disminuye al comienzo de la segunda
etapa y aumenta en la tercera, debido al reformado con vapor del C2H4 y el CH4.
De manera análoga a lo señalado en el reformado con vapor, ha sido
comprobada la actividad de catalizadores de metales nobles, que ofrecen alta
conversión y selectividad, aunque con la limitación del coste de los metales, que
motiva el mayor interés de los catalizadores de metales de transición, especialmente
Ni y Co. El catalizador de Ni-γAl 2O3 soportado en monolitos de cordierita ha sido
ensayado para la oxidación parcial de etanol operando con bajos tiempos espaciales y
relación O2/Etanol= 0.3 (Rodrigues y Schmal, 2011). Este catalizador promueve la
deshidrogenación de etanol a baja temperatura, mientras que a alta promueve las
reacciones de reformado. A 375 ºC la conversión de etanol es 20 % y la relación
H2/CO de 1.06, mientras que a 675 ºC la conversión es de 80 % y la relación
H2/CO=0.25. En ensayos realizados con etanol acuoso (95 % de etanol), se observó
que aumenta la selectividad de H2, ya que se favorece la deshidrogenación del etanol.

46
Capítulo 1
Además, el aumento de la relación O2/etanol de 0.3 a 0.5, favorece la conversión de
etanol y la selectividad de H2.
Kraleva y cols. (2013) estudiaron el comportamiento de catalizadores de Ni y
Co soportados en Al2O3, ZnO y AlZn, a 600 ºC y relación O2/Etanol = 0.75,
determinando para los catalizadores de Co el siguiente orden de actividad de acuerdo
con los soportes: Al2O3 < ZnO < AlZn. Para el caso del Ni: ZnO < Al2O3 < AlZn. Es
destacable que en las condiciones de reacción indicadas estos catalizadores no
promueven la formación de etileno, acetaldehído ni acido acético, y además con los
catalizadores de Ni se reduce notablemente la formación de CH4. Con el soporte de
AlZn se obtiene una selectividad de H2 de 89 % con el catalizador de Ni, y de 79 %
con el de Co; y selectividades de CO de 78 y 76 %, respectivamente. Los mismos
autores (Kraleva y cols., 2014), han comprobado que el soporte de AlZn tiene una
estructura de tipo espinela ZnAl2O4, sobre la que se soporta la fase metálica, como
Co3O4 u NiO. Los resultados de selectividad muestran que para temperaturas entre 400
y 500 ºC el catalizador con mejor comportamiento es el de Co, mientras que por
encima de 600 ºC es mejor el de Ni. Así, a 750 ºC con el de NiAlZn se obtienen: SH2 =
95 % y SCO = 90 %, mientras que con el catalizador de CoAlZn los valores
correspondientes son 90 % y 83 %, respectivamente. Estas diferencias se atribuyen al
grado de reducibilidad de los metales y a la capacidad de almacenamiento de O2 en el
soporte.
Entre los trabajos con catalizadores de metales nobles, Teng y Chiu (2013)
obtuvieron conversiones altas (entre 97.5 y 98.3 %), con los catalizadores de Pt
soportados en CNT (nanotubos de carbono), Al2O3, ZrO2 y CeO2, a 400 ºC y relación
O2/Etanol = 0.5. El orden de la selectividad de H2 es: Pt/CNTs (97.5 %) > Pt/ZrO2
(72.6 %) > Pt/CeO2 (67.5 %) > Pt/Al2O3 (52.6 %). Por otro lado, determinaron un 5 %
de Pt como óptimo, porque un mayor contenido disminuye la selectividad de H2.
1.4.5.2. Reformado oxidativo y autotérmico
El reformado oxidativo de etanol consiste en la integración de los procesos de
reformado con vapor y oxidación parcial, razón por la cual se realiza con la

1. Introducción 47
Carolina Montero Calderón
alimentación conjunta de etanol, agua y aire (u oxígeno), con la siguiente
estequiometría:
C2H5OH + 2H2O + 0.5O2 → 5H2 + 2CO2 =∆ 0298H -50.7 kJ/mol (1.29)
En este proceso integrado el calor generado por la oxidación parcial (que es
exotérmica) puede compensar el requerimiento del reformado con vapor
(endotérmico), y con el adecuado ajuste de la relación molar H2O/O2/etanol de forma
que la entalpia neta sea cero. Estas condiciones corresponden al régimen autotérmico
(ATR) (Graschinsky y cols., 2012):
C2H5OH + 2.28H2O + 0.36O2 → 5.28H2 + 2CO2 ≈∆ 0298H 0 kJ/mol (1.30)
C2H5OH + 1.78H2O + 0.61O2 → 4.78H2 + 2CO2 ≈∆ 0298H 0 kJ/mol (1.31)
Los estudios termodinámicos predicen que los compuestos presentes en
condiciones de equilibrio son: H2, CO, CO2 y CH4. Relaciones H2O/etanol mayores de
3 favorecen la formación de H2, mientras que el aumento en la relación O2/etanol
desfavorece esta formación. La formación de CO disminuye al aumentar la
concentración de O2 en el medio, debido a que se favorecen las reacciones de
oxidación, y por debajo de 430 ºC no se forma. El aumento de las concentraciones de
H2O y O2 desfavorece la formación de coque, y relaciones H2O/etanol > 3 y O2/etanol
> 1 en la alimentación, lo evitan completamente. Para una relación O2/etanol = 0.5, se
obtienen los rendimientos máximos de H2 operando entre 550 y 670 ºC. Así, para una
relación H2O/etanol = 6 el rendimiento sería ~67 %. Además, se han realizado
estudios que consideran la adición de aire en lugar de O2 puro para disminuir costes.
El reformado oxidativo de etanol ha sido estudiado principalmente con
catalizadores metálicos soportados, comprobándose la existencia de diferentes
mecanismos de reacción según la naturaleza del metal (Hung y cols., 2012). Los
metales Cu, Ag y Au activan la reacción secundaria de oxidación de etanol a
acetaldehído, mientras que Co, Ni, Pd y Pt activan la deshidratación de etanol a
etileno, el cual es precursor del coque encapsulante. A diferencia de los anteriores, Ru,
Rh e Ir son selectivos para la ruptura de enlaces del etanol y la posterior oxidación
hacia CO y CO2, con el máximo rendimiento de H2. Con el catalizador de Rh/Al2O3 se

48
Capítulo 1
obtiene un rendimiento de H2 del 94 % a 600 ºC y con una relación
H2O/O2/etanol=3/0.3/1. Además, la capacidad de almacenamiento de O2 del CeO2,
aconseja su uso como soporte, aumentando la actividad del catalizador para el
reformado oxidativo.
1.4.5.3. Reformado con CO2 (Reformado seco)
El CO2 actúa como oxidante:
C2H5OH + CO2 → 3H2 + 3CO =∆ 0298H 339.6 kJ/mol (1.32)
Como se puede deducir de la estequiometría, el rendimiento máximo de H2 es la
mitad del correspondiente al reformado con vapor. Sin embargo, el proceso tiene
interés como ruta alternativa a la gasificación para la producción de gas de síntesis y
para la valorización del CO2.
Estudios termodinámicos realizados por Wang y Wang (2009) señalan que las
condiciones de equilibrio óptimas están entre 900 y 1000 °C (aunque en bibliografía la
mayoría de trabajos se realizan entre 450-750 °C), con una relación molar de CO2/
etanol de 1.2 a 1.3, y que la corriente de productos estaría integrada por H2 (RH2 ~ 94.5
%), CO y CH4. Termodinámicamente solo se formarían productos como el C2H4, y el
acetaldehído para bajas relaciones CO2/etanol (< 1.2) y bajas temperaturas. Estos
productos serían precursores de la formación de coque (como nanotubos y nanofibras),
lo cual causaría la desactivación de los catalizadores utilizados en este proceso.
Entre los catalizadores estudiados, resulta interesante la utilización de Rh por la
capacidad de disociación del CO2. Con el catalizador de Rh/CeO2 en lecho fijo,
presión atmosférica y relación CO2/etanol = 1, Da Silva y cols. (2011) han obtenido un
rendimiento de H2 del 95 %, que disminuye hasta ~ 50 % tras 24 h de reacción. Sin
embargo, la conversión de CO2 es relativamente baja (~12 %) y se mantiene constante
con el tiempo de reacción, lo que indica poca reactividad entre el etanol y el CO2.
Comparado con el reformado con vapor en condiciones similares, el reformado seco
tiene menor selectividad de H2 y mayor de CO, lo que es atribuido a la reacción WGS
inversa. Por otro lado, la desactivación de los catalizadores por coque es más rápida

1. Introducción 49
Carolina Montero Calderón
que en el reformado con vapor, si bien es atenuada por el aumento de la relación CO2/
etanol y de la temperatura, que favorecen la reacción de Boudouard inversa. A partir
del seguimiento por espectroscopia de los intermedios de reacción, mediante DRIFTS
(Diffuse reflectance infrared spectroscopy) Da Silva y cols. (2011) han propuesto el
mecanismo de la Figura 1.9. La adsorción disociativa del etanol sobre el CeO2 da
lugar a especies etoxi y enlaces de tipo OH, la oxidación de las especies etoxi se
produce por la adición de O2, y el Rh promueve la desmetanación del acetato a
carbonato y el reformado seco de las especies CHx.
CO + H2
C2H6O + CO2
Ce=Ce+4 yCe=Ce+3 + e-
Figura 1.9. Mecanismo de reacción del reformado con CO2 de etanol con catalizador de Rh/CeO2 (Da Silva y cols., 2011).
En una publicación reciente (Zawadzki y cols., 2014) se ha estudiado la
actividad de los catalizadores de Ni (5 %) preparados por impregnación húmeda, con
metanol como disolvente, y soportados en Al2O3, CeO2, MgO y ZrO. A partir de los
resultados en un reactor de lecho fijo, a 700 °C y con una relación CO2/etanol = 1.5, se
ha establecido el siguiente orden para la conversión de etanol: Ni/Al2O3 (80.6 %) <
Ni/MgO (95 %) < Ni/ZrO2 (99.7 %) < Ni/CeO2 (99.7 %). Con el catalizador de
Ni/CeO2 el rendimiento de H2 es 2.5 mol H2/mol etanol.


2. EXPERIMENTAL


53
Carolina Montero Calderón
2. EXPERIMENTAL
En este apartado se describen en primer lugar los métodos de preparación de los
catalizadores (Apartado 2.1), y posteriormente las técnicas de caracterización de los
mismos (Apartado 2.2), y el equipo de reacción y de análisis, así como la metodología
de análisis de datos (Apartado 2.3).
2.1. SÍNTESIS DE LOS CATALIZADORES
Como se ha comentado en el Apartado 1.5.2., el reformado con vapor de etanol
ha sido estudiado en la bibliografía sobre gran variedad de catalizadores de diversos
metales. De entre ellos, los de Ni son los más interesantes para la aplicación industrial
a gran escala, ya que presenta una elevada actividad y selectividad de H2, junto con un
coste moderado.
El catalizador de Ni soportado en La2O3-αAl2O3 presenta una gran estabilidad
comparado con el soportado solo en Al2O3 (Vicente, 2012), dado que la presencia de
La2O3 atenúa la deposición de coque (Sánchez-Sánchez y cols., 2007; Valle y cols.,
2014b, Gayubo y cols., 2014). Atendiendo a su buen comportamiento, ha sido elegido
para realizar el estudio cinético.
El catalizador Ni/La2O3-αAl2O3 (con valor nominal de 10% de Ni y 10% de
La2O3 en -Al2O3) ha sido sintetizado mediante impregnación a humedad incipiente,
basándose en el método descrito por Sánchez-Sánchez y cols. (2007). Para la
preparación del catalizador se han utilizado los siguientes reactivos:
-Al2O3 proporcionada por Derivados del Flúor, con diámetro de partícula entre
150 y 250 μm.
La(NO3)3.6H2O, Alfa Aesar 99.9%
Ni(NO3)2.6H2O, Panreac 99%
Inicialmente se prepara el soporte, para lo cual en un rotavapor Buchi R-114 se
impregna el La en solución sobre la α-Al2O3 a 70 ºC y a vacio. El sólido obtenido se

54
Capítulo 2
seca a 110 ºC durante 24 h y se calcina durante 3 h a 900 ºC (Fatsikotas y Verykios,
2004).
Posteriormente el Ni se impregna sobre este soporte a partir de una disolución
acuosa de Ni con la concentración deseada. Los sólidos obtenidos se secan a 110 ºC y
se calcinan a 550 ºC durante 2h (5 ºC/min), obteniéndose el catalizador denominado
10Ni/LaAl-F (donde la F hace referencia al catalizador “fresco”, tal cual se sintetiza).
Esta temperatura de calcinación ha sido la determinada como óptima tras un estudio
del comportamiento cinético del catalizador calcinado a diferentes temperaturas, que
se detalla en un apartado posterior.
Con la finalidad de garantizar un comportamiento reproducible del catalizador
al operar en ciclos sucesivos de reacción-regeneración, en un trabajo anterior (Vicente,
2012) se planteó la necesidad de someter al catalizador a un proceso de estabilización
o “equilibrado”, consistente en un ciclo previo de reacción-regeneración. Las
condiciones en que se ha llevado a cabo este equilibrado del catalizador han sido las
siguientes:
Reacción de reformado: relación vapor/etanol=3 molar, tiempo espacial de 0.34
gcath/gEtOH, escalones sucesivos de temperatura a 700-600-500 ºC, de 2 h de
duración cada uno.
Regeneración: combustión con aire (150 ml/min) a 550 ºC durante 2 h, para
garantizar la eliminación total del coque.
Tras la etapa de estabilización se obtiene el catalizador denominado 10Ni/LaAl-
E que es el que finalmente ha sido utilizado para los estudios paramétrico y cinético,
dado que presenta un comportamiento reproducible, tal como se muestra en un
apartado posterior.

2. Experimental 55
Carolina Montero Calderón
2.2. CARACTERIZACIÓN DE LOS CATALIZADORES
Los catalizadores (fresco, equilibrado, en diferentes estados de desactivación y
regenerado) han sido caracterizados mediante técnicas que han permitido determinar
sus características: físicas (superficie específica, volumen y diámetro de poros),
químicas (composición), morfológicas, metálicas (grado de reducibilidad de las
especies metálicas, superficie metálica, dispersión y tamaño de cristal y distribución
de especies en la superficie). Además, se ha analizado el contenido y naturaleza del
coque depositado en los catalizadores desactivados, por medio de la oxidación a
temperatura programada (TPO), espectroscopía infrarroja (FTIR) y espectroscopía
Raman.
2.2.1. Estructura porosa
Las isotermas de adsorción-desorción de N2 a temperaturas criogénicas por
métodos volumétricos, permiten determinar el área superficial, distribución del
volumen de poros y diámetro medio de poros de los catalizadores. Este procedimiento
ha sido realizado en un equipo Autosorb iQ2 de Quantacrome, utilizando una celda de
cuarzo de 9 mm de diámetro con una varilla interna para evitar volúmenes muertos.
Para garantizar la adsorción-desorción completa se requiere que la muestra represente
por lo menos entre 15 y 20 m2, con una cantidad mínima de 50 mg.
Previamente a la adsorción de N2 se ha realizado una desgasificación de la
muestra, para eliminación de las posibles impurezas absorbidas sobre la superficie de
los catalizadores, calentando a 150 ºC por medio de una manta termoeléctrica y con un
vacío de 10-3
mmHg, durante un tiempo mínimo de 6 h.
La adsorción de N2 (Air liquid, 99.9995%) se ha realizado a la temperatura de
N2 líquido (77 K) y en múltiples etapas de equilibrio hasta la saturación de la muestra,
recogiendo los valores de presión de equilibrio alcanzados en cada etapa tras añadir un
volumen conocido de N2. Posteriormente se ha llevado a cabo la desorción en el
intervalo de presiones relativas de 0.01 a 1.

56
Capítulo 2
La medida de la superficie específica se ha realizado por el método de BET
multipuntos, en la rama de adsorción y en un intervalo de presiones relativas entre
0.05 y 0.35. El volumen total de poros así como el radio medio de poro, han sido
calculados a partir de la distribución del tamaño de poros, por el método propuesto
Barrett, Joyner y Halenda (BJH). Los resultados han sido obtenidos directamente con
el software ASiQWin de Quantacrome.
2.2.2. Composición química
La composición química se ha cuantificado mediante espectroscopia de emisión
atómica por plasma de acoplamiento inductivo (ICP-AES), técnica que permite
cuantificar la mayoría de elementos de la tabla periódica a niveles de traza en muestras
disgregadas en una solución acuosa.
La solución se prepara mediante ataque multi-ácido (proporción 1:2 de HNO3:
HF) a 50 mg de muestra en recipientes cerrados, a los que se les mantiene en placa
calentadora a 90 ºC durante 24 h. Posteriormente se procede a la adición de HClO4 y
evaporación mediante placa calentadora y epirradiador (equipo para hacer ensayos con
calor radiante), seguida de la adición de HCl calentándolo nuevamente a 90 ºC durante
24 h más. Luego se evapora el HCl y se retoma la muestra en HNO3. Previamente al
análisis, la muestra ha sido diluida hasta un factor de dilución de 1:2500.
Los análisis se han realizado en el Servicio de Geocronología y Geoquímica
Isotópica de los Servicios Generales (SGIker) de la UPV-EHU, con un espectrómetro
de masas cuadrupolar con fuente de plasma (Q-ICP-MS) marca Thermo, modelo X7-
II, equipado con una interface Xt, antorcha apantallada y nebulizador concéntrico.
2.2.3. Morfología del catalizador
La morfología de los catalizadores (fresco, estabilizado (o equilibrado) y
desactivado) ha sido analizada por microscopía electrónica de barrido (SEM, Scanning
Electron Microscopy), en los Servicios Generales de Investigación (SGIker), con un

2. Experimental 57
Carolina Montero Calderón
microscopio electrónico de barrido SEM, del tipo JEOL/JSM-7000f equipado con
accesorios de Espectroscopia por dispersión de energía, EDS y operado a 25 kV.
El principio básico del SEM es escanear la muestra con un haz de electrones
que atraviesan una columna que ha sido sometida a alto vacio (aprox. 10-7
torr) y
enfocado con energía del orden del keV. En esta columna el haz inicial es concentrado
por una serie de lentes electromagnéticas que van disminuyendo su diámetro hasta que
el haz es puntual. La imagen se forma punto a punto por la recolección de datos
provenientes de las señales elegidas en zonas de la muestra, y se logra con la
sincronización entre haz y por la modulación del brillo en el tubo catódico.
Las principales utilidades del SEM son: i) la alta resolución (~100 Å); ii) la gran
profundidad de campo, que le da la apariencia tridimensional a las imágenes y iii) la
sencilla preparación de las muestras, ya que solo requieren la dispersión con
disolvente, que es generalmente etanol.
2.2.4. Propiedades metálicas
2.2.4.1. Difracción de rayos X (XRD) (Estructura cristalina)
La difracción de rayos X (XRD) es una técnica no destructiva que permite
analizar la estructura de sólidos cristalinos, proporcionado información sobre la
orientación cristalográfica, identificación de las fases, tamaño de los cristalitos y
polimorfismo, entre otros. En este método, un haz monocromático de rayos X incide
sobre el material cristalino y la intensidad del haz dispersado elásticamente
(difractado) es medida como una función del ángulo difractado 2 θ.
Esta técnica se ha utilizado para determinar el tamaño medio de cristal de los
centros activos metálicos del catalizador, aplicando la formula de Debye-Scherrer.
cos
k=dMO (2.1)
donde dMO es el tamaño medio del cristalito; k, la constante de Scherrer cuyo valor
general es de 0.9; λ, la longitud de onda de la radiación utilizada (kα media=1.541874

58
Capítulo 2
Å); θ, es la posición del pico de difracción correspondiente al metal a analizar (en este
caso Ni); ß, el ancho de la anchura media del pico de difracción de la muestra, el cual
debe corregirse teniendo en cuenta la contribución del equipo de medida (0.08 º).
Los análisis de XRD se han realizado en los Servicios Generales de
Investigación (SGIker), con un difractómetro PHILIPS X´PERT PRO, operando a 40
kV y 40 mA, en configuración theta-theta, con un monocromador secundario con
radiación de CuKα1 a una longitud de onda de 1.5418 Å. Se ha empleado un detector
de energía dispersiva PIXcel con longitud activa en 2θ = 3.347 Å. La toma de datos se
ha realizado en modo continuo, de 10 a 80 º, paso de 0.02606 º en 2θ y tiempo de paso
de 300 s. Además, se ha usado una rendija fija anti-dispersión, dando un volumen
constante de iluminación a la muestra.
Mediante el programa informático FULLPROF incluido en el software
WinPLOTR, se ha realizado el afinamiento de los diagramas de difracción, variando
los parámetros de celda, el desplazamiento de la muestra respecto al origen, la relación
entre gaussiana y lorentziana de la forma de los máximos, la evolución con 2θ de la
anchura a media altura y la asimetría de los máximos de difracción. El afinamiento ha
conducido a un buen ajuste entre los diagramas observado y calculado, obteniéndose
unos factores de ajuste satisfactorios.
2.2.4.2. Espectroscopía fotoelectrónica de rayos X (XPS) (Interacción metal-soporte)
La interacción superficial entre el Ni y el soporte ha sido analizada por XPS.
Debido a su alta sensibilidad, esta técnica permite conocer la naturaleza y el estado de
oxidación, así como la composición de la superficie del catalizador. Cabe mencionar,
que esta técnica también ha sido utilizada para estudiar la composición del coque
depositado, completando con ello las técnicas de análisis de coque descritas en el
Apartado 2.2.5.
La técnica XPS está basada en la interpretación cuántica del efecto
fotoeléctrico, en la cual se asume que la energía cinética de los electrones emitidos en
un proceso de fotoemisión, EK, puede calcularse mediante la diferencia entre la

2. Experimental 59
Carolina Montero Calderón
energía del fotón incidente, hv, y la energía de enlace de los electrones en la muestra,
EB (binding energy):
BK E-hv=E (2.2)
La energía de enlace de un electrón es característica del átomo y del orbital en
el que se encontraba el electrón que ha sido emitido; solo los electrones emitidos o que
han interaccionado con la superficie de la muestra pueden alcanzar el detector para ser
analizados, motivo por el que la técnica XPS es superficial y brinda información sobre
tres o cuatro capas de átomos en la superficie de la muestra.
Cada elemento de la tabla periódica cuenta con uno o más niveles
característicos de energía de ligadura, por lo cual esta técnica permite la determinación
de la composición de las superficies de las muestras (para composiciones mayores al
0.1 % en peso). Además, analizando el espectro se observa que el valor máximo de los
picos depende del entorno químico del átomo responsable del pico, de modo que las
energías de enlace aumentan a medida que el estado de oxidación se hace más
positivo.
Las medidas se han realizado en los Servicios Generales de Investigación
(SGIker), en un sistema SPECS equipado con analizador Phoibos 150 1D-DLD y
fuente de radiación monocromática Al Kα (1486.6 eV). Se realiza inicialmente un
espectro de reconocimiento de baja resolución (survey scan) para detectar el tipo de
elementos presentes, con las siguientes condiciones: incrementos de energía de 1 eV;
tiempo de residencia de 0.1 s y salto de energía de 80 eV. Posteriormente en cada una
de las zonas donde se identifican los elementos se realizan espectros locales de alta
resolución (detail scan), para cuantificación a partir de las áreas de cada pico,
realizándose este análisis en las siguientes condiciones: incrementos de energía de 0.1
eV, tiempo de residencia de 0.1 s, salto de energía de 30 eV y con un ángulo de salida
de electrones de 90 º. El pico 1s del carbono se ha establecido en 284.6 eV para
corregir el efecto de carga y el espectrómetro fue previamente calibrado con el pico
Ag 3d 5/2 (368.28 eV). Los espectros fueron ajustados mediante el software CasaXPS
2.3.16, que modeliza las contribuciones Gauss-Lorentzian, después de una
substracción del fondo (tipo Shirley).

60
Capítulo 2
2.2.4.3. Reducción a temperatura programada (TPR) Reducibilidad de especies
metálicas
La reducción a temperatura programada (TPR) ha sido empleada para
determinar la temperatura de reducción así como para diferenciar las diferentes fases
metálicas del catalizador fresco y estabilizado (equilibrado).
Al ser una técnica con alta sensibilidad permite estudiar el proceso de reducción
de un sólido en una corriente reductora (H2 diluido en un gas inerte). A medida que el
gas fluye a través de la muestra, ésta se calienta a velocidad constante y el H2 es
consumido durante la reducción. Este consumo del gas reductor genera variaciones en
la conductividad térmica, que se cuantifica por medio de un detector TCD.
Este análisis se ha realizado en un equipo Autochem 2920 de Micromeritics,
con una corriente de 10 % en volumen de H2 en He (50 cm3/min) sobre
aproximadamente 100 mg de muestra de catalizador, que es cargada en un reactor de
cuarzo en forma de “U”. Se realiza una desgasificación previa manteniendo la muestra
a 400 ºC; a continuación la muestra se calienta hasta 1000 ºC a una velocidad de
calentamiento constante de 10 ºC/min, mientras se registran continuamente las señales
de temperatura y del detector TCD, con lo cual se obtienen los perfiles de reducción
de los catalizadores.
2.2.4.4. Quimisorción de H2 (Superficie metálica, dispersión y tamaño de cristal)
La superficie metálica, dispersión y el tamaño de cristal de los catalizadores han
sido determinadas por medio de la quimisorción de H2, técnica cuyo objetivo principal
es determinar el número de centros activos presentes en una muestra.
La medición de la capacidad de quimisorción de los catalizadores a diferentes
presiones de adsorción permite estimar la cantidad de adsorbato necesaria para formar
una monocapa de gas quimisorbido, valor que por medio de correlaciones como la de
Lagmuir, es utilizado en el cálculo del área de superficie activa (superficie metálica),
porcentaje de dispersión metálica y tamaño promedio del cristal.

2. Experimental 61
Carolina Montero Calderón
Experimentalmente, esta técnica conlleva los siguientes pasos:
Preparación de la muestra; consiste en la estabilización y limpieza de la superficie
antes del análisis, sometiendo la muestra a alta temperatura bajo un flujo de gas
seco, que en la mayoría de experimentos es H2, de modo que se produce la
reducción de la fase metálica.
Generación de la isoterma; una vez finalizada la preparación, la muestra se enfría
(a vacío) a la temperatura de adsorción deseada. Posteriormente, se añaden
secuencialmente a la muestra cantidades preseleccionadas del gas a adsorber, con
lo que se generan las isotermas (volumen de gas adsorbido en función de la presión
de equilibrio).
La primera isoterma recogida tras la preparación de la muestra representa la
contribución combinada de la quimisorción (en los centros fuertes) y la adsorción
física (si existe) en la muestra.
Correcciones por la adsorción física; se realizan con la finalidad de eliminar su
contribución a la adsorción total de gas en la muestra. La adsorción física y
química varía en función de la temperatura de adsorción (así como la presión, el
tratamiento previo, etc.) y ambas disminuyen al aumentar la temperatura. A
temperaturas por encima de 0 ºC el aporte de la fisisorción es tan pequeño que la
cantidad adsorbida por quimisorción es mayoritaria. Sin embargo, a temperaturas
muy altas la quimisorción también disminuye, por lo que en los experimentos es
deseable seleccionar una temperatura que minimice las captaciones de fisisorción
manteniendo una notable capacidad de quimisorción.
Las moléculas de gas adsorbidas formando una única capa sobre el metal
constituyen el volumen de la monocapa (Vm, cm3/gcat), que se determina por
extrapolación a presión cero de la recta de la isoterma. Conocido este valor y la
estequiometria de adsorción (Sq, que en el caso del H2 es 1) puede calcularse el
número de átomos superficiales (Nm, mol/g) que a su vez permiten determinar el área
de centros activos o superficie metálica activa (AM, m2/gcat)

62
Capítulo 2
Para los catalizadores metálicos soportados, es necesario conocer la fracción de
átomos metálicos activos que están expuestos, debido a que los átomos que se ubican
en el interior de partículas de metal no son activos. La dispersión metálica (D), se
define como la fracción de átomos metálicos que se encuentra en la superficie (Nm); y
se calcula en %:
100L
MSN=D
qm(2.3)
siendo M, el peso molecular del metal y L el contenido en masa de metal en el
catalizador.
El tamaño de partícula metálica se calcula a partir de la densidad (ρM) y
superficie metálica activa (AM), considerando que las partículas son esféricas:
MM
mM
.A
6=d (2.4)
Estos ensayos se han realizado en un equipo Autosorb iQ2 de Quantacrome, en
modo Chemisorption. El procedimiento ha consistido en una desgasificación previa de
la muestra (aprox. 1 g) seguido de reducción de la fase metálica en atmósfera de 10 %
de H2 en He, a 700 ºC (con rampa de calentamiento de 5 ºC/min), y finalmente
evacuación de la superficie a 35 ºC.
2.2.5. Análisis de la naturaleza y el contenido de coque
2.2.5.1. Oxidación a temperatura programada (TPO)
La determinación del contenido de coque depositado sobre la superficie del
catalizador, causante de la desactivación del mismo durante la etapa de reacción, se ha
realizado mediante oxidación a temperatura programada (TPO), con aire. Con esta
técnica seguimos el comportamiento térmico del coque en condiciones de oxidación,
determinando el contenido y naturaleza del coque y su cinética de combustión.
Tras finalizar cada reacción y previamente al análisis, el catalizador desactivado
es sometido a un barrido con He a 300 ºC (30 min) en el propio reactor de lecho

2. Experimental 63
Carolina Montero Calderón
fluidizado, para desorber los productos volátiles que hubieran quedado adsorbidos, y
que no contribuyen a la desactivación.
El análisis se ha realizado en un equipo TGA Q5000TA (Thermo Scientific),
conectado en línea mediante una línea termostatizada con un espectrómetro de masas
Thermostar (Balzers instruments), que permite conocer la evolución de los gases de
combustión, principalmente CO2. Se ha llevado a cabo el siguiente procedimiento:
barrido con He (10 ml/min) para eliminar impurezas; estabilización de la temperatura
a 50 ºC para tener una temperatura inicial constante en todos los ensayos; combustión
con aire (50 ml/min) hasta 800 ºC siguiendo una rampa de temperatura de 10 ºC/min;
una vez alcanzada esa temperatura se mantiene constante durante 20 min para asegurar
la completa combustión del coque; finalmente se enfría a temperatura ambiente, 20 ºC.
Generalmente, la cantidad total de coque depositado se determina por diferencia
entre la masa inicial y final de la muestra tras el ensayo TPO. Sin embargo, para este
catalizador no puede aplicarse este método de cuantificación, ya que al ser metálico se
oxida en paralelo con la combustión, y el aumento de masa que ello conlleva
enmascara la pérdida de masa debida a la combustión. Por tal motivo, para determinar
bajos contenidos de coque (< 10 % en peso) se ha utilizado un espectrómetro de masas
acoplado en línea con la termobalanza, que registra las señales de masa 14, 18, 28, 44
correspondientes al N2, H2O, CO y CO2, respectivamente. El calibrado de la señal de
CO2 se ha realizado siguiendo la descomposición de carbonato cálcico. Para el cálculo
del contenido de coque solo se ha considerado la señal del CO2, debido a que: i) no se
puede diferenciar el agua formada durante la combustión del coque respecto a la
humedad y; ii) la combustión es completa, siendo el contenido de CO insignificante,
debido a que se oxida inmediatamente a CO2 (reacción activada por la función
metálica de los catalizadores). El contenido de coque se calcula:
100M
g 44
g 12.M
=(%) Ccat
CO2
CCO
c
2
(2.5)

64
Capítulo 2
2.2.5.2. Espectroscopía infrarroja por transformada de Fourier (FTIR)
La espectrometría de infrarrojos es un tipo de espectrometría de absorción que
utiliza la región infrarroja del espectro electromagnético, y puede ser utilizada para
identificar un compuesto o investigar la composición de una muestra. En este caso ha
sido utilizada para determinar la composición del coque en catalizadores desactivados.
Esta técnica se fundamenta en que los enlaces químicos de las sustancias tienen
frecuencias de vibración específicas, que corresponden a los niveles de energía de la
molécula.
Con el fin de hacer medidas en una muestra, se transmite un rayo monocromo
de luz infrarroja a través de la muestra, y se registra la cantidad de energía absorbida.
Esta técnica funciona casi exclusivamente en enlaces covalentes y proporciona un
espectro de reflexión de las bandas de los grupos funcionales de las sustancias
inorgánicas y orgánicas, por lo cual es posible realizar una identificación de los
materiales.
Los análisis de los componentes del coque mediante FTIR se han llevado a cabo
en un espectrómetro FTIR Nicolet 6700. El procedimiento experimental consiste, en
primer lugar, en preparar una pastilla prensada de catalizador desactivado (3-5 mg)
sobre un soporte de KBr (300 mg, pureza > 99 %), aplicando una fuerza equivalente a
10 t cm-2
durante 10 min. Previo al análisis se graba un espectro IR como referencia o
background de la muestra y luego los espectros IR son tomados directamente tras
incidir el láser en la muestra.
2.2.5.3. Espectroscopía Raman
La espectrometría Raman es una técnica espectroscópica utilizada para el
estudio de los modos vibracionales, rotacionales y otros de baja frecuencia en un
sistema. Se basa en la dispersión inelástica, o dispersión Raman, de la luz
monocromática, que por lo general procede de un láser en el rango visible, infrarrojo
cercano, o ultravioleta cercano. La luz láser interactúa con fotones u otras excitaciones
en el sistema, por lo que la energía de los fotones láser se desplaza hacia arriba o hacia
abajo. Normalmente, la muestra se ilumina con un rayo láser. La luz del punto

2. Experimental 65
Carolina Montero Calderón
iluminado se recoge con una lente y se envía a través de un monocromador. Las
longitudes de onda cercanas a la línea láser, debidas a la dispersión elástica de
Rayleigh, son filtradas, mientras que el resto de la luz recogida se dispersa en un
detector.
La dispersión Raman espontánea es generalmente muy débil, y como resultado
la principal dificultad de la espectrometría Raman es separar la luz débil dispersada
inelásticamente de la luz intensa láser por dispersión de Rayleigh. La instrumentación
moderna emplea filtros de muesca o borde para rechazar el láser, así como
espectrógrafos y detectores CCD.
La espectroscopía Raman se ha llevado a cabo en los Servicios Generales
(SGIker) utilizando un láser (para realizar la excitación) con una longitud de onda de
514 nm y un microscopio confocal Renishaw.
La muestra de catalizador (3-5 mg) es depositada en el recipiente y colocada en
la trayectoria del láser. Mediante un microscopio óptico se direcciona el láser hacia
puntos representativos de la muestra. Es necesario realizar varios análisis en diferentes
partes de la muestra para poder obtener mayor reproducibilidad, evitando siempre el
contacto con aire (atmósfera de N2) para evitar la oxidación del coque. La
fluorescencia de las muestras se elimina utilizando una línea base de 4 puntos.

66
Capítulo 2
2.3. EQUIPO DE REACCIÓN Y ANÁLISIS
Los ensayos cinéticos de reformado se han llevado a cabo en un equipo
Microactivity Reference, provisto de un reactor isotermo de lecho fluidizado en línea
con un microcromatógrafo de gases Agilent 3000, para el análisis de la corriente de
productos de reacción. En los siguientes apartados se describen las dos unidades,
conjuntamente con la metodología utilizada para verificar el funcionamiento de la
unidad de reacción y para validar los ensayos cromatográficos. Además, se describen
las condiciones de operación y los índices de reacción que se utilizan para analizar los
resultados cinéticos.
2.3.1. Equipo de reacción
La unidad está diseñada para realizar estudios cinéticos heterogéneos,
permitiendo trabajar a alta presión (hasta 100 bar) y alta temperatura (hasta 800 ºC)
tanto con alimentaciones gaseosas como líquidas. Tiene alto grado de automatización
(posibilidad de operar con secuencias de variables de operación, y en sucesivos ciclos
de reacción-regeneración) y un alto nivel de seguridad (posee un sistema anti-fallo de
la corriente eléctrica, un sistema de alarmas de temperatura, presión, nivel y flujo, y
una sesión de inhibición que se activa al saltar alguna de las alarmas, interrumpiendo
el desarrollo del experimento).
En la Figura 2.1 se muestra un esquema del equipo de reacción, el cual se divide
en cinco zonas: i) alimentación de reactantes; ii) precalentamiento de los gases,
evaporación y mezcla de los reactantes; iii) reacción; iv) muestreo y; v) salida de
productos.
i) Zona de alimentación de los reactantes
Se subdivide en una zona de alimentación de gases y una de alimentación de
líquidos. En este proceso se han utilizado cuatro líneas de entrada de gases que
corresponden a: gas inerte (He), gas oxidante (aire), gas reductor (H2) y gas auxiliar o
carrier (He). Cada línea dispone de una válvula de corte y un medidor-controlador de
flujo másico Bronkhosrt High-Tech para regular el caudal. Este tipo de medidores son

2. Experimental 67
Carolina Montero Calderón
idealmente independientes de cambios en la presión y la temperatura, y muestran los
datos en condiciones estándar (25 ºC y 1 atm). Ante un posible cambio en la dirección
del flujo de los gases, los controladores están protegidos con restrictores sellados con
teflón. Las líneas de alimentación de los gases, excepto la del gas auxiliar, se unen
después de pasar por sus correspondientes restrictores en un distribuidor, del que sale
una única línea de alimentación gaseosa que entra al horno. Del mismo modo, la línea
del gas auxiliar entra en el horno para su calentamiento.
Figura 2.1. Esquema del equipo de reacción.
La zona de alimentación de los líquidos, en el presente caso mezcla agua-etanol,
consta de un depósito y una bomba de pistón Gilson 307 HPLC con un cabezal SC.
Este sistema de bombeo permite trabajar con flujos de 0.010 a 5 ml/min, hasta 600 bar
y hasta 40 ºC. El líquido bombeado pasa por un restrictor que genera una sobrepresión
en el cabezal de unos 30 bar respecto del sistema de reacción, por un antirretorno de
bajo volumen muerto y por un filtro de 10 m, antes de su entrada al horno.

68
Capítulo 2
ii) Zona de precalentamiento de gases, evaporación y mezcla de los reactantes
Los gases y la alimentación líquida acceden a un horno calentado (caja caliente)
a 160 ºC, que es controlado por medio de un controlador de temperatura PID digital
Toho TTM-005 Series. Los gases, tras precalentarse y homogenizarse, se unen a la
alimentación líquida mediante una conexión del tipo “T”, térmicamente aislada y
rellena de un material inerte de alta porosidad para conseguir la evaporación uniforme
de la alimentación. La mezcla gaseosa formada por los gases y por la mezcla líquida
evaporada llega a una válvula de 6 puertos, que permite cortocircuitar el reactor.
iii) Zona de reacción
Se ha trabajado con un reactor de lecho fluidizado de acero inoxidable 316
(Dinterior= 22 mm) con un volumen de ~ 43 cm3, longitud total de 460 mm, de los
cuales 116 mm corresponden a la zona de reacción, y que dispone de una placa porosa
para soportar el catalizador y distribuir adecuadamente los gases reactantes.
El control de la temperatura interior del reactor se realiza por medio de un
controlador de temperatura digital Toho TTM-005 Series. La lectura de la misma se
lleva a cabo con termopares tipo K que están en contacto directo con el lecho del
catalizador (1-2 mm sobre la placa). El reactor se aloja en un horno que consiste en
una carcasa de acero inoxidable rellena internamente por material refractario, y en
cuyo interior se encuentra la resistencia eléctrica.
iv) Zona de muestreo de productos
Tras la salida del reactor, los productos de reacción pasan por dos filtros en
serie, el primero para partículas de hasta 20 m y el segundo de alta eficacia para
partículas de hasta 2 m, para evitar el arrastre de finos de catalizador a la válvula de 6
puertos y a la línea de muestreo. A continuación los gases pasan por una válvula de
aguja micrométrica que está controlada por un controlador de presión PID digital
Toho TTM-005 Series, siendo el medidor de presión del tipo Sensor-Technik-
Wiedemann.

2. Experimental 69
Carolina Montero Calderón
El muestreo se lleva a cabo en continuo, es decir, el gas auxiliar arrastra y
diluye un pequeño caudal de la corriente de salida del reactor, que es enviada al
microcromatógrafo de forma continua. Se logra de este modo que la muestra sea más
representativa y estable que en un muestreo discontinuo (Mier y cols., 2007). La
corriente de muestra está calorifugada para evitar puntos fríos de posible
condensación.
v) Zona de salida de productos
La corriente principal de productos de reacción se enfría mediante una célula
Peltier a 0 ºC, con la finalidad de separar los compuestos menos volátiles por
condensación. El nivel del líquido en el condensador está controlado por un
controlador PID digital Toho TTM-005 Series, que valora la señal medida por un
sensor capacitivo situado en el interior del separador. Los líquidos condensados se
recogen en un depósito, mientras que los gases se envían a venteo tras pasar por un
elemento mecánico que crea la sobrepresión necesaria para el sistema de muestreo en
continuo.
Sistema de control y adquisición de datos
La supervisión y control de la unidad, así como la adquisición de datos a tiempo
real, se lleva a cabo mediante el software de control de procesos Process@. La
comunicación entre la unidad y el computador se realiza mediante un cable de red. La
conexión bilateral entre el ordenador central y los terminales permite enviar valores de
punto consigna a los dispositivos de control, así como leer los valores actuales del
proceso. Sin embargo, el verdadero control del proceso se lleva a cabo por medio de
los dispositivos correspondientes (controladores, variadores de frecuencia, etc.) cuyos
parámetros pueden ser modificados manual y automáticamente. La adquisición de
datos se realiza cada 10 s, aunque el usuario puede definir la frecuencia de muestreo.
El sistema de seguridad del equipo se encuentra integrado por un microprocesador
independiente del ordenador; de esta forma, las señales de alarma de los diferentes
lazos de control se centralizan en el microprocesador, que actúa frente a los diferentes
contratiempos del sistema.

70
Capítulo 2
La operación del equipo es sencilla: se puede trabajar manualmente con el
equipo adquiriendo valores con el software, o bien se puede crear una tabla de
sesiones, que son las etapas en las que se puede dividir el experimento. En este caso
las sesiones utilizadas han sido las siguientes:
Sesión 1: Arranque del proceso, en el cual se establece la comunicación entre el
equipo y el software.
Sesión 2: Estabilización previa de todas las variables de reacción (temperatura del
reactor y de la caja caliente, presión, caudales de inerte y gas de arrastre) y
encendido de la célula de Peltier, así como de la línea calorifugada al
cromatógrafo.
Sesión 3: Reducción del catalizador, para lo cual se activa la entrada de un caudal
de H2 y se procede al calentamiento hasta la temperatura de reducción con una
rampa de temperatura.
Sesión 4: Barrido con gas inerte para eliminar posibles trazas de H2, y a su vez
enfriamiento del reactor a la temperatura de reacción.
Sesión 5: Actuación sobre la válvula neumática de 6 puertos cambiando de la
configuración de modo reactor a modo bypass. Se activa la bomba de alimentación
líquida (mezcla etanol y agua) para homogenizar los caudales de entrada de los
reactantes. La composición real de la mezcla que ingresa se comprueba durante 30
min en modo bypass con el cromatógrafo.
Sesión 6: Reacción en las condiciones de operación. Actuación sobre la válvula
neumática de 6 puertos retornándola a la configuración modo reactor.
Sesión 7: Barrido con inerte, siguiendo una rampa de 10 ºC/min desde la
temperatura de reacción a 300 ºC, que se mantiene durante 30 min.
Sesión 8: Dependiendo del experimento, se incluye una etapa de regeneración del
catalizador, por medio de la combustión del coque a 550 ºC, para lo cual se activa
la entrada de un caudal definido de aire al reactor. Una vez concluida esta etapa se
somete nuevamente a una etapa de barrido.
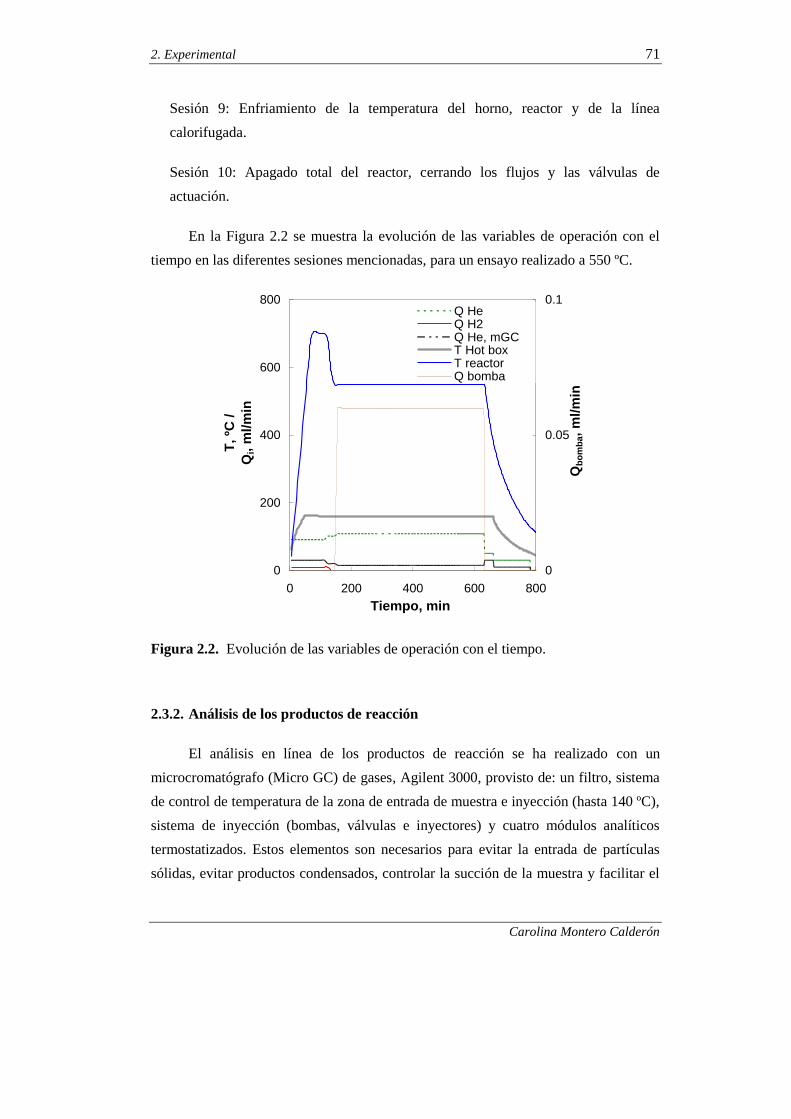
2. Experimental 71
Carolina Montero Calderón
Sesión 9: Enfriamiento de la temperatura del horno, reactor y de la línea
calorifugada.
Sesión 10: Apagado total del reactor, cerrando los flujos y las válvulas de
actuación.
En la Figura 2.2 se muestra la evolución de las variables de operación con el
tiempo en las diferentes sesiones mencionadas, para un ensayo realizado a 550 ºC.
Figura 2.2. Evolución de las variables de operación con el tiempo.
2.3.2. Análisis de los productos de reacción
El análisis en línea de los productos de reacción se ha realizado con un
microcromatógrafo (Micro GC) de gases, Agilent 3000, provisto de: un filtro, sistema
de control de temperatura de la zona de entrada de muestra e inyección (hasta 140 ºC),
sistema de inyección (bombas, válvulas e inyectores) y cuatro módulos analíticos
termostatizados. Estos elementos son necesarios para evitar la entrada de partículas
sólidas, evitar productos condensados, controlar la succión de la muestra y facilitar el
0
200
400
600
800
0 200 400 600 800
Tiempo, min
T, ºC
/
Qi,
ml/
min
0
0.05
0.1
Qb
om
ba,
ml/
min
Q HeQ H2Q He, mGCT Hot boxT reactorQ bomba
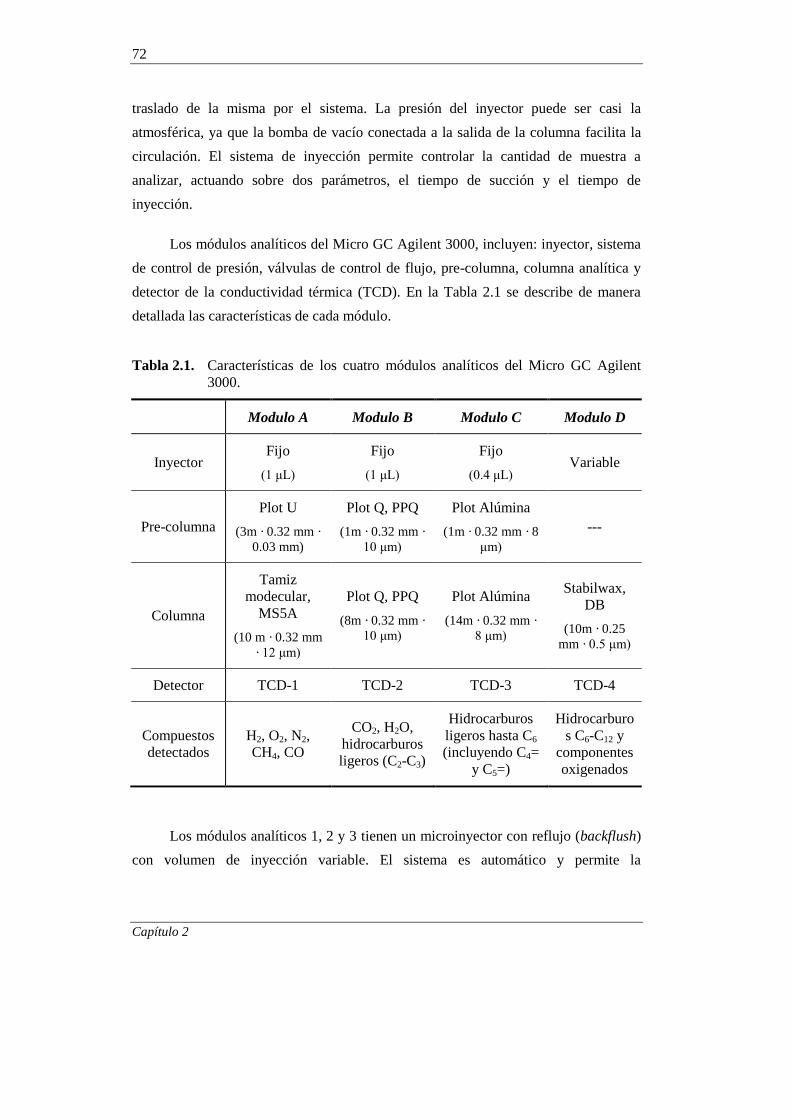
72
Capítulo 2
traslado de la misma por el sistema. La presión del inyector puede ser casi la
atmosférica, ya que la bomba de vacío conectada a la salida de la columna facilita la
circulación. El sistema de inyección permite controlar la cantidad de muestra a
analizar, actuando sobre dos parámetros, el tiempo de succión y el tiempo de
inyección.
Los módulos analíticos del Micro GC Agilent 3000, incluyen: inyector, sistema
de control de presión, válvulas de control de flujo, pre-columna, columna analítica y
detector de la conductividad térmica (TCD). En la Tabla 2.1 se describe de manera
detallada las características de cada módulo.
Tabla 2.1. Características de los cuatro módulos analíticos del Micro GC Agilent
3000.
Modulo A Modulo B Modulo C Modulo D
Inyector Fijo
(1 μL)
Fijo
(1 μL)
Fijo
(0.4 μL) Variable
Pre-columna
Plot U
(3m · 0.32 mm ·
0.03 mm)
Plot Q, PPQ
(1m · 0.32 mm ·
10 μm)
Plot Alúmina
(1m · 0.32 mm · 8
μm)
---
Columna
Tamiz
modecular,
MS5A
(10 m · 0.32 mm
· 12 μm)
Plot Q, PPQ
(8m · 0.32 mm ·
10 μm)
Plot Alúmina
(14m · 0.32 mm ·
8 μm)
Stabilwax,
DB
(10m · 0.25
mm · 0.5 μm)
Detector TCD-1 TCD-2 TCD-3 TCD-4
Compuestos
detectados
H2, O2, N2,
CH4, CO
CO2, H2O,
hidrocarburos
ligeros (C2-C3)
Hidrocarburos
ligeros hasta C6
(incluyendo C4=
y C5=)
Hidrocarburo
s C6-C12 y
componentes
oxigenados
Los módulos analíticos 1, 2 y 3 tienen un microinyector con reflujo (backflush)
con volumen de inyección variable. El sistema es automático y permite la

2. Experimental 73
Carolina Montero Calderón
programación de flujos, así como la inversión de flujo en cada columna para evitar la
entrada de compuestos no deseados. Consiste en una pre-columna y una columna
analítica, las dos acopladas a un punto de presión, que hace posible invertir la
dirección de flujo de gas portador a través de la pre-columna a un tiempo prefijado,
denominado tiempo de backflush. Cuando los compuestos deseados son transferidos a
la columna analítica, en la que se lleva a cabo la separación de los componentes, la
válvula backflush se acciona. Esto invierte el flujo en la pre-columna, con lo que los
componentes que permanecen en ella son enviados a venteo.
La comunicación entre el Micro GC y el PC se realiza mediante un cable
cruzado de red. El control del Micro GC, así como la adquisición y tratamiento de los
datos se realiza mediante el software EZ Chrome. Este programa informático permite
que el usuario defina el método de análisis cromatográfico, así como el método de
integración de los cromatogramas recopilados. El método de análisis cromatográfico
utilizado se detalla en la Tabla 2.2, mientras que los cromatogramas tipo obtenidos se
muestran en la Figura 2.3.
La identificación de los productos de reacción se ha llevado a cabo mediante la
inyección de compuestos puros o mezclas patrones de gases conocidos, determinando
el tiempo de retención para cada uno de los componentes. La cuantificación de los
compuestos a partir de los cromatogramas se ha realizado por transformación del área
resultante de la integración en su valor molar, mediante multiplicación por el factor de
respuesta de cada compuesto. Finalmente, la fracción molar de cada componente se
determina como la cantidad molar del compuesto i dividido entre la cantidad molar de
todos los compuestos detectados.
Los factores de respuesta de los componentes detectados (Tabla 2.3) se han
calculado mediante calibrado con patrones de composición conocida, tomando como
referencia la señal de CO2. Los factores de algunos compuestos de la columna de
alúmina, que no formaban parte de la composición de las muestras patrón, se han
calculado en base a la experiencia en estudios anteriores, aplicando la misma relación
existente entre dichos factores y el factor de un compuesto conocido.
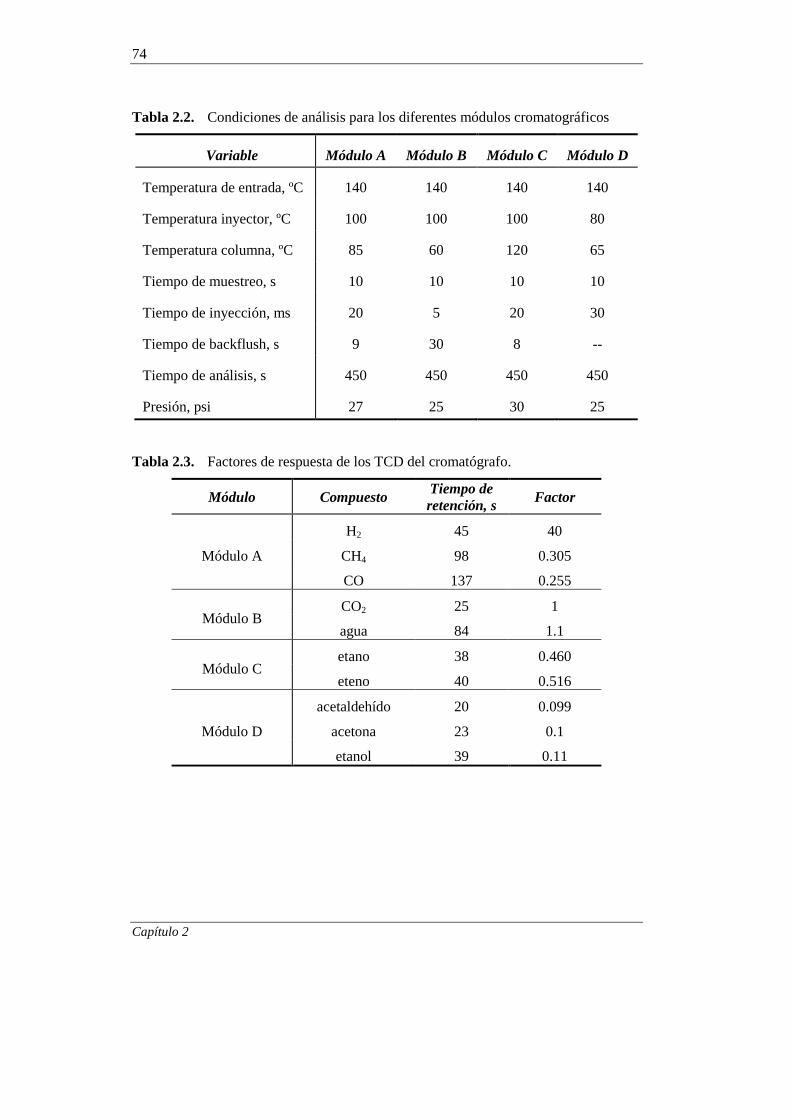
74
Capítulo 2
Tabla 2.2. Condiciones de análisis para los diferentes módulos cromatográficos
Variable Módulo A Módulo B Módulo C Módulo D
Temperatura de entrada, ºC 140 140 140 140
Temperatura inyector, ºC 100 100 100 80
Temperatura columna, ºC 85 60 120 65
Tiempo de muestreo, s 10 10 10 10
Tiempo de inyección, ms 20 5 20 30
Tiempo de backflush, s 9 30 8 --
Tiempo de análisis, s 450 450 450 450
Presión, psi 27 25 30 25
Tabla 2.3. Factores de respuesta de los TCD del cromatógrafo.
Módulo Compuesto Tiempo de
retención, s Factor
Módulo A
H2 45 40
CH4 98 0.305
CO 137 0.255
Módulo B CO2 25 1
agua 84 1.1
Módulo C etano 38 0.460
eteno 40 0.516
Módulo D
acetaldehído 20 0.099
acetona 23 0.1
etanol 39 0.11
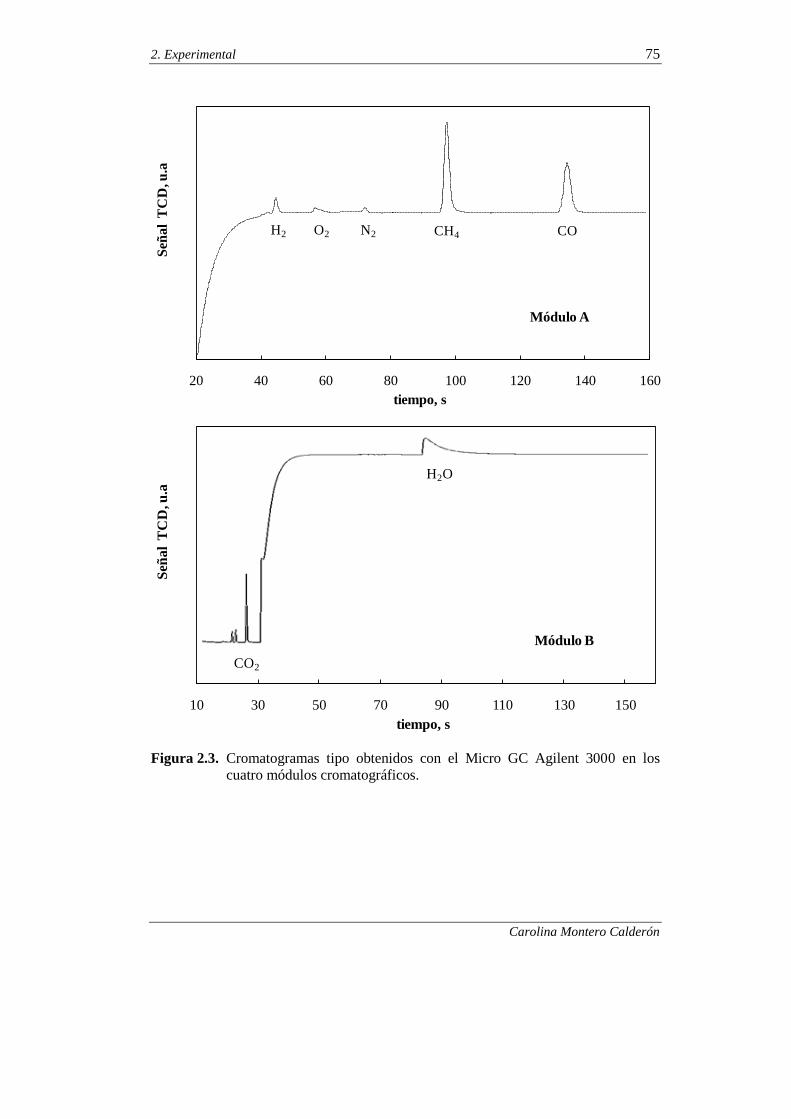
2. Experimental 75
Carolina Montero Calderón
Figura 2.3. Cromatogramas tipo obtenidos con el Micro GC Agilent 3000 en los
cuatro módulos cromatográficos.
20 40 60 80 100 120 140 160
Señ
al
TC
D, u
.a
tiempo, sSeconds
20 30 40 50 60 70 80 90 100 110 120 130 140 150 160
uV
-2500
-2000
-1500
-1000
-500
0
500
1000
1500
2000
2500
uV
-2500
-2000
-1500
-1000
-500
0
500
1000
1500
2000
2500TCD - Channel A
Módulo A
H2 O2 N2 CH4 CO
10 30 50 70 90 110 130 150
Señ
al
TC
D, u
.a
tiempo, s
Módulo B
H2O
CO2
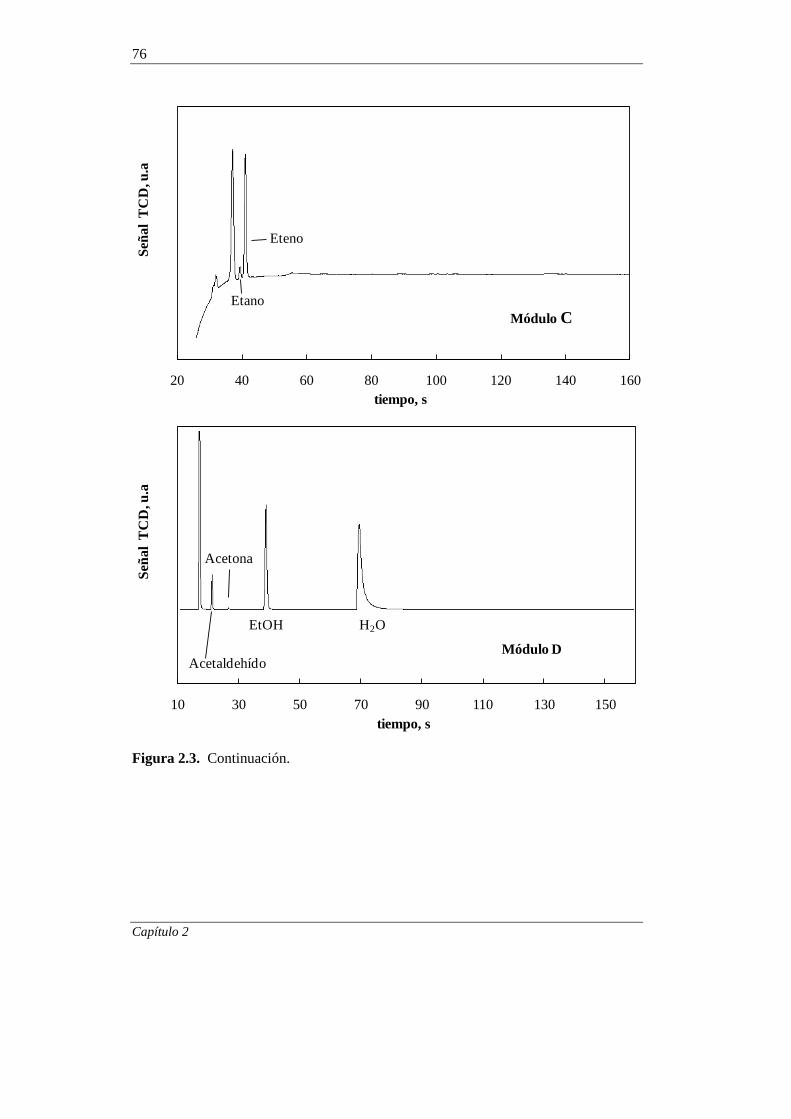
76
Capítulo 2
Figura 2.3. Continuación.
20 40 60 80 100 120 140 160
Señ
al
TC
D, u
.a
tiempo, s
Módulo CEtano
Eteno
10 30 50 70 90 110 130 150
Señ
al
TC
D, u
.a
tiempo, s
Módulo D
EtOH H2OEtOH H2O
Acetaldehído
Acetona

2. Experimental 77
Carolina Montero Calderón
2.3.3. Condiciones fluidodinámicas
Se ha llevado a cabo la experimentación en lecho fluidizado, ya que se
minimizan los gradientes de temperatura tanto en las partículas como en el reactor,
logrando mantener la isotermicidad, y minimizar la desactivación del catalizador por
deposición de coque. Sin embargo, para mantener las condiciones de correcta
fluidización (flujo ideal de pistón en el gas y de mezcla perfecta en el sólido) y para
evitar zonas muertas y formación de burbujas (slugs) es necesario establecer
limitaciones en los valores de los caudales de gases, así como la masa de sólido en el
lecho.
En trabajos previos del grupo de investigación (Remiro, 2012; Vicente, 2012) se
ha establecido que para asegurar el régimen cinético, y sin limitación de la difusión
interna, se debe trabajar con un tamaño de partícula del catalizador entre 150-250 m;
y que para garantizar la correcta fluidización se requiere la dilución del lecho con un
sólido inerte de buenas propiedades fluidodinámicas (CSi), lo que contribuye además
a evitar la utilización de una elevada cantidad de catalizador para tener una altura
suficiente de lecho catalítico. El CSi es inerte en el proceso y su utilización de tamaño
inferior al del catalizador (37 m), facilita su separación por medio de tamizado, para
el posterior estudio y reutilización de los dos sólidos. En estos trabajos previos se
determinó que para estas mezclas catalizador (Ni/La2O3-αAl2O3) - inerte (CSi), la
velocidad mínima de fluidización se encuentra en el intervalo 0.4-0.6 cm/s. En
consecuencia con esta información, para obtener la estabilidad del lecho y la
reproducibilidad de los ensayos cinéticos se ha trabajado con las condiciones
fluidodinámicas de la Tabla 2.4:
Tabla 2.4. Condiciones fluidodinámicas de los ensayos cinéticos de reformado con
vapor de etanol.
Velocidad mínima de fluidización, cm/s 0.4
Caudal de fluidización mínima, cm3/s 1.26
Velocidad lineal de gases/ velocidad mínima de fluidización ~ 6
Caudal total a la temperatura de reacción, cm3/s ~ 7.6
Altura del lecho/diámetro del lecho 2
Masa de catalizador, g 0.02-1.05

78
Capítulo 2
2.3.4. Condiciones de operación
Previamente a los ensayos cinéticos, y simultáneamente a la etapa de
calentamiento, se lleva a cabo la activación del catalizador mediante la reducción de
los centros metálicos con una corriente del 10 % en volumen de H2 diluido en He,
hasta la temperatura de reducción de 700 ºC, definida por los perfiles de TPR.
Para la realización de los ensayos cinéticos se han considerado los siguientes
intervalos de las variables de operación, descritos en la Tabla 2.5:
Tabla 2.5. Condiciones de operación en los ensayos cinéticos de reformado con
vapor de etanol.
Relación molar agua/ etanol (S/E) 3, 6, 9
Temperatura, ºC 500-700
P, atm 1.2
PEtOH, atm 0.08
Tiempo espacial, gcat.h/gEtOH 0.02-0.0.69
2.3.5. Índices de reacción
A partir de los balances de materia llevados a cabo para validar el análisis
(Anexo A), se puede determinar el caudal molar total de productos de reacción, que
junto con los valores de fracción molar determinados para cada componente y con los
caudales conocidos de reactantes a la entrada, permiten calcular los índices de
reacción:
La conversión (XEtOH), que se define como la fracción de etanol reaccionado y
se calcula a partir de los caudales molares de etanol a la entrada (FEtOH,e) y a la
salida (FEtOH, s) del reactor:
e,EtOH
s,EtOHe,EtOHEtOH
F
FFX (2.6)

2. Experimental 79
Carolina Montero Calderón
El rendimiento de cada producto (Ri), que se define como la relación entre el
caudal molar del compuesto i producido, Fi; y el caudal molar de etanol
alimentado, considerando la estequiometría de cada reacción de
transformación de etanol:
e,EtOH
1ii
iF
FR (2.7)
donde i = 6 para el H2, i = 2 para el CO2, CO y CH4, y i = 1 para el
acetaldehído, e hidrocarburos, principalmente etileno.
La selectividad (Si) se define como el caudal molar del compuesto i producido
entre el caudal molar total de productos formados:
ii
i
s,O2Hs,oxigenadoT
Tii
F
F
FFF
FxS (2.8)
donde FT, es el caudal molar total a la salida del reactor; FH2O, es el caudal
molar de agua a la salida del reactor.


3. OPTIMACIÓN DE LAS CONDICIONES
DE PREPARACIÓN DEL CATALIZADOR


83
Carolina Montero Calderón
3. OPTIMACIÓN DE LAS CONDICIONES DE PREPARACIÓN
DEL CATALIZADOR
En este apartado se aborda la selección de las condiciones de preparación del
catalizador, adecuadas para: 1) proporcionar una elevada actividad, selectividad de H2
y estabilidad en el proceso SRE, y; 2) garantizar un comportamiento reproducible del
catalizador para la operación en ciclos sucesivos de reacción-regeneración. Este
segundo requerimiento exige la utilización de un binomio (condiciones de operación +
catalizador) que garantice que la desactivación sea reversible (por deposición de
coque), evitando, por tanto, la pérdida irreversible de actividad por sinterización de la
función metálica. Los datos cinéticos obtenidos en estas condiciones reproducibles
permitirán desarrollar modelos cinéticos para la reacción y para la desactivación, que
serán de aplicabilidad general para el diseño del reactor operando con diferentes
estrategias.
En un trabajo anterior (Valle y cols., 2014b) se ha puesto de manifiesto que el
comportamiento cinético en el reformado de bio-oil del catalizador de Ni/La2O3-
αAl2O3 depende en gran medida del tipo de fases metálicas resultantes tras la
calcinación, lo que a su vez depende de la temperatura de calcinación. Por otro lado,
en otro estudio previo (Vicente, 2012) se puso de manifiesto que los catalizadores de
Ni soportados en αAl2O3 o en La2O3-αAl2O3 tienen un comportamiento reproducible
para el reformado de etanol (llevado a cabo a 500 ºC) tras ser sometidos a un
tratamiento consistente en un ciclo de reacción-regeneración (con reacción a 500 ºC y
regeneración por combustión del coque con aire a 550 ºC).
Con esta experiencia previa, en esta Tesis se ha planteado, por un lado, analizar
el efecto de la temperatura de calcinación del catalizador Ni/La2O3-αAl2O3 sobre su
actividad, estabilidad y selectividad de H2 en el proceso SRE y, por otro lado, analizar
el efecto de un tratamiento de equilibrado o estabilización del catalizador (mediante un
ciclo de reacción-regeneración) sobre su comportamiento cinético, así como sobre sus
propiedades físico-químicas, para determinar la causa de la estabilización observada
con dicho tratamiento.

84
Capítulo 3
3.1. EFECTO DE LA TEMPERATURA DE CALCINACIÓN EN EL
COMPORTAMIENTO CINÉTICO DEL CATALIZADOR
Previamente al estudio de la estabilización del catalizador, se ha realizado un
estudio para determinar la temperatura más adecuada de calcinación, en aras de
obtener el mejor compromiso entre rendimiento de H2 y estabilidad del catalizador.
Para ello, se han preparado catalizadores calcinados a tres temperaturas (TC):
550, 650 y 850 ºC. Previamente a los ensayos cinéticos los catalizadores han sido
reducidos in situ, a 700 ºC (temperatura seleccionada en base a los resultados de TPR,
mostrados en un apartado posterior) durante 2 h en una atmósfera de 10 % de H2 en
He. Tras la reducción, se ha analizado su comportamiento cinético en las siguientes
condiciones:
Temperatura de reformado: 700 ºC
Presión parcial de etanol: 0.083 bar
Relación molar vapor de agua/etanol: 6
Tiempo espacial: 0.09 gcat h/gEtOH
Tiempo de reacción: 2 h.
3.1.1. Índices de reacción a tiempo cero
El efecto de la temperatura de calcinación sobre los valores a tiempo cero de
conversión de etanol y del rendimiento de los productos de reformado con vapor (H2 y
CO2) se muestra en la Figura 3.1. Como se observa, en las condiciones estudiadas, se
obtienen altos valores de conversión de etanol a tiempo cero, la cual es completa para
el catalizador calcinado a 550 ºC, y disminuye a medida que aumenta la temperatura
de calcinación, especialmente por encima de 650 ºC. El rendimiento a tiempo cero de
H2 es también muy elevado para bajas temperaturas de calcinación, y disminuye
notablemente para la temperatura de calcinación de 850 ºC, a pesar de que la
conversión es elevada, lo que pone de manifiesto que para este catalizador las
reacciones secundarias del esquema cinético cobran mayor importancia que el
reformado con vapor.
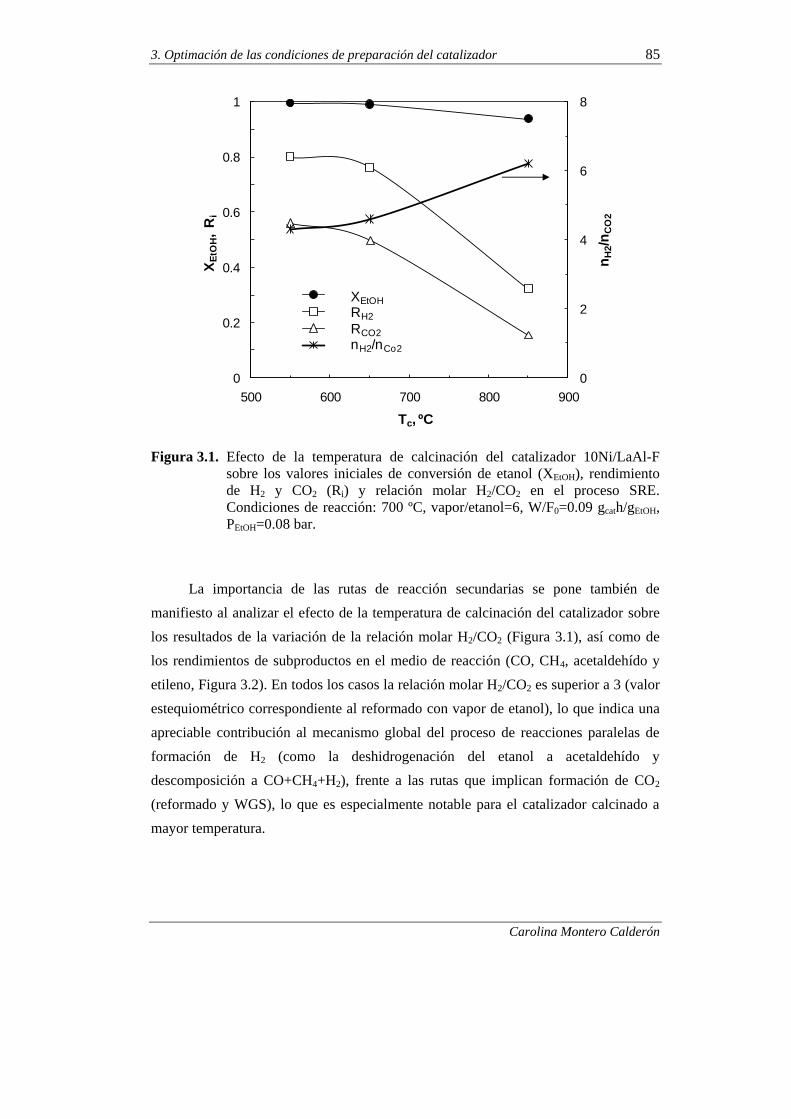
3. Optimación de las condiciones de preparación del catalizador 85
Carolina Montero Calderón
Figura 3.1. Efecto de la temperatura de calcinación del catalizador 10Ni/LaAl-F
sobre los valores iniciales de conversión de etanol (XEtOH), rendimiento
de H2 y CO2 (Ri) y relación molar H2/CO2 en el proceso SRE.
Condiciones de reacción: 700 ºC, vapor/etanol=6, W/F0=0.09 gcath/gEtOH,
PEtOH=0.08 bar.
La importancia de las rutas de reacción secundarias se pone también de
manifiesto al analizar el efecto de la temperatura de calcinación del catalizador sobre
los resultados de la variación de la relación molar H2/CO2 (Figura 3.1), así como de
los rendimientos de subproductos en el medio de reacción (CO, CH4, acetaldehído y
etileno, Figura 3.2). En todos los casos la relación molar H2/CO2 es superior a 3 (valor
estequiométrico correspondiente al reformado con vapor de etanol), lo que indica una
apreciable contribución al mecanismo global del proceso de reacciones paralelas de
formación de H2 (como la deshidrogenación del etanol a acetaldehído y
descomposición a CO+CH4+H2), frente a las rutas que implican formación de CO2
(reformado y WGS), lo que es especialmente notable para el catalizador calcinado a
mayor temperatura.
0
2
4
6
8
0
0.2
0.4
0.6
0.8
1
500 600 700 800 900
nH
2/n
CO
2
XE
tOH,
Ri
Tc, ºC
XEtOH
RH2
RCO2
nH2/nCo2
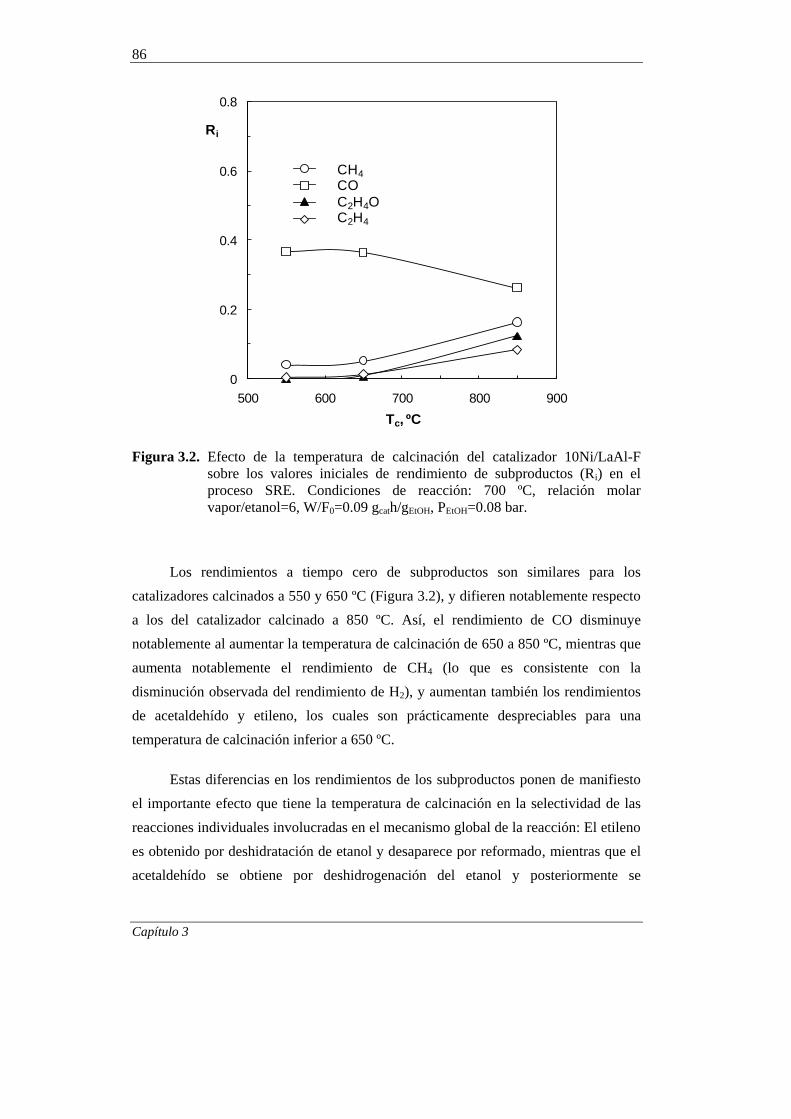
86
Capítulo 3
Figura 3.2. Efecto de la temperatura de calcinación del catalizador 10Ni/LaAl-F
sobre los valores iniciales de rendimiento de subproductos (Ri) en el
proceso SRE. Condiciones de reacción: 700 ºC, relación molar
vapor/etanol=6, W/F0=0.09 gcath/gEtOH, PEtOH=0.08 bar.
Los rendimientos a tiempo cero de subproductos son similares para los
catalizadores calcinados a 550 y 650 ºC (Figura 3.2), y difieren notablemente respecto
a los del catalizador calcinado a 850 ºC. Así, el rendimiento de CO disminuye
notablemente al aumentar la temperatura de calcinación de 650 a 850 ºC, mientras que
aumenta notablemente el rendimiento de CH4 (lo que es consistente con la
disminución observada del rendimiento de H2), y aumentan también los rendimientos
de acetaldehído y etileno, los cuales son prácticamente despreciables para una
temperatura de calcinación inferior a 650 ºC.
Estas diferencias en los rendimientos de los subproductos ponen de manifiesto
el importante efecto que tiene la temperatura de calcinación en la selectividad de las
reacciones individuales involucradas en el mecanismo global de la reacción: El etileno
es obtenido por deshidratación de etanol y desaparece por reformado, mientras que el
acetaldehído se obtiene por deshidrogenación del etanol y posteriormente se
0
0.2
0.4
0.6
0.8
500 600 700 800 900
Ri
Tc, ºC
CH4
CO
C2H4OC2H4

3. Optimación de las condiciones de preparación del catalizador 87
Carolina Montero Calderón
descompone en CO y CH4, los cuales a su vez se transforman mediante la reacción
WGS y reformado, respectivamente (no estando favorecida la ruta de metanación del
CO a la temperatura de reformado estudiada). Los altos rendimientos de CO obtenidos
con los catalizadores calcinados a baja temperatura indican que las rutas de reacción
de reformado directo y posterior formación de CO por reacción WGS reversa están
más favorecidas que otras posibles rutas. Sin embargo, los altos rendimientos de
etileno y acetaldehído obtenidos para el catalizador calcinado a 850 ºC indican que
para este catalizador cobran importancia las reacciones de deshidratación y
deshidrogenación del etanol, mientras que están menos favorecidas tanto el reformado
directo a CO2+H2 como la posterior descomposición del acetaldehído y el reformado
de etileno y de metano (lo que justifica una relación molar H2/CO2 muy superior a 3).
La diferencia en el comportamiento del catalizador calcinado a diferentes
temperaturas puede explicarse por la diferente naturaleza de las especies metálicas de
Ni resultantes de la calcinación. Según han observado Valle y cols. (2014b), al
aumentar la temperatura de calcinación del catalizador Ni/La2O3-αAl2O3 disminuye la
reducibilidad del catalizador debido a la formación de NiAl2O4, que tiene más fuerte
interacción con el soporte que los óxidos NiOx. La presencia de estructuras tipo
NiAl2O4 en el catalizador calcinado favorece la formación de cristalitas de Ni0
mayores (y menos activas) tras la reducción. Por el contrario, la calcinación por
debajo de 700 ºC evita la formación de clústeres, separando los cristales de Ni, con lo
que aumenta la dispersión metálica y la reducibilidad.
3.1.2. Desactivación
La evolución con el tiempo de los índices de reacción para los catalizadores
calcinados a diferentes temperaturas se muestra en la Figura 3.3 (conversión de etanol
y rendimiento de H2 (gráfica a) y relación molar H2/CO2 (gráfica b)) y en la Figura 3.4.
(rendimiento de subproductos). La conversión (Figura 3.3a) se mantiene casi constante
en 2 h de reacción para el catalizador calcinado a la menor temperatura, mientras que
disminuye apreciablemente con el tiempo para los catalizadores calcinados a 650 y
850 ºC, lo que indica una mayor velocidad de desactivación al aumentar la
temperatura de calcinación del catalizador.
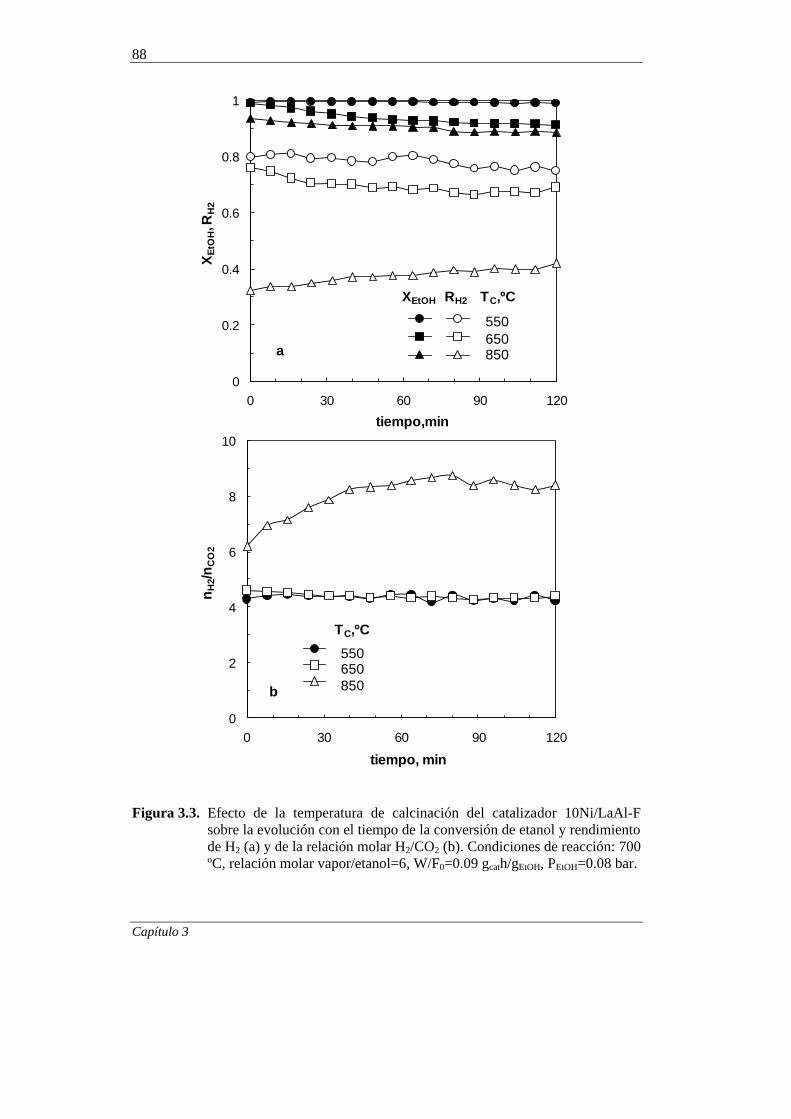
88
Capítulo 3
Figura 3.3. Efecto de la temperatura de calcinación del catalizador 10Ni/LaAl-F
sobre la evolución con el tiempo de la conversión de etanol y rendimiento
de H2 (a) y de la relación molar H2/CO2 (b). Condiciones de reacción: 700
ºC, relación molar vapor/etanol=6, W/F0=0.09 gcath/gEtOH, PEtOH=0.08 bar.
0
0.2
0.4
0.6
0.8
1
0 30 60 90 120
XE
tOH, R
H2
tiempo,min
XEtOH RH2 TC,ºC
550
650850a
0
2
4
6
8
10
0 30 60 90 120
nH
2/n
CO
2
tiempo, min
TC,ºC
550650
850b
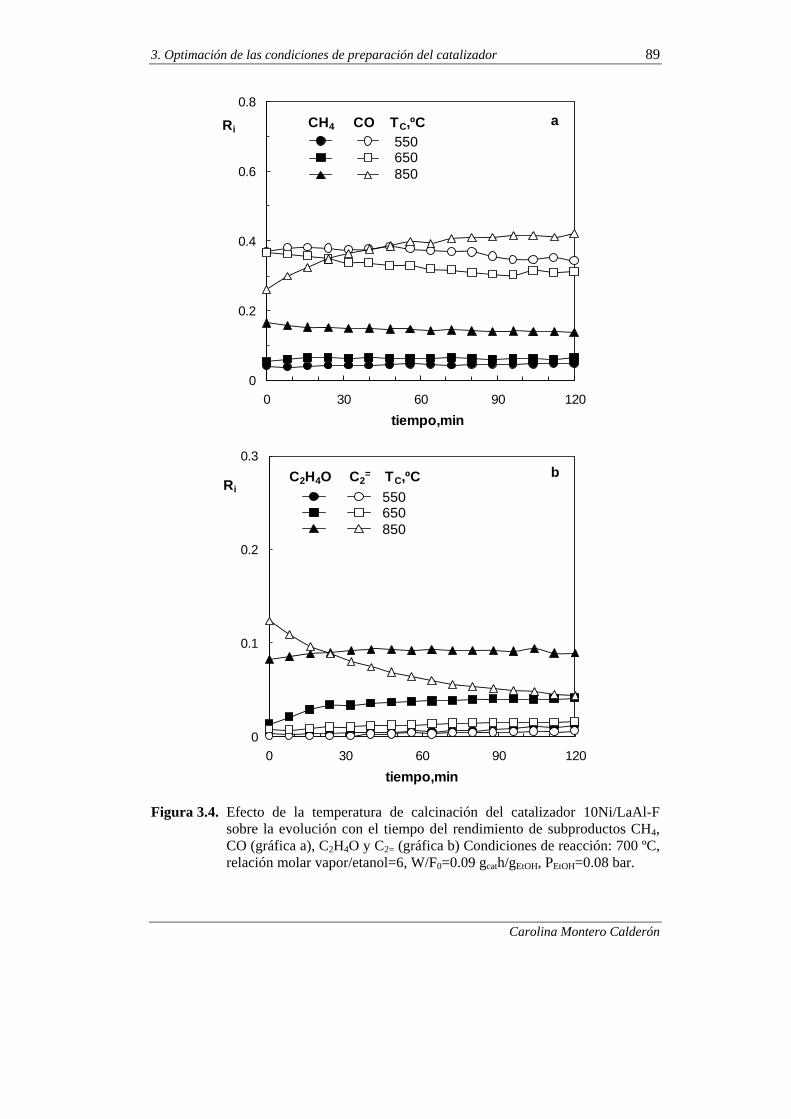
3. Optimación de las condiciones de preparación del catalizador 89
Carolina Montero Calderón
Figura 3.4. Efecto de la temperatura de calcinación del catalizador 10Ni/LaAl-F
sobre la evolución con el tiempo del rendimiento de subproductos CH4,
CO (gráfica a), C2H4O y C2= (gráfica b) Condiciones de reacción: 700 ºC,
relación molar vapor/etanol=6, W/F0=0.09 gcath/gEtOH, PEtOH=0.08 bar.
0
0.2
0.4
0.6
0.8
0 30 60 90 120
Ri
tiempo,min
CH4 CO TC,ºC
550 650
850
a
0
0.1
0.2
0.3
0 30 60 90 120
Ri
tiempo,min
C2H4O C2= TC,ºC
550 650
850
b

90
Capítulo 3
En el periodo de tiempo estudiado, los rendimientos de los productos de
reacción varían muy poco para los catalizadores calcinados a 550 y 650 ºC,
observándose tan solo una ligera disminución en el rendimiento de H2 y de CO,
mientras que los de acetaldehído y etileno aumentan ligeramente y el de metano
permanece casi constante. Sin embargo, para el catalizador calcinado a 850 ºC los
rendimientos de los productos de reacción varían notablemente, especialmente los de
H2, CO y etileno, lo que pone de manifiesto la notablemente más rápida desactivación
de este catalizador. La rápida disminución del rendimiento de etileno con el tiempo
indica que la ruta de deshidratación de etanol está más afectada por la desactivación
que otras rutas del mecanismo, como la deshidrogenación, el propio reformado (tanto
de etanol, como de metano o etileno) o la reacción WGS, lo que es coherente con el
aumento observado con el tiempo de reacción de los rendimientos de H2 y de CO y de
la relación molar H2/CO2.
Para correlacionar la disminución de los índices de reacción con la
desactivación por deposición de coque, se ha cuantificado el contenido de coque
mediante oxidación a temperatura programada (TPO), trabajando en las condiciones
explicadas en el Apartado 2.2.5.1. Los resultados se muestran en la Tabla 3.1.
Tabla 3.1. Contenido de coque (% en peso) depositado sobre el catalizador
10Ni/LaAl-F calcinado a diferentes temperaturas. Condiciones de
reacción: Igual a Figura 3.3
TC,ºC Coque, % peso
550 0.22
650 0.28
850 3.73
Como se observa en la Tabla 3.1, el contenido de coque es muy similar para los
catalizadores calcinados a 550 y 650 ºC, pero aumenta notablemente para el
catalizador calcinado a 850 ºC. Este elevado contenido de coque, que es coherente con
la más rápida desactivación observada para este catalizador, es probablemente
consecuencia de la presencia de partículas de Ni0 más grandes (Valle y cols., 2014b),
las cuales promueven la formación y crecimiento de depósitos carbonosos (Bengaard

3. Optimación de las condiciones de preparación del catalizador 91
Carolina Montero Calderón
y cols., 2002). El mayor contenido de coque para el catalizador calcinado a 850 ºC es
también coherente con la notable formación de etileno como subproducto, dado que el
etileno es considerado un importante precursor de la formación de coque, mediante
reacciones de deshidrogenación, aromatización y condensación (Bartholomew, 2001;
Verykios, 2003). Es igualmente coherente con la mayor concentración en el medio de
reacción, para este catalizador, de otros posibles precursores de coque, como metano
(por descomposición) y acetaldehído (Vicente y cols., 2014a).
Como conclusión de los resultados mostrados en este apartado, puede
establecerse que la temperatura más adecuada de calcinación es 550 ºC, dado que
proporciona el mayor rendimiento de H2 y la menor deposición de coque. Esto es
consecuencia de que la calcinación a 550 ºC minimiza la formación de espinela
NiAl2O4 y produce mayor cantidad de óxidos de Ni dispersos, que se reducen para
formar nanopartículas de Ni0 altamente dispersas, lo que da lugar a un catalizador más
activo y más estable.

92
Capítulo 3
3.2. EQUILIBRADO DEL CATALIZADOR
En una Tesis anterior (Vicente, 2012) se comprobó que en la operación en
sucesivos ciclos de reacción-regeneración en el proceso SRE con catalizador de
Ni/La2O3-Al2O3, se producía una variación de los índices de reacción (conversión de
etanol y rendimientos de productos) entre el 1er y el 2º ciclo de reacción, pero se
alcanzaba un comportamiento reproducible del catalizador a partir de la segunda etapa
de reacción. Por ello, de cara a la obtención de datos cinéticos para abordar
posteriormente el modelado cinético del proceso operando en condiciones
reproducibles, en esta Tesis se ha sometido al catalizador sintetizado a un proceso de
equilibrado, que ha consistido en un ciclo previo de reacción-regeneración, bajo las
siguientes condiciones de operación:
Reacción: Reformado con vapor con relación molar vapor/etanol=3, tiempo
espacial=0.34 gcat h/gEtOH, escalones sucesivos de temperatura a 700-600-500 ºC
de 2 h de duración cada uno.
Regeneración: Combustión con aire (150 ml/min) a 550 ºC durante 2 h, para
garantizar la eliminación total del coque.
Al catalizador obtenido tras el proceso de equilibrado se le ha denominado
10Ni/LaAl-E, y es el que se ha utilizado para el posterior estudio paramétrico del
efecto de las variables de operación y para el modelado cinético.
En este apartado se presentan de manera conjunta los resultados de
comportamiento cinético en la operación cíclica del catalizador sin equilibrar
(10Ni/LaAl-F) y equilibrado (10Ni/LaAl-E), para verificar la eficacia del tratamiento
de equilibrado sobre la reproducibilidad del comportamiento cinético. Se incluyen
además resultados de la caracterización de los dos catalizadores, para explicar el
comportamiento cinético observado.
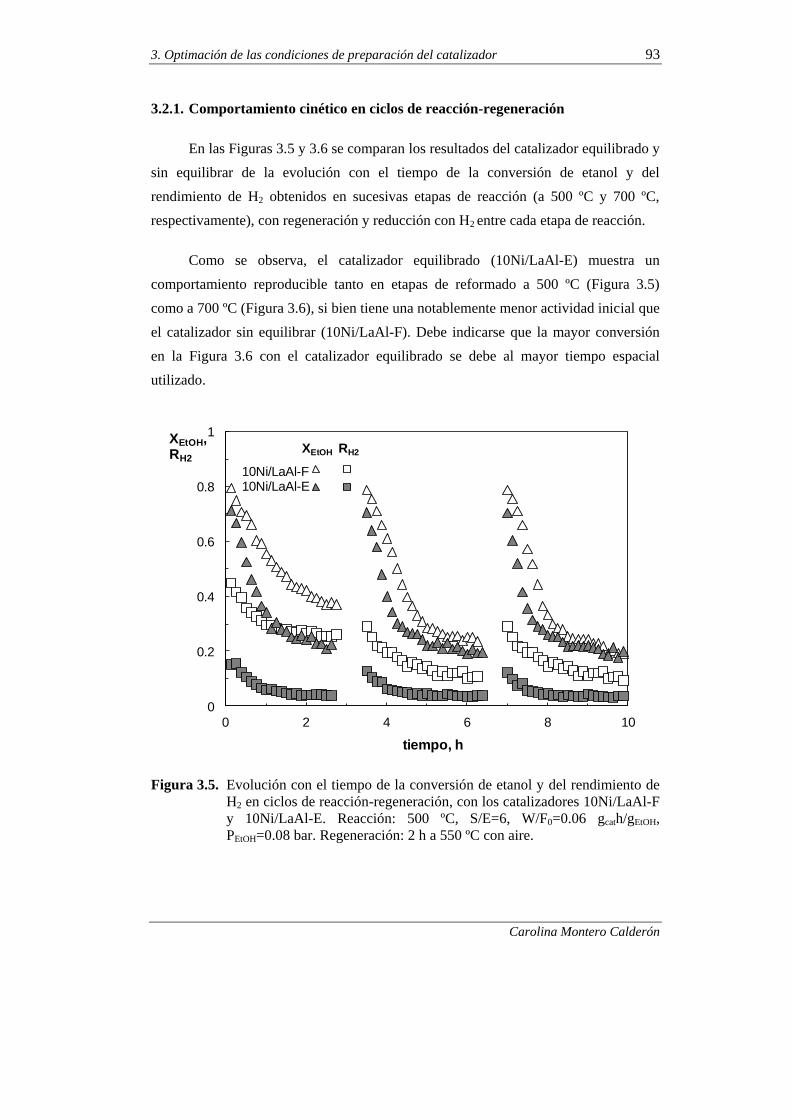
3. Optimación de las condiciones de preparación del catalizador 93
Carolina Montero Calderón
3.2.1. Comportamiento cinético en ciclos de reacción-regeneración
En las Figuras 3.5 y 3.6 se comparan los resultados del catalizador equilibrado y
sin equilibrar de la evolución con el tiempo de la conversión de etanol y del
rendimiento de H2 obtenidos en sucesivas etapas de reacción (a 500 ºC y 700 ºC,
respectivamente), con regeneración y reducción con H2 entre cada etapa de reacción.
Como se observa, el catalizador equilibrado (10Ni/LaAl-E) muestra un
comportamiento reproducible tanto en etapas de reformado a 500 ºC (Figura 3.5)
como a 700 ºC (Figura 3.6), si bien tiene una notablemente menor actividad inicial que
el catalizador sin equilibrar (10Ni/LaAl-F). Debe indicarse que la mayor conversión
en la Figura 3.6 con el catalizador equilibrado se debe al mayor tiempo espacial
utilizado.
Figura 3.5. Evolución con el tiempo de la conversión de etanol y del rendimiento de
H2 en ciclos de reacción-regeneración, con los catalizadores 10Ni/LaAl-F
y 10Ni/LaAl-E. Reacción: 500 ºC, S/E=6, W/F0=0.06 gcath/gEtOH,
PEtOH=0.08 bar. Regeneración: 2 h a 550 ºC con aire.
0
0.2
0.4
0.6
0.8
1
0 2 4 6 8 10
XEtOH,RH2
tiempo, h
XEtOH RH2
10Ni/LaAl-F10Ni/LaAl-E
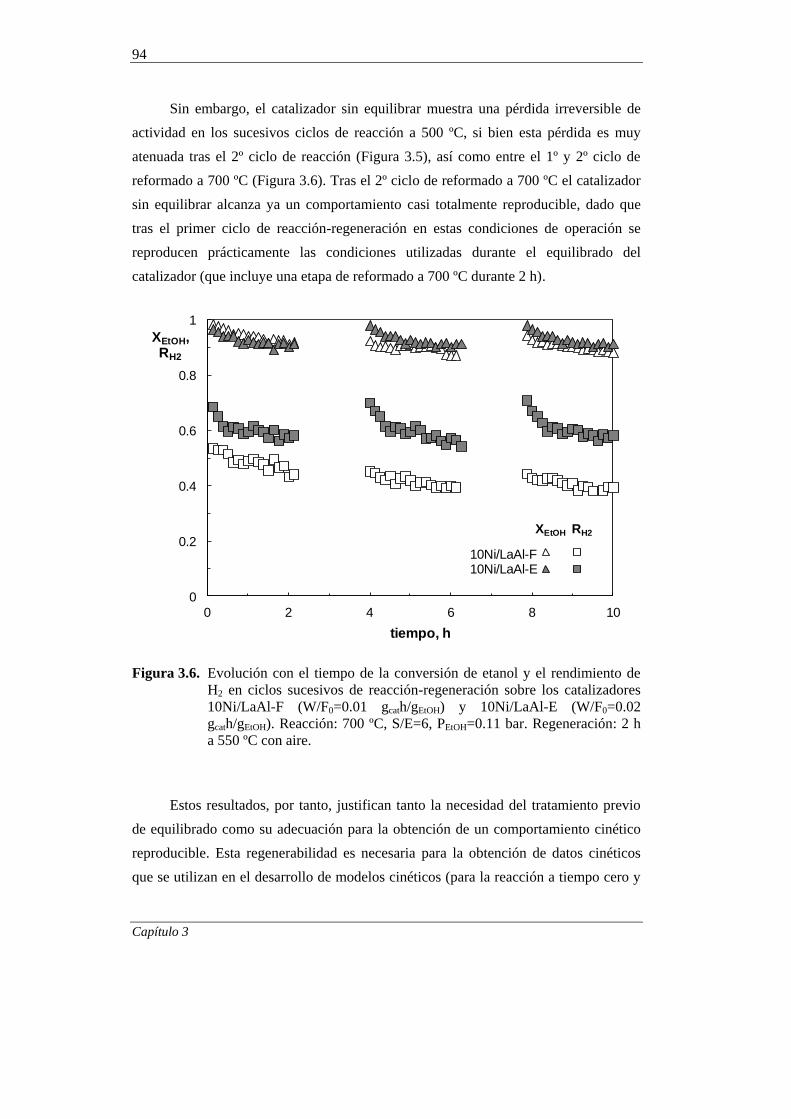
94
Capítulo 3
Sin embargo, el catalizador sin equilibrar muestra una pérdida irreversible de
actividad en los sucesivos ciclos de reacción a 500 ºC, si bien esta pérdida es muy
atenuada tras el 2º ciclo de reacción (Figura 3.5), así como entre el 1º y 2º ciclo de
reformado a 700 ºC (Figura 3.6). Tras el 2º ciclo de reformado a 700 ºC el catalizador
sin equilibrar alcanza ya un comportamiento casi totalmente reproducible, dado que
tras el primer ciclo de reacción-regeneración en estas condiciones de operación se
reproducen prácticamente las condiciones utilizadas durante el equilibrado del
catalizador (que incluye una etapa de reformado a 700 ºC durante 2 h).
Figura 3.6. Evolución con el tiempo de la conversión de etanol y el rendimiento de
H2 en ciclos sucesivos de reacción-regeneración sobre los catalizadores
10Ni/LaAl-F (W/F0=0.01 gcath/gEtOH) y 10Ni/LaAl-E (W/F0=0.02
gcath/gEtOH). Reacción: 700 ºC, S/E=6, PEtOH=0.11 bar. Regeneración: 2 h
a 550 ºC con aire.
Estos resultados, por tanto, justifican tanto la necesidad del tratamiento previo
de equilibrado como su adecuación para la obtención de un comportamiento cinético
reproducible. Esta regenerabilidad es necesaria para la obtención de datos cinéticos
que se utilizan en el desarrollo de modelos cinéticos (para la reacción a tiempo cero y
0
0.2
0.4
0.6
0.8
1
0 2 4 6 8 10
XEtOH,RH2
tiempo, h
XEtOH RH2
10Ni/LaAl-F10Ni/LaAl-E
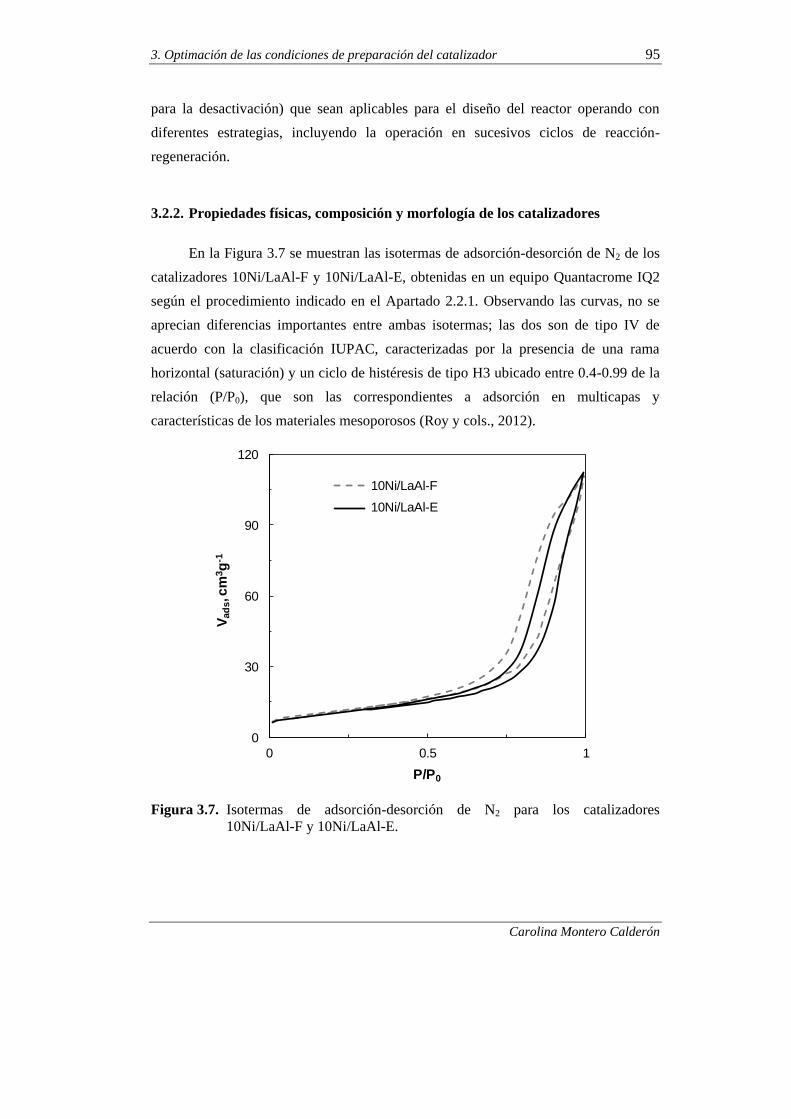
3. Optimación de las condiciones de preparación del catalizador 95
Carolina Montero Calderón
para la desactivación) que sean aplicables para el diseño del reactor operando con
diferentes estrategias, incluyendo la operación en sucesivos ciclos de reacción-
regeneración.
3.2.2. Propiedades físicas, composición y morfología de los catalizadores
En la Figura 3.7 se muestran las isotermas de adsorción-desorción de N2 de los
catalizadores 10Ni/LaAl-F y 10Ni/LaAl-E, obtenidas en un equipo Quantacrome IQ2
según el procedimiento indicado en el Apartado 2.2.1. Observando las curvas, no se
aprecian diferencias importantes entre ambas isotermas; las dos son de tipo IV de
acuerdo con la clasificación IUPAC, caracterizadas por la presencia de una rama
horizontal (saturación) y un ciclo de histéresis de tipo H3 ubicado entre 0.4-0.99 de la
relación (P/P0), que son las correspondientes a adsorción en multicapas y
características de los materiales mesoporosos (Roy y cols., 2012).
Figura 3.7. Isotermas de adsorción-desorción de N2 para los catalizadores
10Ni/LaAl-F y 10Ni/LaAl-E.
0
30
60
90
120
0 0.5 1
Vad
s, c
m3g
-1
P/P0
10Ni/LaAl-F
10Ni/LaAl-E
96
Capítulo 3
En la Tabla 3.2 se han relacionado los resultados de superficie específica (SBET),
radio medio de poros (rporo) y volumen total de mesoporos (Vporos), así como los
contenidos metálicos (expresados en porcentaje del elemento en la muestra)
determinados mediante ICP-AES, para el catalizador no equilibrado y equilibrado. Los
resultados no revelan cambios físicos significativos entre los dos catalizadores; la
etapa de equilibrado previo realizado al catalizador provoca una ligera disminución de
la superficie BET, pero sin disminución apreciable en el volumen de poros del
catalizador, probablemente asociado a la ligera disminución de contenido de La en el
soporte.
Tabla 3.2. Propiedades físicas y contenido metálico de los catalizadores 10Ni/LaAl-
F y 10Ni/LaAl-E.
Catalizador SBET, m
2/g
Vporos, cm
3/g
rporo,
Å
Ni, %
La, %
10Ni/LaAl-F 38 0.18 47 8.73 ± 0.18 6.94 ± 0.14
10Ni/LaAl-E 35 0.18 62 8.82 ± 0.18 6.79 ± 0.14
El análisis elemental por ICP indica diferencias entre la carga nominal y la
carga metálica real (10 % y 9 % en peso nominal, respectivamente, de Ni y La en el
catalizador), posiblemente debidas a la dificultad para depositar átomos de gran peso
atómico (como el La) sobre soportes de baja área superficial específica como la α-
Al2O3, y a las dificultades propias del método de cuantificación, por tener que
disgregar la muestra (lo que puede conllevar la posible precipitación del La). El
tratamiento de equilibrado previo no provoca cambios significativos en la cantidad de
Ni y La presentes en el catalizador.
Para apreciar las posibles variaciones en la morfología del catalizador con la
etapa de equilibrado se ha realizado el análisis de ambos catalizadores mediante
microscopia electrónica de barrido SEM. En la Figura 3.8 se muestran las imágenes
SEM tomadas con un aumento de 250 veces el tamaño real. En las fotografías se
aprecia que las partículas de los dos catalizadores presentan forma esférica, con una
ligera deformación en el catalizador equilibrado.

3. Optimación de las condiciones de preparación del catalizador 97
Carolina Montero Calderón
Figura 3.8. Imágenes SEM de los catalizadores 10Ni/LaAl-F (a) y 10Ni/LaAl-E (b).
a
b
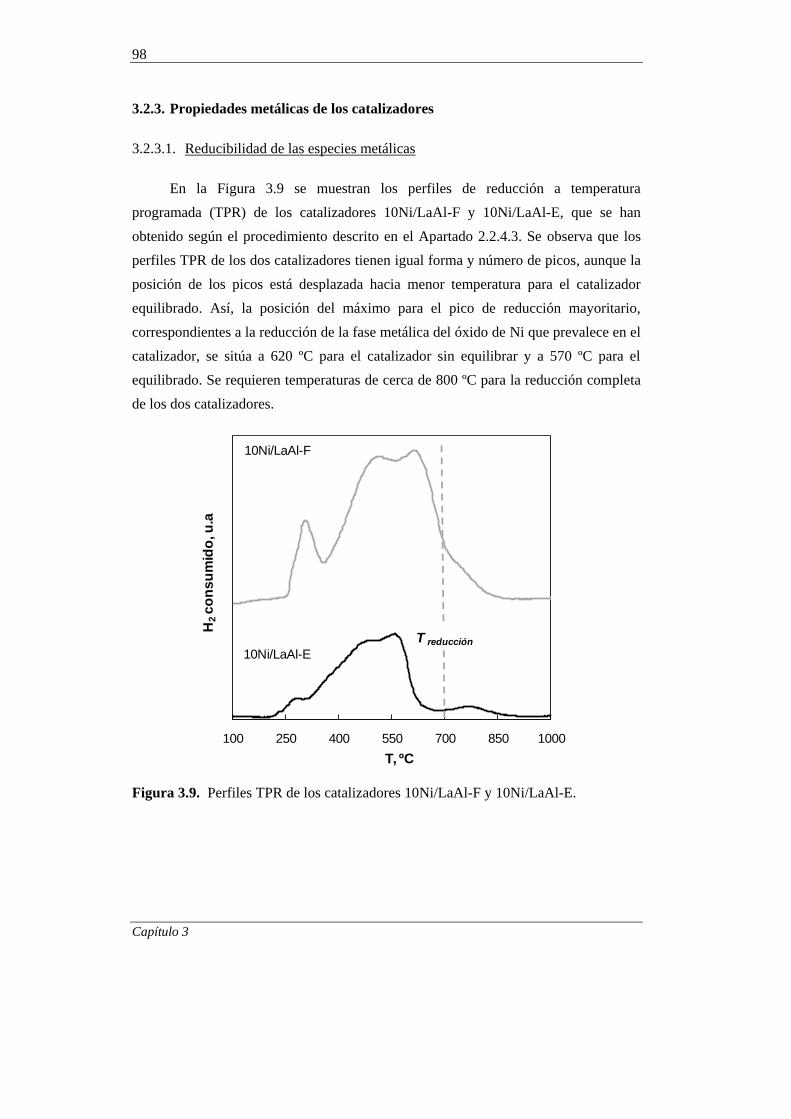
98
Capítulo 3
3.2.3. Propiedades metálicas de los catalizadores
3.2.3.1. Reducibilidad de las especies metálicas
En la Figura 3.9 se muestran los perfiles de reducción a temperatura
programada (TPR) de los catalizadores 10Ni/LaAl-F y 10Ni/LaAl-E, que se han
obtenido según el procedimiento descrito en el Apartado 2.2.4.3. Se observa que los
perfiles TPR de los dos catalizadores tienen igual forma y número de picos, aunque la
posición de los picos está desplazada hacia menor temperatura para el catalizador
equilibrado. Así, la posición del máximo para el pico de reducción mayoritario,
correspondientes a la reducción de la fase metálica del óxido de Ni que prevalece en el
catalizador, se sitúa a 620 ºC para el catalizador sin equilibrar y a 570 ºC para el
equilibrado. Se requieren temperaturas de cerca de 800 ºC para la reducción completa
de los dos catalizadores.
Figura 3.9. Perfiles TPR de los catalizadores 10Ni/LaAl-F y 10Ni/LaAl-E.
100 250 400 550 700 850 1000
H2
co
ns
um
ido
, u
.a
T, ºC
10Ni/LaAl-F
10Ni/LaAl-E
T reducción

3. Optimación de las condiciones de preparación del catalizador 99
Carolina Montero Calderón
El perfil de TPR sugiere que más de una especie metálica contribuye a la
reducción global, observándose para ambos catalizadores tres fases que se reducen en
intervalos específicos de temperaturas (Valle y cols., 2014b):
Intervalo I, para temperaturas menores de 380 ºC: Este pico correspondería a la
reducción del NiO másico (clúster, de acuerdo con Natesakhawat y cols.
(2005a), si bien ellos lo señalan a 400 ºC) o con baja interacción con el soporte.
Li y Chen (1995), en su estudio de TPR de NiO, sin soporte, señalan que el pico
a baja temperatura corresponde a NiO másico, de modo que el pico observado
correspondería a partículas grandes de NiO, que se encuentran dispersas en el
soporte pero no se forman enlaces suficientemente fuertes con la αAl2O3. Este
pico es más pronunciado para el catalizador sin equilibrar, siendo la mayor
diferencia en los perfiles TPR de ambos catalizadores.
Intervalo II, entre 400-700 ºC. Estos picos son atribuidos a la reducción de las
especies NiOx, que estarían mejor dispersas e interactúan con el soporte (Valle y
cols., 2014b). Natesakhawat y cols. (2005a) señalan que este tipo de NiOx es
posiblemente amorfo. El desplazamiento del máximo hacia menor temperatura
sería indicativo de que existe mayor reducibilidad de las especies de Ni, como
apuntan Natesakhawat y cols. (2005) al comparar un catalizador de Ni
soportado en La2O3-Al2O3 respecto al catalizador soportado en Al2O3.
Intervalo III, para temperaturas mayores a 700 ºC. Corresponde a la fase
espinela de NiAl2O4, posiblemente formada por la migración de los átomos de
Ni sobre la Al2O3. En el catalizador equilibrado se identifica este pico, mientras
que en el catalizador sin equilibrar se observa como un hombro. La presencia en
los dos catalizadores de este pico, ya sea como hombro o como pico
diferenciado, indica la formación de la fase espinela, de manera más
pronunciada en el catalizador equilibrado (Li y Chen, 1995).
Los resultados de TPR muestran, por tanto, que el catalizador equilibrado tiene
mayor reducibilidad que el catalizador sin equilibrar.

100
Capítulo 3
3.2.3.2. Superficie metálica, dispersión y tamaño de cristal
Para determinar la dispersión metálica en los dos catalizadores se ha empleado
la quimisorción de H2 en un equipo Quantacrome IQ2. Se ha realizado un primer
análisis considerando la adsorción reversible e irreversible de manera combinada
(fisisorción+quimisorción) y un segundo análisis solo de adsorción reversible
(fisisorción), lo que permite por diferencia cuantificar la quimisorción. La dispersión
del Ni se ha calculado asumiendo que un átomo de hidrógeno es adsorbido en cada
átomo superficial de Ni (Natesakhswat y cols., 2005a).
En la Figura 3.10 se han representado las isotermas de adsorción asociadas a la
quimisorción de H2 para los catalizadores sin equilibrar y equilibrado. Se observa que
el catalizador sin equilibrar adsorbe mayor cantidad de H2 que el equilibrado, lo que es
indicativo de su mayor superficie metálica, tal como muestran los resultados
cuantificados en la Tabla 3.3.
Tabla 3.3. Propiedades del Ni en los catalizadores 10Ni/LaAl-F y 10NiLaAl-E.
Catalizador AM,
m2/gcat
Dispersión, %
dMm
, Å
10Ni/LaAl-F 3.41 5.86 172.54
10Ni/LaAl-E 1.99 3.39 298.62
Los bajos valores de dispersión y superficie metálica son similares a los
obtenidos por Natesakhawat y cols. (2005a) para el catalizador 20Ni-5La/Al2O3 (3.3
% y 4.5 m2/g, respectivamente). Estos autores señalan que la baja dispersión puede
deberse a que para altas cantidades, el La puede bloquear los centros de Ni.
Comparando los resultados para los catalizadores no equilibrado y equilibrado
se observa que la superficie metálica disminuye con el tratamiento de equilibrado y,
por ende, también disminuye la dispersión de Ni en el soporte.
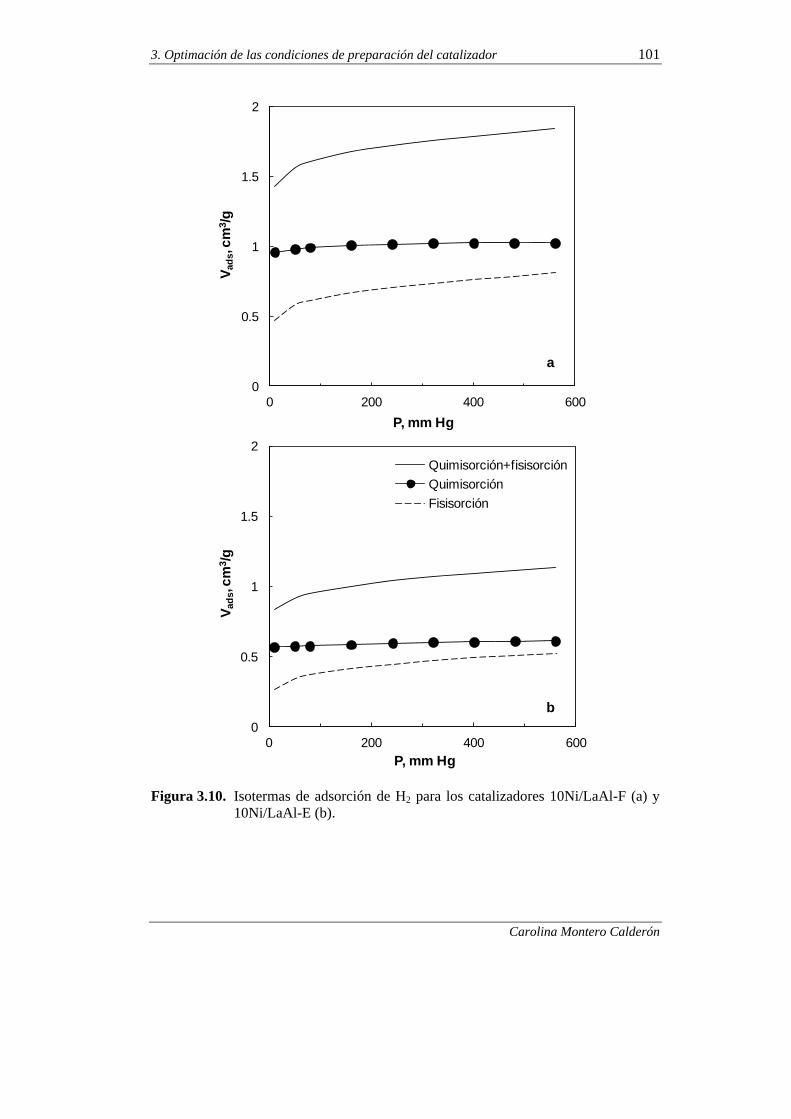
3. Optimación de las condiciones de preparación del catalizador 101
Carolina Montero Calderón
Figura 3.10. Isotermas de adsorción de H2 para los catalizadores 10Ni/LaAl-F (a) y
10Ni/LaAl-E (b).
0
0.5
1
1.5
2
0 200 400 600
Vad
s, c
m3/g
P, mm Hg
a
0
0.5
1
1.5
2
0 200 400 600
Vad
s, c
m3/g
P, mm Hg
Quimisorción+fisisorción
Quimisorción
Fisisorción
b
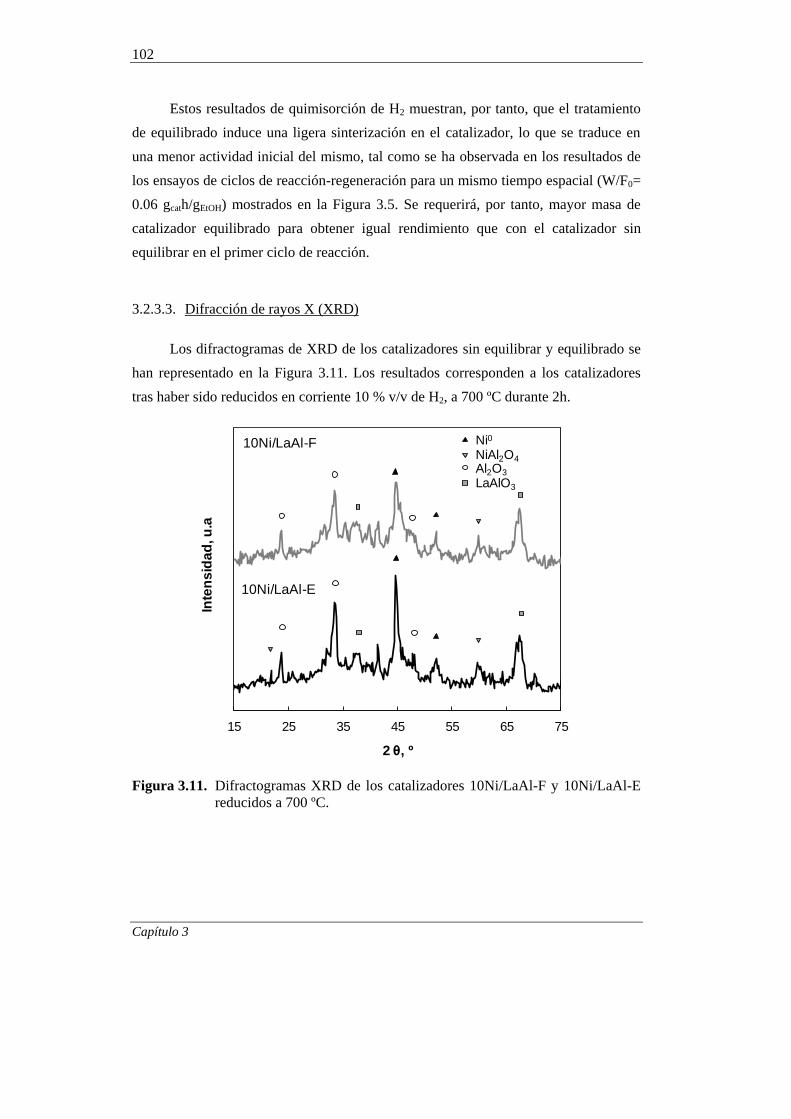
102
Capítulo 3
Estos resultados de quimisorción de H2 muestran, por tanto, que el tratamiento
de equilibrado induce una ligera sinterización en el catalizador, lo que se traduce en
una menor actividad inicial del mismo, tal como se ha observada en los resultados de
los ensayos de ciclos de reacción-regeneración para un mismo tiempo espacial (W/F0=
0.06 gcath/gEtOH) mostrados en la Figura 3.5. Se requerirá, por tanto, mayor masa de
catalizador equilibrado para obtener igual rendimiento que con el catalizador sin
equilibrar en el primer ciclo de reacción.
3.2.3.3. Difracción de rayos X (XRD)
Los difractogramas de XRD de los catalizadores sin equilibrar y equilibrado se
han representado en la Figura 3.11. Los resultados corresponden a los catalizadores
tras haber sido reducidos en corriente 10 % v/v de H2, a 700 ºC durante 2h.
Figura 3.11. Difractogramas XRD de los catalizadores 10Ni/LaAl-F y 10Ni/LaAl-E
reducidos a 700 ºC.
15 25 35 45 55 65 75
Inte
ns
ida
d, u
.a
2 θ, º
10Ni/LaAl-F
10Ni/LaAl-E
Ni0
NiAl2O4
Al2O3
LaAlO3

3. Optimación de las condiciones de preparación del catalizador 103
Carolina Montero Calderón
La presencia de picos anchos y deformes indica que tanto el soporte como el Ni
tienen baja cristalinidad en los dos catalizadores. En ambos difractogramas se
observan los picos correspondientes al soporte: Al2O3 (2θ=23.7, 33.7 y 41.8º) y
LaAlO3, (2θ=37.5 y 67.5º), este último asociado a la combinación de La2O3 y α-Al2O3,
que también ha sido observado por Barrera y cols. (2007) y Valle y cols. (2014b).
Además, para los dos catalizadores se observa la presencia de NiAl2O4 que no
se ha reducido completamente, que aparece en los dos catalizadores en el ángulo de
difracción 2θ=60º, y para el catalizador equilibrado se observa además esta especie en
posición 2θ=21.7º. Este resultado es coherente con los perfiles mostrados TPR (Figura
3.9), que mostraban la necesidad de utilizar temperaturas muy altas (por encima de
800 ºC) para reducir completamente las especies de Ni de tipo espinela NiAl2O4.
Los picos correspondientes al Ni0 (derivados de la reducción del NiO) se
identifican claramente en 2θ = 44.8 y 52º (Natesakhawat y cols., 2005a,b; Nichele y
cols., 2014; Valle y cols., 2014b). Con este último pico se calcula el valor del tamaño
de cristal de Ni0 en el plano (2 0 0) mediante la ecuación de Scherrer, Ec. (2.10), valor
que se recoge en la Tabla 3.4 para el catalizador sin equilibrar y equilibrado. Como se
observa, el tamaño de cristal aumenta tras el tratamiento de equilibrado, lo que indica
que este tratamiento origina cierta sinterización del Ni, resultado que es coherente con
los resultados de quimisorción de H2. Ambas técnicas revelan que el tamaño ha
aumentado ~ 1.7 veces.
Tabla 3.4. Tamaño de cristal de Ni0, medido en el plano Ni (2 0 0) para 2 = 52º.
Catalizador dMO (Ni0), Å
10Ni/LaAl-F 66
10Ni/LaAl-E 106
3.2.3.4. Espectroscopía Fotoelectrónica de Rayos X (XPS)
El análisis de XPS se ha realizado para determinar la composición química de la
superficie de los catalizadores una vez reducidos (2 h a 700 º C en 10% H2/Ar). Esta
técnica permite cuantificar las energías de enlace características de cada elemento en

104
Capítulo 3
la superficie, y por medio del análisis de las áreas de cada uno de los picos establecer
una composición centesimal para los dos catalizadores.
Las muestras han sido inicialmente analizadas con un espectro de
reconocimiento de baja resolución (survey scan) que permite la identificación de los
elementos en la superficie. Posteriormente, en cada una de las zonas donde se
identifican los elementos se realizan espectros locales de alta resolución (detail scan)
que permite la cuantificación en función de las áreas de los picos de cada elemento
identificado.
Durante el survey scan del catalizador 10Ni/LaAl-F se identificaron únicamente
los elementos constituyentes del catalizador: Ni, La, Al, O. Sin embargo, en el
catalizador 10Ni/LaAl-E se identificó C y Si, en lugar de Ni. La presencia de estos
elementos recubriendo la superficie de la muestra puede atribuirse a que durante el
tratamiento de equilibrado del catalizador se utiliza CSi como sólido inerte para
mantener las condiciones fluidodinámicas adecuadas, como se explica en el Apartado
2.3.3. Por este motivo, para determinar la composición superficial del catalizador
10Ni/LaAl-E se ha realizado una análisis de perfil destructivo (depth profile), ensayo
que consiste en inducir la colisión de iones de un gas noble (Ar), que son acelerados
por la diferencia en el potencial eléctrico existente, limpiando así la superficie, lo que
permite que los átomos de Ni de la superficie reaparezcan y pueden ser entonces
analizador por detail scan (Matienzo, 1996).
En la Tabla 3.5 se presentan los resultados del análisis elemental de la
superficie por medio de XPS, del catalizador sin equilibrar (detail scan) y de
catalizador equilibrado (depth profile+detail scan). Los resultados revelan que en la
muestra equilibrada el contenido de Ni es menor y el de Al mayor, lo cual estaría en
concordancia con la disminución de la dispersión cuantificada por quimisorción
(Tabla 3.3), mientras que el crecimiento del cristal de Ni, evidenciado por XRD (Tabla
3.4), puede ser la causa de la ligera disminución en el contenido de La medido en la
superficie, al poder estar éste cubierto por las partículas más grandes de Ni.
3. Optimación de las condiciones de preparación del catalizador 105
Carolina Montero Calderón
Tabla 3.5. Composición atómica en la superficie de los catalizadores sin equilibrar
(10Ni/LaAl-F) y equilibrado (10Ni/LaAl-E).
10Ni/LaAl-F 10Ni/LaAl-E
Elemento Orbital Energía de
enlace, eV
Concentración,
%
Energía de
enlace, eV Concentración,%
Ni Ni2p
854.8 9.0
19.0
852.0 2.7
6.5
861.7 3.6 867.5 1.7
872.0 4.6 856.3 1.1
879.7 1.8
871.0 0.5
858.2 0.3
874.2 0.2
La La3d 5/2
834.5 2.3
6.1
834.7 1.3
3.4 838.0 1.3 837.9 0.7
851.2 1.6 850.4 0.9
853.9 0.9 853.2 0.5
Al Al2p 73.8 18.4 18.4 73.4 33.3 33.3
O O1s 531.0 36.8
56.5 531.7 24.5
56.8 529.1 19.7 529.6 32.3
Se ha realizado el análisis de los picos del espectro de Ni 2p, que se muestra en la
Figura 3.12, para identificar la naturaleza de las especies de Ni en la superficie de los
catalizadores reducidos. Previamente a la deconvolución de los picos se ha corregido
el efecto de carga en los espectros en base al pico 1s del carbono en posición 284.6
eV.
En los dos catalizadores se observa las señales características atribuidas al Ni0 y
a Ni2+, atribuible este último posiblemente a la presencia de NiAl2O4 o remanentes del
NiOx que no ha sido reducido.
El catalizador 10Ni/LaAl-E presenta mayor número e intensidad de espectros
asignados al Ni0 (852.0, 867.5, 858.2, 874.2 eV) comparado con el catalizador
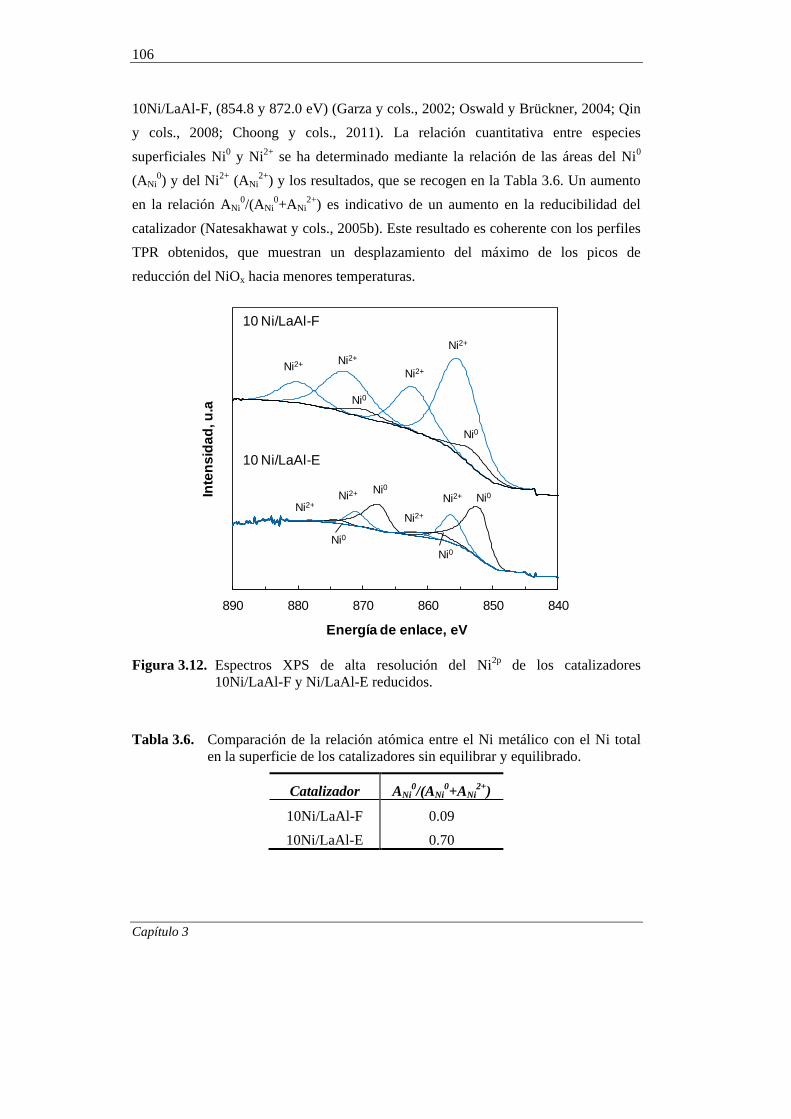
106
Capítulo 3
10Ni/LaAl-F, (854.8 y 872.0 eV) (Garza y cols., 2002; Oswald y Brückner, 2004; Qin
y cols., 2008; Choong y cols., 2011). La relación cuantitativa entre especies
superficiales Ni0 y Ni2+ se ha determinado mediante la relación de las áreas del Ni0
(ANi0) y del Ni2+ (ANi
2+) y los resultados, que se recogen en la Tabla 3.6. Un aumento
en la relación ANi0/(ANi
0+ANi2+) es indicativo de un aumento en la reducibilidad del
catalizador (Natesakhawat y cols., 2005b). Este resultado es coherente con los perfiles
TPR obtenidos, que muestran un desplazamiento del máximo de los picos de
reducción del NiOx hacia menores temperaturas.
Figura 3.12. Espectros XPS de alta resolución del Ni2p de los catalizadores
10Ni/LaAl-F y Ni/LaAl-E reducidos.
Tabla 3.6. Comparación de la relación atómica entre el Ni metálico con el Ni total
en la superficie de los catalizadores sin equilibrar y equilibrado.
Catalizador ANi0/(ANi
0+ANi
2+)
10Ni/LaAl-F 0.09
10Ni/LaAl-E 0.70
840850860870880890
Inte
ns
ida
d, u
.a
Energía de enlace, eV
10 Ni/LaAl-F
10 Ni/LaAl-E
Ni2+ Ni2+
Ni2+
Ni0
Ni2+
Ni0
Ni2+
Ni0
Ni2+ Ni0
Ni2+
Ni2+ Ni0
Ni0

3. Optimación de las condiciones de preparación del catalizador 107
Carolina Montero Calderón
3.2.4. Discusión de los resultados
Los resultados de caracterización mostrados en los apartados anteriores indican
que la principal diferencia entre el catalizador sin equilibrar y el sometido a
tratamiento de equilibrado es la menor dispersión y superficie metálica, así como el
mayor grado de reducibilidad del catalizador equilibrado. La menor dispersión
metálica indica que el tratamiento de equilibrado ha inducido un cierto nivel de
sinterización del Ni, el cual es responsable de la menor actividad del catalizador
equilibrado respecto del catalizador sin equilibrar (como se ha observado en la Figura
3.5), a pesar de la mayor reducibilidad de las especies de Ni restantes en el catalizador
equilibrado.
La falta de reproducibilidad en la operación cíclica del catalizador sin equilibrar
debe atribuirse a que en las sucesivas etapas de reacción-regeneración está teniendo
lugar una progresiva sinterización del Ni (probablemente por las altas temperaturas
locales alcanzadas durante la combustión del coque en la etapa de regeneración en las
condiciones de la Figura 3.5, dado que la temperatura de reacción en baja, inferior a la
de calcinación). En estas condiciones, la pérdida de superficie específica por
sinterización es progresiva y acumulada en los sucesivos ciclos hasta alcanzar un
comportamiento estable.
Sin embargo, en las condiciones de la Figura 3.6, con reformado a elevada
temperatura, 700 ºC, el primer ciclo de reacción-regeneración está actuando como
tratamiento de equilibrado (en el cual tiene lugar la sinterización de parte de los
centros metálicos), por lo que alcanza un comportamiento reproducible a partir del 2º
ciclo de reacción. El catalizador equilibrado en estas condiciones (con una fracción de
centros ya sinterizados), muestra, por tanto, un comportamiento menos activo que el
catalizador sin equilibrar, pero ya reproducible a partir del primer ciclo de reacción-
regeneración, lo que resulta adecuado para el estudio cinético.


4. EFECTO DE LAS CONDICIONES
DE OPERACIÓN


111
Carolina Montero Calderón
4. EFECTO DE LAS CONDICIONES DE OPERACIÓN
En este Capítulo se presentan los resultados de los ensayos cinéticos llevados a
cabo con el catalizador 10Ni/LaAl-E, con la finalidad de analizar el efecto de las
diferentes variables de operación sobre su comportamiento cinético, así como para
obtener suficientes datos cinéticos para abordar con rigor el modelado cinético del
proceso de reformado con vapor de etanol.
Se ha analizado el efecto de cada variable de operación (temperatura, relación
molar vapor de agua/etanol, tiempo espacial) sobre los índices de reacción a tiempo
cero, así como sobre la evolución de los índices de reacción con el tiempo en
reacciones de 20 h de duración. También se han realizado varios ensayos de muy larga
duración (hasta 200 h), para verificar la estabilidad del catalizador en diferentes
condiciones. En todos los casos se ha mantenido constantes las condiciones
fluidodinámicas y se ha trabajo en condiciones que evitan limitaciones difusionales.
No se ha profundizado en el estudio del efecto de la presión parcial del etanol, ya que
de acuerdo con las conclusiones de trabajos previos desarrollados dentro del grupo
(Vicente, 2012), el aumento de la presión parcial del etanol favorece las reacciones de
descomposición de oxigenados frente al reformado. Las condiciones de operación han
sido:
Temperatura: entre 500-650 ºC
Presión: 1.4 bar (sobrepresión generada por el dispositivo de muestreo en
continuo).
Presión parcial del etanol: 0.083 bar
Relación molar agua/etanol alimentada: 3-9
Tiempo espacial: 0.02-0.69 gcath/gEtOH (W=0.03-1.05 g)
Caudales: QHe=25-115 cm3 (CN)/min, QH2O=0.03-0.10 ml/min
Tamaño de partícula del catalizador: 0.15-0.25 mm.
Altura del lecho: 4 cm.
Dilución del catalizador con CSi (tamaño de partícula 0.04 nm): superior a 8/1
Relación u/umf: 6

112
Capítulo 4
4.1. CONTRIBUCION DE LAS RUTAS TÉRMICAS EN EL SRE
Para delimitar la temperatura a partir de la cual es importante la contribución de
las posibles rutas térmicos en el mecanismo de reacción, se han realizado ensayos “en
blanco”, es decir, sin catalizador, operando con un lecho constituido únicamente por
carburundum (CSi) y manteniendo las mismas condiciones fluidodinámicas que en los
estudios con catalizador. Estos ensayos se han realizado con relación molar S/E de 3, 6
y 9, y en el intervalo de temperatura de 500 - 700 ºC. Los resultados de composición
de la corriente de productos a la salida del reactor (expresados como fracción molar en
base seca) se muestran en la Figura 4.1 (H2 y etanol), Figura 4.2 (acetaldehído y
etileno) y Figura 4.3 (CO2, CO, CH4).
Se observa que a 500 ºC la corriente está mayoritariamente formada por etanol
(~95 %), detectándose para todas las relaciones S/E pequeñas cantidades de H2 (~3
%), acetaldehído (~2 %), y etileno (~0.1 %). A esta temperatura no se observan
diferencias por el efecto de la relación S/E.
El aumento de temperatura hasta 600 ºC supone solo un ligero aumento de la
conversión de etanol, mientras que por encima de esa temperatura se produce un
rápido aumento de la conversión de etanol (Figura 4.1a), y un notable aumento
también del rendimiento de H2 (Figura 4.1b). El aumento de la relación molar S/E
también supone en general una mayor conversión de etanol y un mayor rendimiento
de H2, principalmente entre S/E =3 y 6 y más atenuadamente para una mayor relación
S/E, aunque este parámetro tiene menor influencia en los resultados que la
temperatura. Se alcanza una fracción molar (en base seca) de 0.36 de H2 a 700 ºC y
relación molar S/E de 9.
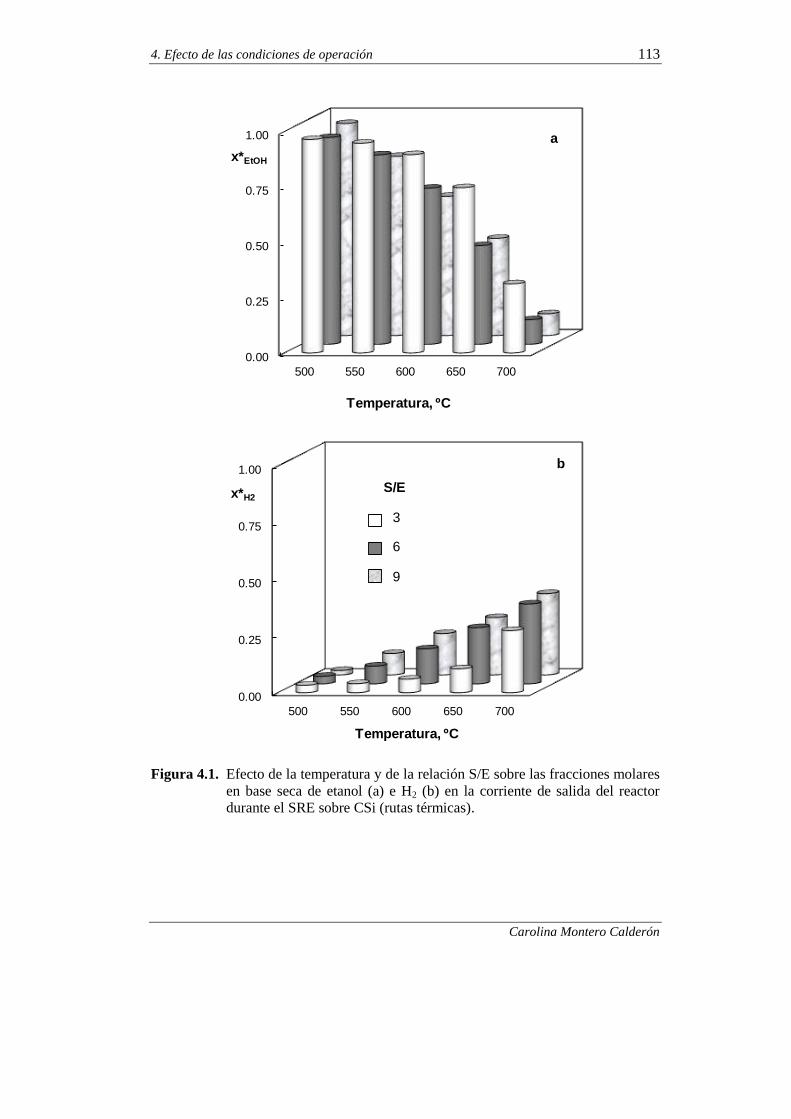
4. Efecto de las condiciones de operación 113
Carolina Montero Calderón
Figura 4.1. Efecto de la temperatura y de la relación S/E sobre las fracciones molares
en base seca de etanol (a) e H2 (b) en la corriente de salida del reactor
durante el SRE sobre CSi (rutas térmicas).
0.00
0.25
0.50
0.75
1.00
500 550 600 650 700
x*EtOH
Temperatura, ºC
a
0.00
0.25
0.50
0.75
1.00
500 550 600 650 700
x*H2
Temperatura, ºC
S/E
3
6
9
b
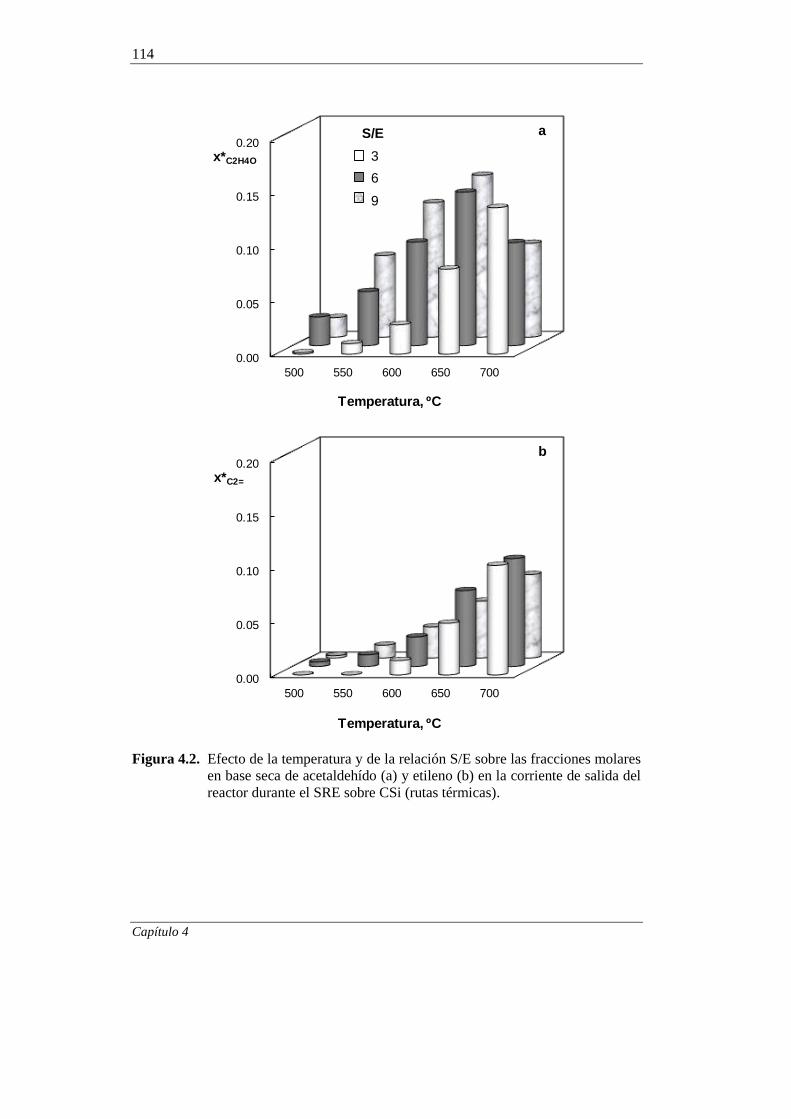
114
Capítulo 4
Figura 4.2. Efecto de la temperatura y de la relación S/E sobre las fracciones molares
en base seca de acetaldehído (a) y etileno (b) en la corriente de salida del
reactor durante el SRE sobre CSi (rutas térmicas).
0.00
0.05
0.10
0.15
0.20
500 550 600 650 700
x*C2H4O
Temperatura, ºC
aS/E
3
6
9
0.00
0.05
0.10
0.15
0.20
500 550 600 650 700
x*C2=
Temperatura, ºC
b
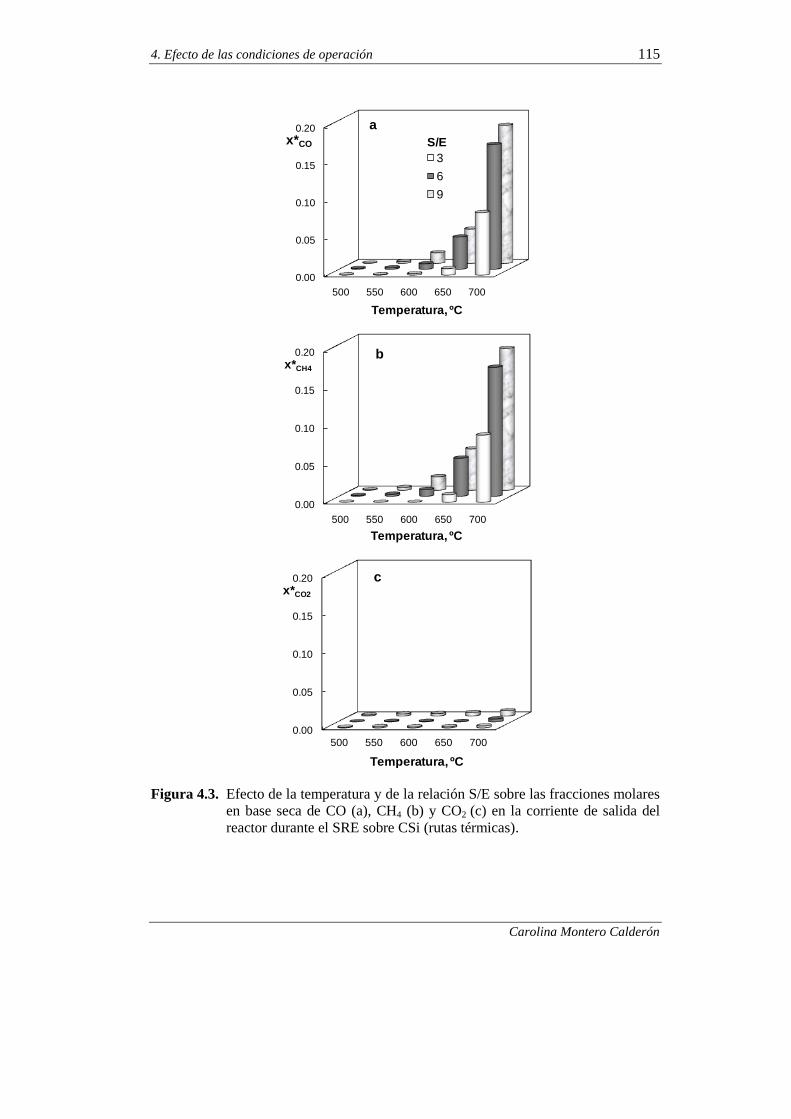
4. Efecto de las condiciones de operación 115
Carolina Montero Calderón
Figura 4.3. Efecto de la temperatura y de la relación S/E sobre las fracciones molares
en base seca de CO (a), CH4 (b) y CO2 (c) en la corriente de salida del
reactor durante el SRE sobre CSi (rutas térmicas).
0.00
0.05
0.10
0.15
0.20
500 550 600 650 700
x*CO
Temperatura, ºC
3
6
9
a
S/E
0.00
0.05
0.10
0.15
0.20
500 550 600 650 700
x*CH4
Temperatura, ºC
b
0.00
0.05
0.10
0.15
0.20
500 550 600 650 700
x*CO2
Temperatura, ºC
c

116
Capítulo 4
En cuanto a los productos carbonados (Figuras 4.2 y 4.3), son mayoritarios los
hidrocarburos con 2 átomos de C, acetaldehído y etileno, especialmente el primero,
para una temperatura inferior a 650 ºC. La formación de CO y de CH4 aumenta
exponencialmente por encima de 600 ºC, siendo casi insignificante la formación de
CO2. Esta distribución de productos indica que el aumento de temperatura es capaz de
promover en primer lugar la reacción de deshidrogenación de etanol para formar
acetaldehído (Ec. 1.9) (cuya presencia ya es significativa a 500 ºC), y a continuación
la deshidratación para generar etileno (Ec. 1.10) (cuya formación es apreciable a partir
de 550 ºC), mientras que las reacciones de descomposición bien del etanol (Ec. 1.11) o
del acetaldehído (Ec. 1.16) son importantes solo a partir de 650 ºC y se promueven
exponencialmente con el aumento de la temperatura. La reacción WGS (Ec. 1.19), el
reformado con vapor de metano (Ec. 1.21) y el reformado directo con vapor del etanol
(Ec. 1.8) no están promovidas por el aumento de temperatura en el intervalo 500-700
ºC, como se deduce de las similares concentraciones de CO y CH4 obtenidas a altas
temperaturas, así como de la casi insignificante formación de CO2.
En general, todas estas reacciones de transformación únicamente por efecto
térmico está promovidas por el aumento de la relación S/E de 3 a 6, si bien un
posterior aumento de esta variable afecta menos significativamente. Contenidos muy
elevados de H2O si parecen promover bien la reacción WGS o el reformado directo de
etanol a temperatura muy elevada, dado que para S/E=9 por encima de 650 ºC se
observa una presencia significativa de CO2 (si bien solo de hasta un 1 % molar en base
seca).
En la bibliografía son escasos los trabajos que muestren resultados de ensayos
sin catalizador para analizar las rutas térmicas del reformado con vapor. Fatsikostas y
Verykios (2004) realizaron un estudio “en blanco” de SRE mediante TPSR
(Temperature-programmed surface reaction), consistente en realizar un ensayo TPD
bajo condiciones de reacción, con relación S/E=3 en el intervalo 500-800 ºC, y
concluyeron que la transformación de etanol se activa a partir de 600 ºC, siendo
significativa por encima de 700 ºC. Señalan que a temperaturas bajas predomina la
deshidrogenación mientras que a mayor temperatura la deshidratación y el craqueo o
disociación son las reacciones principales, en concordancia con los resultados
obtenidos en esta Tesis.

4. Efecto de las condiciones de operación 117
Carolina Montero Calderón
Barratini y cols. (2009) realizaron un ensayo en reactor de lecho fijo utilizando
cuarzo como inerte, en el intervalo entre 300 y 840 ºC y con relación S/E=3.
Obtuvieron que la conversión de etanol se promueve de manera significativa a partir
de 430 ºC, llegando a ser completa a 790 ºC. A baja temperatura (400 ºC) los
productos principales fueron etileno y acido acético, mientras que a temperaturas
mayores (730 ºC) obtuvieron pequeñas cantidades de CO, CH4 e H2 y la selectividad
del acido acético se desfavorece. La selectividad de etileno se mantuvo estable con el
cambio de temperatura y no detectaron acetaldehído, lo que contrasta con los
resultados obtenidos en este trabajo, así como con los obtenidos por Fatsikostas y
Verykios (2004).
Melchor-Hernández y cols. (2013) llevaron a cabo un ensayo de reformado sin
catalizador de mezcla S/E=3 en reactor de lecho fijo. Si bien no presentan resultados
de distribución de productos, señalan que la conversión de etanol es significativa para
temperaturas mayores de 600 ºC.
Se puede concluir, por tanto, que para temperaturas de reformado muy elevadas
(a partir de 650 ºC), habrá que tener en cuenta la posible contribución de las rutas
térmicas a los valores de conversión y rendimientos de productos obtenidos en los
ensayos catalíticos, siendo esta contribución prácticamente despreciable por debajo de
600 ºC. Por otro lado, la mayor velocidad de reacción de las etapas catalizadas
minimizarán la contribución de las reacciones de origen térmico.
En base a estos resultados, para el estudio del efecto de las variables de
operación que se muestra en el siguiente apartado, el intervalo de temperatura de
reformado analizado se ha fijado entre 500-650 ºC, con objeto de tener un intervalo
suficientemente amplio de operación, pero evitando una contribución excesiva de las
reacciones térmicas, para poder extraer conclusiones relativas a la actividad,
selectividad y estabilidad propias del catalizador utilizado, 10Ni/LaAl equilibrado.

118
Capítulo 4
4.2. EFECTO DE LAS CONDICIONES DE REACCIÓN EN EL
COMPORTAMIENTO DEL CATALIZADOR ESTABILIZADO
Se muestra en este Apartado el efecto de las variables de operación sobre el
comportamiento cinético a tiempo cero del catalizador de Ni equilibrado (10Ni/LaAl-
E), así como sobre su estabilidad en el proceso SRE, analizando los resultados
obtenidos en ensayos de 20 h de duración, así como en ensayos de muy larga duración
(hasta 200 h). Los índices de reacción estudiados (conversión, rendimiento y
selectividad de productos) se han definido en el Apartado 2.3.5 (Ecs. (2.6)-
(2.8)).Tanto la conversión como los rendimientos se expresan en tanto por 1. El efecto
de las variables de operación sobre la deposición de coque se analiza en un Capítulo
posterior.
4.2.1. Comportamiento cinético a tiempo cero
4.2.1.1. Efecto de la temperatura
El efecto de la temperatura de reacción en el SRE para catalizadores de Ni ha
sido ampliamente estudiado en la bibliografía (Fatsikostas y cols., 2004; Llera y cols.,
2012; Patel y cols., 2013; Melchor-Hernández y cols., 2013; Zhang y cols., 2014b;
Dan y cols., 2015). Esta variable tiene gran influencia en el comportamiento cinético
del catalizador, aumentando notablemente la conversión y el rendimiento de H2 al
aumentar la temperatura, debido al aumento de la velocidad de reacción y porque el
proceso es favorecido al ser muy endotérmico.
Los resultados del efecto de la temperatura se muestran en las Figuras 4.4-4.5,
en las que, a modo de ejemplo, se han representado los valores de conversión de
etanol y rendimiento de H2 (Figura 4.4) y rendimiento de productos carbonados
(Figura 4.5), para relación molar S/E=3 (la relación estequiométrica del proceso SRE)
y dos valores del tiempo espacial, bajo (0.04 gcath/gEtOH) y alto (0.35 gcath/gEtOH), con la
finalidad de establecer claramente el efecto de la temperatura en condiciones alejadas
del equilibrio (régimen cinético), y en condiciones cercanas al equilibrio (régimen
termodinámico), cuyas concentraciones se analizan en detalle en el Anexo B.
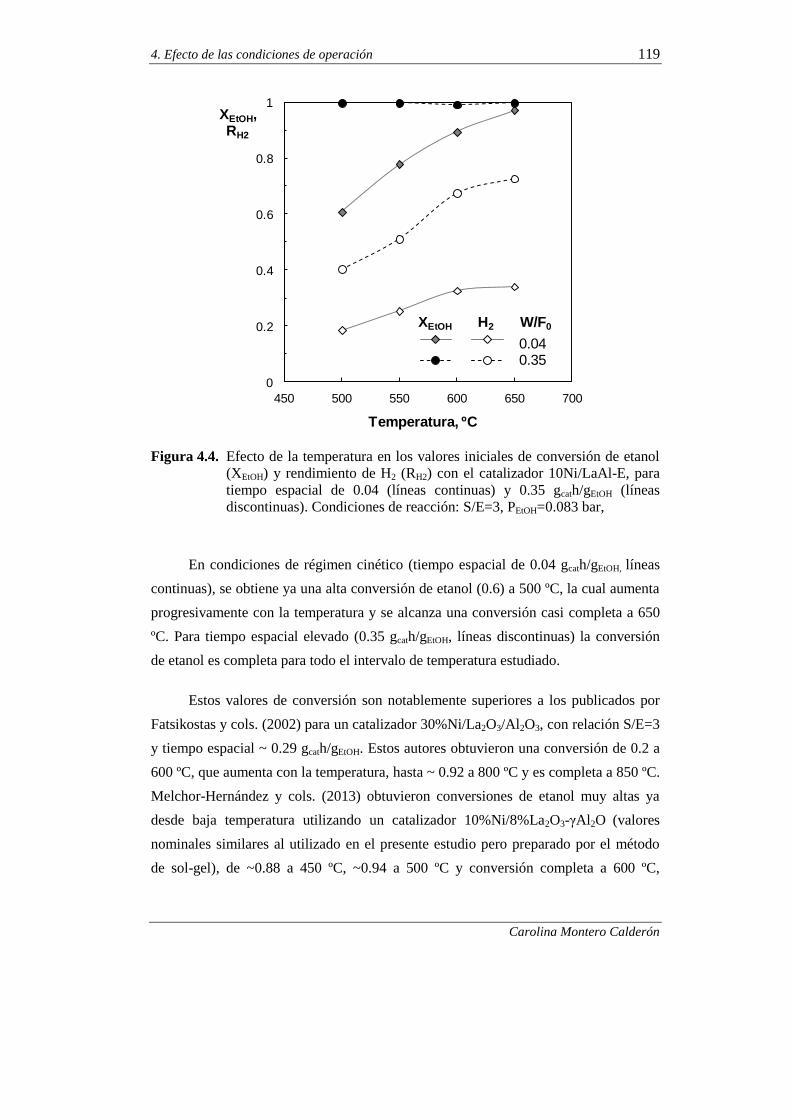
4. Efecto de las condiciones de operación 119
Carolina Montero Calderón
Figura 4.4. Efecto de la temperatura en los valores iniciales de conversión de etanol
(XEtOH) y rendimiento de H2 (RH2) con el catalizador 10Ni/LaAl-E, para
tiempo espacial de 0.04 (líneas continuas) y 0.35 gcath/gEtOH (líneas
discontinuas). Condiciones de reacción: S/E=3, PEtOH=0.083 bar,
En condiciones de régimen cinético (tiempo espacial de 0.04 gcath/gEtOH, líneas
continuas), se obtiene ya una alta conversión de etanol (0.6) a 500 ºC, la cual aumenta
progresivamente con la temperatura y se alcanza una conversión casi completa a 650
ºC. Para tiempo espacial elevado (0.35 gcath/gEtOH, líneas discontinuas) la conversión
de etanol es completa para todo el intervalo de temperatura estudiado.
Estos valores de conversión son notablemente superiores a los publicados por
Fatsikostas y cols. (2002) para un catalizador 30%Ni/La2O3/Al2O3, con relación S/E=3
y tiempo espacial ~ 0.29 gcath/gEtOH. Estos autores obtuvieron una conversión de 0.2 a
600 ºC, que aumenta con la temperatura, hasta ~ 0.92 a 800 ºC y es completa a 850 ºC.
Melchor-Hernández y cols. (2013) obtuvieron conversiones de etanol muy altas ya
desde baja temperatura utilizando un catalizador 10%Ni/8%La2O3-γAl2O (valores
nominales similares al utilizado en el presente estudio pero preparado por el método
de sol-gel), de ~0.88 a 450 ºC, ~0.94 a 500 ºC y conversión completa a 600 ºC,
0
0.2
0.4
0.6
0.8
1
450 500 550 600 650 700
XEtOH, RH2
Temperatura, ºC
XEtOH H2 W/F0
0.040.35

120
Capítulo 4
operando con relación S/E= 3 y tiempo espacial de ~0.07 gcath/gEtOH, aunque con bajo
rendimiento de H2 (~0.04 a 400 ºC, que aumenta hasta 0.50 para ~600 ºC y aún más al
aumentar el contenido de La2O3 en el soporte). Para una relación S/E=5.5 con
catalizador de Ni (II)-Al (III)-LDH y operando a tiempos espaciales bajos, Llera y
cols. (2012) observaron que entre 600-650 ºC hay aumento drástico en la conversión
(desde ~0.10 hasta 0.55), mientras que con catalizador Ni/Ce2O3/Zr2O3, para tiempos
espaciales altos y relación S/E=9, Patel y cols. (2013) han obtenido una conversión de
0.95 a 600 ºC y completa a 700 ºC.
En un estudio reciente de Dan y cols. (2015) sobre el efecto de la temperatura
en el reformado con vapor de bioetanol (40 % de etanol), con catalizador
10%Ni/6%La2O3-Al2O3, se alcanza la conversión completa del etanol a 350 ºC, lo cual
puede ser atribuido principalmente a la alta relación S/E (~15).
Respecto al rendimiento de H2 obtenido en esta Tesis (Figura 4.4), en
condiciones de régimen cinético (bajo tiempo espacial), éste aumenta con la
temperatura (entre 0.19 a 500 ºC y 0.33 a 650 ºC) aunque en menor medida que la
conversión de etanol, especialmente por encima de 600 ºC, mientras que en
condiciones de conversión completa el aumento del rendimiento de H2 con la
temperatura es más progresivo en todo el intervalo entre 500 ºC (con valor de 0.40) y
650 ºC (rendimiento de 0.73), si bien el aumento también está atenuado por encima de
600 ºC. Cabe mencionar que la relación S/E=3 utilizada no es la más favorable para
maximizar el rendimiento de H2, y que se obtienen mayores rendimientos al aumentar
la relación S/E (0.88 para relación S/E=9 y 600 ºC), tal como se muestra en un
apartado posterior.
Esta evolución del rendimiento de H2, dispar con la evolución de la conversión
de etanol, se explica en base al diferente efecto de la temperatura sobre las diferentes
reacciones involucradas en el mecanismo global de reacción, lo que conlleva
igualmente un importante cambio en los rendimientos de productos carbonados al
variar la temperatura, como se muestra en la Figura 4.5.
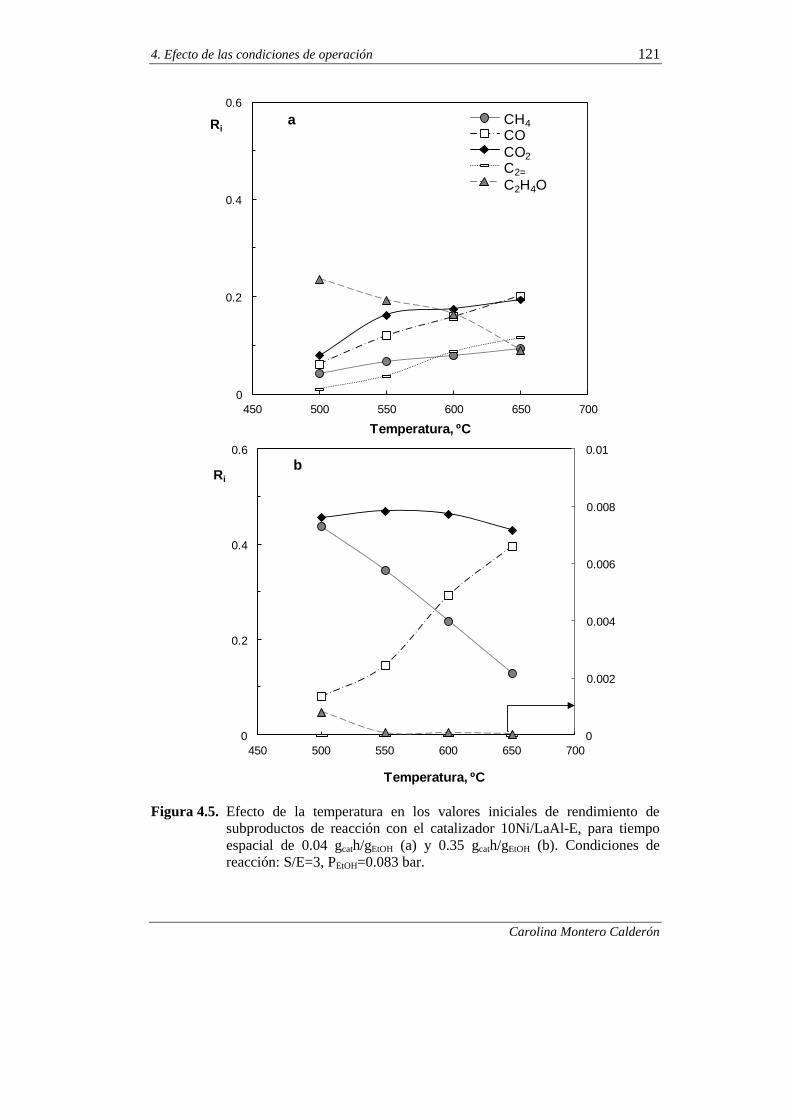
4. Efecto de las condiciones de operación 121
Carolina Montero Calderón
Figura 4.5. Efecto de la temperatura en los valores iniciales de rendimiento de
subproductos de reacción con el catalizador 10Ni/LaAl-E, para tiempo
espacial de 0.04 gcath/gEtOH (a) y 0.35 gcath/gEtOH (b). Condiciones de
reacción: S/E=3, PEtOH=0.083 bar.
0
0.2
0.4
0.6
450 500 550 600 650 700
Ri
Temperatura, ºC
CH4
CO
CO2
C2=
C2H4O
a
0
0.002
0.004
0.006
0.008
0.01
0
0.2
0.4
0.6
450 500 550 600 650 700
Ri
Temperatura, ºC
b

122
Capítulo 4
En condiciones de régimen cinético (bajo tiempo espacial) (Figura 4.5a), a 500
ºC el producto mayoritario es el acetaldehído, cuyo rendimiento (0.24) es muy
superior al de CO2 (0.08), el cual a su vez es ligeramente mayor que los rendimientos
de CO y CH4 (siendo estos últimos muy similares entre sí) y el etileno es claramente el
producto minoritario (0.01). Es destacable que en estas condiciones la relación molar
H2/CO2 (6.91) es notablemente mayor que el valor estequiométrico de 3
correspondiente al reformado de etanol. Estos resultados indican que a 500 ºC la
reacción más rápida del esquema cinético es la deshidrogenación a acetaldehído, que
es notablemente más rápido que a la de reformado de etanol, y esta a su vez es más
rápida que las reacciones de descomposición (tanto de etanol como de acetaldehído),
siendo lentas las reacciones WGS, reformado de metano y deshidratación a etileno,
especialmente esta última.
Para este bajo valor del tiempo espacial, el rendimiento de acetaldehído
disminuye notablemente al aumentar a temperatura, especialmente por encima de 600
ºC (siendo ligeramente inferior a 0.1 a 650 ºC), mientras que el rendimiento del resto
de productos aumenta con la temperatura, especialmente el de etileno, siendo el
rendimiento de metano el que menos aumenta. La relación molar H2/CO2 disminuye
asintóticamente al aumentar la temperatura (hasta 5.3 a 650 ºC). Estos resultados
ponen de manifiesto que el aumento de temperatura promueve principalmente la
velocidad de la reacción de deshidratación del etanol, así como de las reacciones de
descomposición de etanol y de acetaldehído, o el reformado de este último.
Para elevado tiempo espacial (Figura 4.5b) y, por tanto, en condiciones en que
la conversión de etanol es completa y se alcanza el equilibrio termodinámico, hay
ausencia prácticamente total de etileno en todo el intervalo de temperaturas estudiado
y tampoco se observa la formación de acetaldehído por encima de 550 ºC. Este
resultado indica que ambos compuestos son productos intermedios en el esquema
cinético de reacción, y que las reacciones de transformación de ambos compuestos
(por reformado del primero y descomposición del segundo) son reacciones
irreversibles cuya conversión es completa en las condiciones de estudio. El
rendimiento de metano disminuye de forma casi lineal con la temperatura (desde 0.45
a 500 ºC hasta 0.13 a 650 ºC) y en paralelo tiene lugar un pronunciado aumento del
rendimiento de CO (casi paralelo este último al obtenido para el rendimiento de H2 en

4. Efecto de las condiciones de operación 123
Carolina Montero Calderón
estas condiciones, Figura 4.4a), lo que es indicativo de la importante contribución de
la reacción de reformado con vapor de metano (Ec. 1.21) al esquema global de
reacción, de modo que el notable aumento de su conversión de equilibrio con la
temperatura (por tratarse de una reacción muy endotérmica) afecta notablemente a la
distribución de productos de reacción y contribuye al notable aumento del rendimiento
de H2 con la temperatura. El rendimiento de CO2 es elevado y casi constante (~0.46)
en el intervalo 500-600 ºC, siendo las reacciones de reformado con vapor (de
oxigenados e hidrocarburos presentes) así como la reacción WGS las responsables de
su formación. Este rendimiento disminuye ligeramente por encima de 600 ºC debido a
que el equilibrio termodinámico de la reacción WGS (exotérmica) se desplaza
(potenciando la reacción WGS reversa), motivo que contribuye a atenuar el aumento
del rendimiento de H2 al aumentar la temperatura por encima de 600 ºC.
Fatsikostas y cols. (2002) señalan que el aumento del rendimiento de CO con la
temperatura para un catalizador de Ni/La2O3 también podría atribuirse, además de al
aumento de la velocidad de reformado con vapor de metano, al reformado seco (con
CO2) lo que conllevaría el aumento del rendimiento de CO y contribuiría a mantener
constante el rendimiento de CO2.
Para el etileno, Melchor-Hernández y cols. (2013) obtuvieron un rendimiento
máximo de ~0.35 en el intervalo entre 500-550 ºC, con disminución posterior hasta ~
0.20 a 600 ºC, siendo menor el rendimiento de etileno al aumentar el contenido de La
en el soporte. Este elevado rendimiento de etileno es debido a que utilizan un soporte
ligeramente ácido (-Al2O3), que es muy activo para la reacción de deshidratación del
etanol. Por el contrario, con un catalizador de Ni/SiO2, trabajando en condiciones de
operación iguales a las de esta Tesis, Vicente y cols. (2014b) han determinado la
ausencia total de etileno en la corriente de productos, debido a que la SiO2 es un
soporte no ácido, que minimiza la deshidratación de etileno. Además, la competencia
de las reacciones catalíticas con las térmicas evita la formación de etileno incluso a
elevada temperatura.
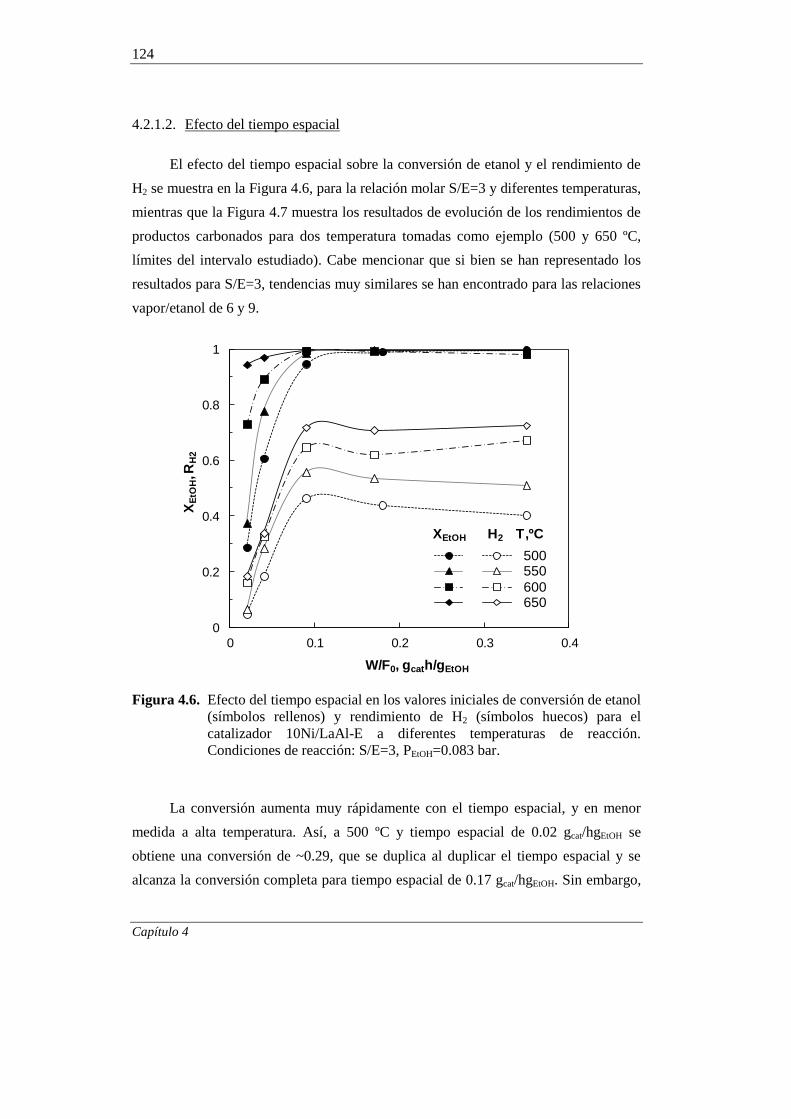
124
Capítulo 4
4.2.1.2. Efecto del tiempo espacial
El efecto del tiempo espacial sobre la conversión de etanol y el rendimiento de
H2 se muestra en la Figura 4.6, para la relación molar S/E=3 y diferentes temperaturas,
mientras que la Figura 4.7 muestra los resultados de evolución de los rendimientos de
productos carbonados para dos temperatura tomadas como ejemplo (500 y 650 ºC,
límites del intervalo estudiado). Cabe mencionar que si bien se han representado los
resultados para S/E=3, tendencias muy similares se han encontrado para las relaciones
vapor/etanol de 6 y 9.
Figura 4.6. Efecto del tiempo espacial en los valores iniciales de conversión de etanol
(símbolos rellenos) y rendimiento de H2 (símbolos huecos) para el
catalizador 10Ni/LaAl-E a diferentes temperaturas de reacción.
Condiciones de reacción: S/E=3, PEtOH=0.083 bar.
La conversión aumenta muy rápidamente con el tiempo espacial, y en menor
medida a alta temperatura. Así, a 500 ºC y tiempo espacial de 0.02 gcat/hgEtOH se
obtiene una conversión de ~0.29, que se duplica al duplicar el tiempo espacial y se
alcanza la conversión completa para tiempo espacial de 0.17 gcat/hgEtOH. Sin embargo,
0
0.2
0.4
0.6
0.8
1
0 0.1 0.2 0.3 0.4
XE
tOH, R
H2
W/F0, gcath/gEtOH
500550
600650
XEtOH H2 T,ºC

4. Efecto de las condiciones de operación 125
Carolina Montero Calderón
a 650 ºC el efecto del tiempo espacial es menos significativo, dada la apreciable
contribución de las rutas térmicas a las reacciones del esquema cinético, de modo que
para un tiempo espacial de 0.02 gcat/hgEtOH ya se obtiene una muy alta conversión
(0.94), la cual aumenta hasta 0.97 al duplicar el tiempo espacial y es completa desde
0.09 gcat/hgEtOH.
Para todas las temperaturas estudiadas el rendimiento de H2 (símbolos huecos;
Figura 4.4) aumenta muy rápidamente con el tiempo espacial entre 0.02 y 0.09
gcat/hgEtOH, intervalo en el que ya se alcanza una elevada conversión de etanol (> 0.94)
a todas las temperaturas. A baja temperatura ( 550 ºC) se alcanza un valor máximo
de rendimiento de H2 para 0.09 gcat/hgEtOH, y el posterior aumento de tiempo espacial
conlleva una ligera disminución del rendimiento de H2, asociada a cambios en el
rendimiento de los subproductos, como se muestra a continuación, alcanzándose el
rendimiento correspondiente al equilibrio termodinámico para un tiempo espacial de
0.35 gcat/hgEtOH. A elevada temperatura ( 600 ºC), también para tiempo espacial de
0.09 gcat/hgEtOH se obtiene un rendimiento máximo de H2, que se mantiene
aproximadamente contante para valores más elevados de tiempo espacial, dado que
corresponde prácticamente al equilibrio termodinámico (Anexo B).
Similares tendencias en la conversión y rendimiento de H2 han sido observadas
para otros catalizadores de Ni. Para el catalizador de Ni (II)-Al (III)-LDH a 650 ºC y
S/E=5.5, Llera y cols. (2012) obtienen una conversión de ~0.50 para un tiempo
espacial de 0.035 gcat/hgEtOH, y se alcanza la conversión es completa para un tiempo
espacial equivalente a 0.14 gcat/hgEtOH. El rendimiento de H2 aumenta con el tiempo
espacial hasta un valor de ~0.80 a 0.14 gcat/hgEtOH, y disminuye ligeramente para
mayor tiempo espacial.
Para el catalizador de Ni esqueletal (no soportado) estudiado por Zhang y cols.
(2014b) se alcanza la conversión completa para un tiempo espacial superior a ~0.15
gcat/hgEtOH, a 450 ºC y con relación S/E=8, mientras que el rendimiento de H2 se
mantiene prácticamente constante (~0.60) en el intervalo de tiempo espacial estudiado,
entre 0.07 y 0.35 gcat/hgEtOH. Patel y cols. (2013) también observan el aumento de la
conversión de etanol con el tiempo espacial, para el catalizador Ni/Ce2O3/Zr2O3 y
S/E=4, si bien no llegan a obtener la conversión completa a 600 ni a 650 ºC, para un

126
Capítulo 4
tiempo espacial elevado de ~0.60 gcat/hgEtOH, alcanzándose un valor de 0.95. El
rendimiento de H2 oscila entre 0.70 y 0.80, ligeramente superior al mostrado en la
Figura 4.5, lo que se debe probablemente a que estos autores operan con mayor
relación S/E.
En la Figura 4.7 se ha representado la evolución con el tiempo espacial del
rendimiento de los subproductos, para baja y alta temperatura de reformado. Al igual
que en la evolución del rendimiento de H2, se identifican dos zonas: i) entre 0.02 y
0.09 gcat/hgEtOH, donde hay una gran influencia del tiempo espacial, y; ii) para tiempos
espaciales mayores a 0.09 gcat/hgEtOH, con la conversión casi completa de etanol y con
pocas variaciones del rendimiento de productos.
En la zona i) se observa un muy rápido aumento del rendimiento de CO2 y de
CO, alcanzándose un máximo en el rendimiento de CO para 0.09 gcat/hgEtOH, con una
posterior ligera disminución en la zona ii), mientras que el rendimiento de CO2
continua aumentando pero de forma muy atenuada para estos tiempos espaciales
elevados, especialmente a 650 ºC, y ambos productos alcanzan las composiciones
correspondientes al equilibrio termodinámico para tiempo espacial de 0.35 gcat/hgEtOH.
A baja temperatura se observa un muy rápido aumento del rendimiento de CH4
en la zona i) que se atenúa notablemente en la zona ii), mientras que a alta temperatura
se obtiene un apreciable rendimiento de CH4 (del 0.09) ya a bajo tiempo espacial (por
la importante contribución de las rutas térmicas a 650 ºC), el cual aumenta muy poco
para mayor tiempo espacial, porque está próximo al correspondiente al equilibrio
termodinámico a esta temperatura. Este resultado indica que las reacciones de
descomposición de oxigenados (que producen CO y CH4) son muy rápidas a 650 ºC.
El acetaldehído y el etileno son los productos mayoritarios a bajo tiempo
espacial, especialmente el primero, lo que corrobora que las reacciones de
deshidrogenación, y en menor medida la deshidratación, son las más rápidas del
esquema cinético de reacción (principalmente a elevada temperatura en el caso de la
deshidratación, lo que justifica el mayor rendimiento de etileno a 650 ºC).
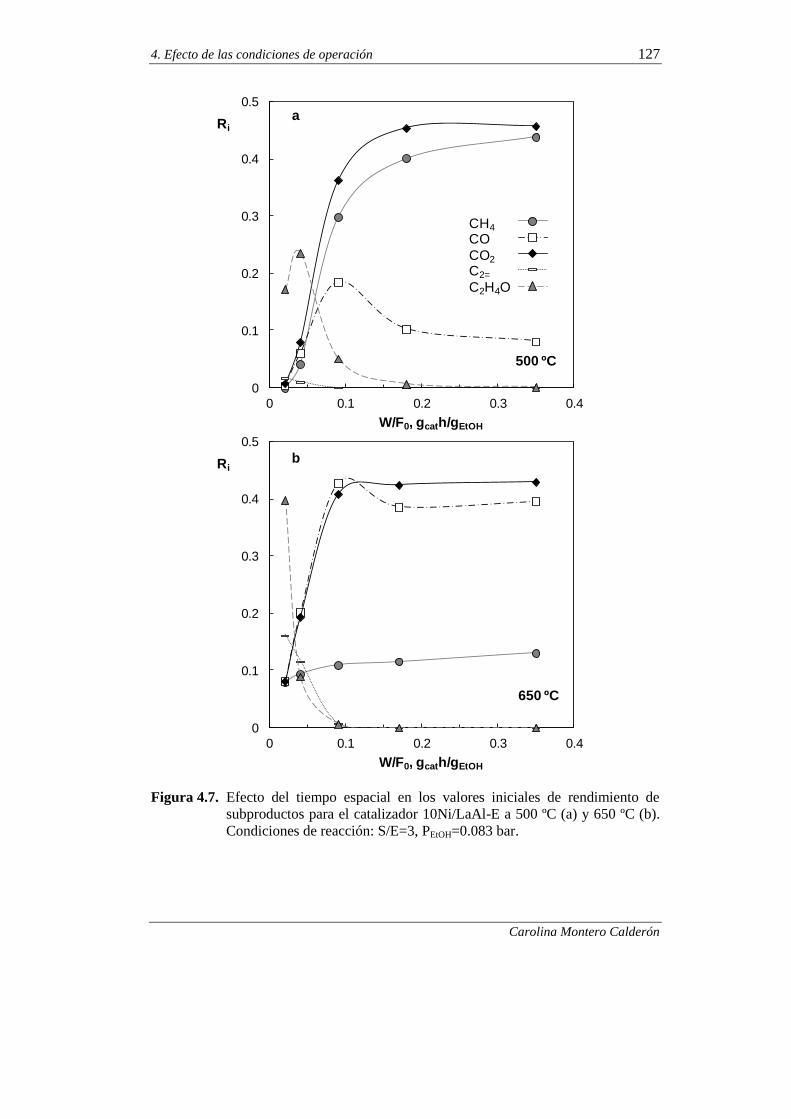
4. Efecto de las condiciones de operación 127
Carolina Montero Calderón
Figura 4.7. Efecto del tiempo espacial en los valores iniciales de rendimiento de
subproductos para el catalizador 10Ni/LaAl-E a 500 ºC (a) y 650 ºC (b).
Condiciones de reacción: S/E=3, PEtOH=0.083 bar.
0
0.1
0.2
0.3
0.4
0.5
0 0.1 0.2 0.3 0.4
Ri
W/F0, gcath/gEtOH
CH4
CO
CO2
C2=
C2H4O
500 ºC
a
0
0.1
0.2
0.3
0.4
0.5
0 0.1 0.2 0.3 0.4
Ri
W/F0, gcath/gEtOH
650 ºC
b

128
Capítulo 4
Similares tendencias en la evolución de los rendimientos de subproductos han
sido observadas por Wu y cols. (2014b) para un catalizador comercial 15%Ni/Al2O3
en el intervalo entre 200 y 600 ºC y para S/E=10, diferenciándose también dos zonas;
una para tiempos de contacto menores a 1 s, y otra a tiempos de contacto mayores. El
CO2 aumenta rápidamente con el tiempo espacial en la primera zona y más
atenuadamente en la segunda zona. El rendimiento de CO presenta un máximo con el
tiempo espacial similar al obtenido en es esta Tesis, aunque dicho máximo es solo
evidente a 600 ºC. El rendimiento de CH4 es igualmente más elevado a baja
temperatura y aumenta con el tiempo espacial en la primera zona (< 1 s). El
acetaldehído disminuye con el tiempo espacial, de forma más acusada a 600 ºC,
aunque a diferencia de lo obtenido en esta Tesis, estos autores siguieron detectando la
formación de acetaldehído para todos los tiempos espaciales estudiados.
4.2.1.3. Efecto de la relación molar vapor/etanol (S/E)
El efecto de esta variable ha sido menos estudiado en la bibliografía que el
efecto de la temperatura. Gran parte de los estudios se han realizado con una relación
molar S/E=3, correspondiente al valor estequiométrico (Haryanto y cols., 2005;
Vaidya y Rodrigues., 2006a; Bshish y cols., 2011, Nahar y Dupont, 2012), y se han
utilizado valores de S/E > 6 principalmente en estudios de reformado con vapor de
bio-etanol crudo, cuyo contenido de etanol es ~14 % en peso (Banach y cols., 2011).
En esta Tesis se ha elegido el intervalo de relación molar S/E entre 3 y 9. La
Figura 4.8 muestra la evolución con la relación molar S/E de la conversión de etanol y
del rendimiento de H2, a 600 ºC y con tiempo espacial de 0.18 gcat/hgEtOH. No se
observa ningún efecto de la relación S/E sobre la conversión de etanol, dado que ésta
es completa para las tres relaciones S/E estudiadas por tratarse de un elevado tiempo
espacial. Ahora bien, tal como predice la termodinámica (Anexo B), se observa un
claro aumento del rendimiento de H2, de forma prácticamente lineal en el intervalo de
valores de S/E estudiado, debido a que el exceso de agua favorece la selectividad de
H2, al desplazar gran parte de las reacciones del esquema cinético que generan H2,
como el reformado de etanol y de metano, así como la reacción WGS.
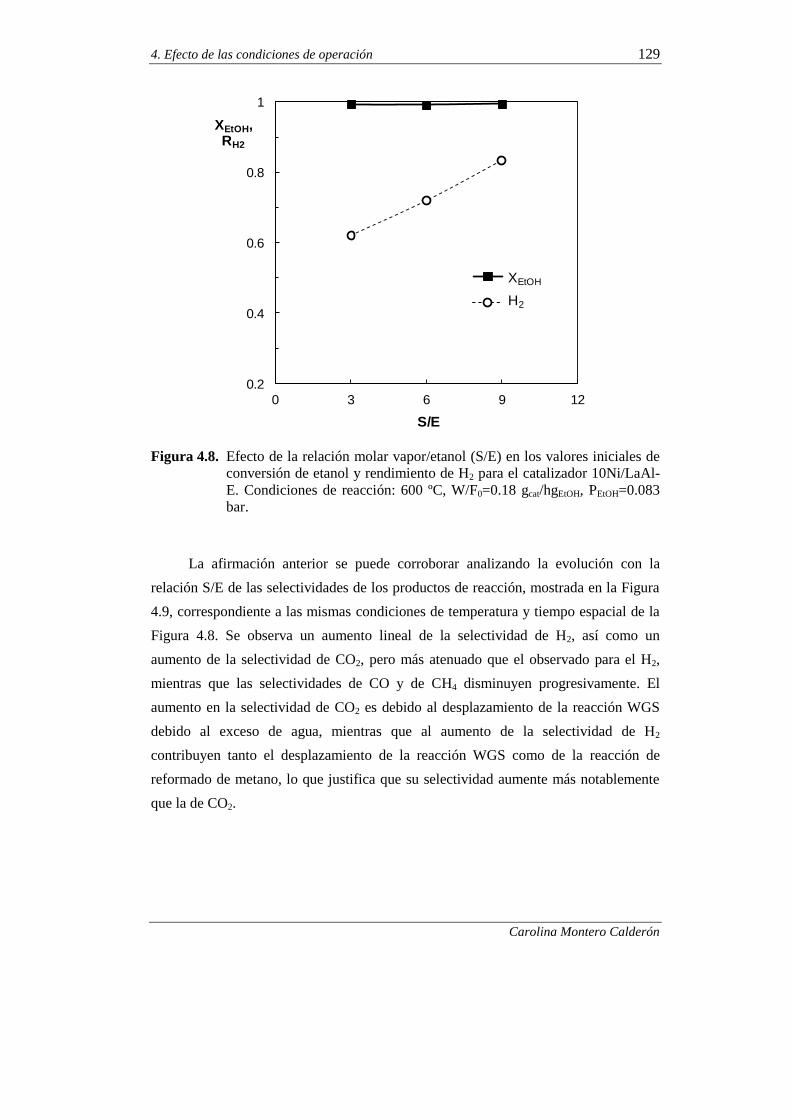
4. Efecto de las condiciones de operación 129
Carolina Montero Calderón
Figura 4.8. Efecto de la relación molar vapor/etanol (S/E) en los valores iniciales de
conversión de etanol y rendimiento de H2 para el catalizador 10Ni/LaAl-
E. Condiciones de reacción: 600 ºC, W/F0=0.18 gcat/hgEtOH, PEtOH=0.083
bar.
La afirmación anterior se puede corroborar analizando la evolución con la
relación S/E de las selectividades de los productos de reacción, mostrada en la Figura
4.9, correspondiente a las mismas condiciones de temperatura y tiempo espacial de la
Figura 4.8. Se observa un aumento lineal de la selectividad de H2, así como un
aumento de la selectividad de CO2, pero más atenuado que el observado para el H2,
mientras que las selectividades de CO y de CH4 disminuyen progresivamente. El
aumento en la selectividad de CO2 es debido al desplazamiento de la reacción WGS
debido al exceso de agua, mientras que al aumento de la selectividad de H2
contribuyen tanto el desplazamiento de la reacción WGS como de la reacción de
reformado de metano, lo que justifica que su selectividad aumente más notablemente
que la de CO2.
0.2
0.4
0.6
0.8
1
0 3 6 9 12
XEtOH,RH2
S/E
XEtOH
H2
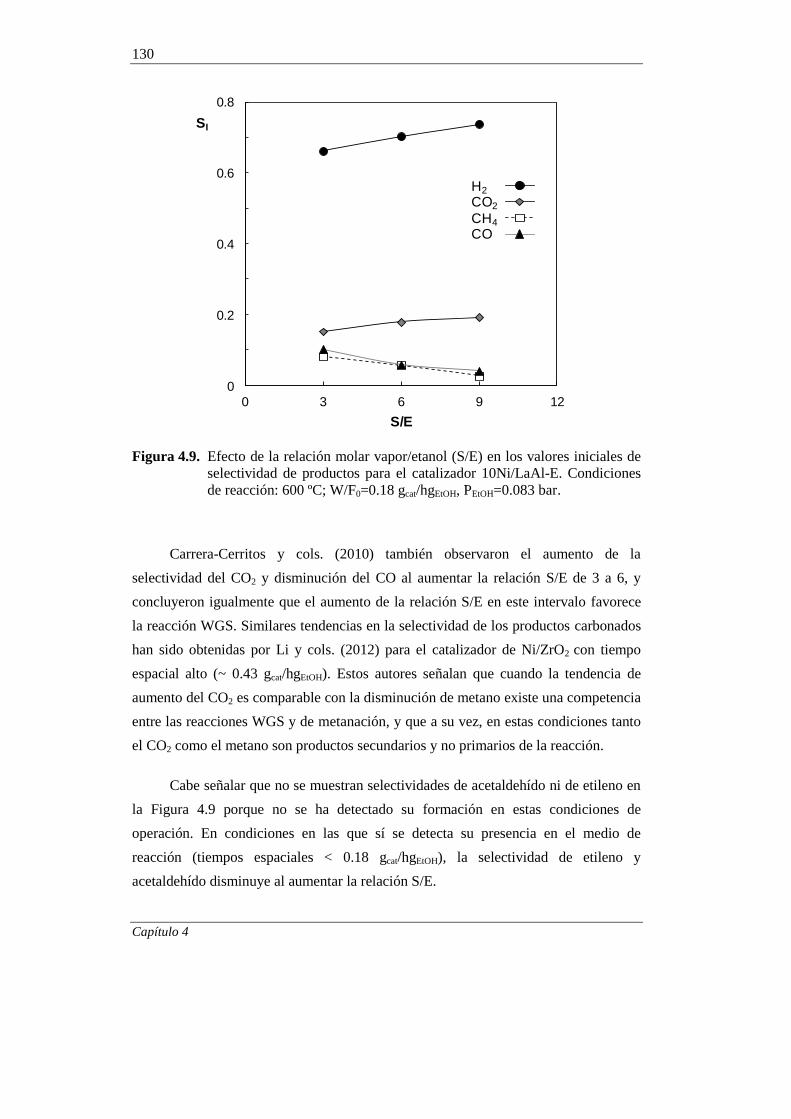
130
Capítulo 4
Figura 4.9. Efecto de la relación molar vapor/etanol (S/E) en los valores iniciales de
selectividad de productos para el catalizador 10Ni/LaAl-E. Condiciones
de reacción: 600 ºC; W/F0=0.18 gcat/hgEtOH, PEtOH=0.083 bar.
Carrera-Cerritos y cols. (2010) también observaron el aumento de la
selectividad del CO2 y disminución del CO al aumentar la relación S/E de 3 a 6, y
concluyeron igualmente que el aumento de la relación S/E en este intervalo favorece
la reacción WGS. Similares tendencias en la selectividad de los productos carbonados
han sido obtenidas por Li y cols. (2012) para el catalizador de Ni/ZrO2 con tiempo
espacial alto (~ 0.43 gcat/hgEtOH). Estos autores señalan que cuando la tendencia de
aumento del CO2 es comparable con la disminución de metano existe una competencia
entre las reacciones WGS y de metanación, y que a su vez, en estas condiciones tanto
el CO2 como el metano son productos secundarios y no primarios de la reacción.
Cabe señalar que no se muestran selectividades de acetaldehído ni de etileno en
la Figura 4.9 porque no se ha detectado su formación en estas condiciones de
operación. En condiciones en las que sí se detecta su presencia en el medio de
reacción (tiempos espaciales < 0.18 gcat/hgEtOH), la selectividad de etileno y
acetaldehído disminuye al aumentar la relación S/E.
0
0.2
0.4
0.6
0.8
0 3 6 9 12
SI
S/E
H2
CO2
CH4
CO
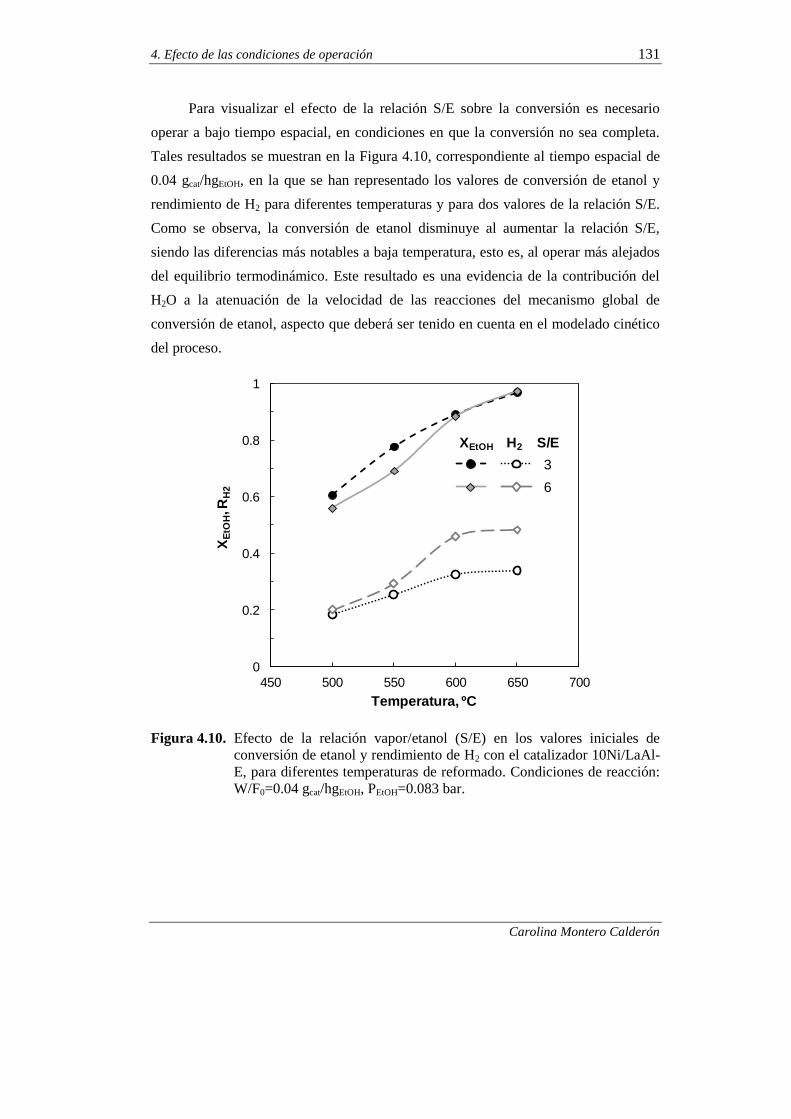
4. Efecto de las condiciones de operación 131
Carolina Montero Calderón
Para visualizar el efecto de la relación S/E sobre la conversión es necesario
operar a bajo tiempo espacial, en condiciones en que la conversión no sea completa.
Tales resultados se muestran en la Figura 4.10, correspondiente al tiempo espacial de
0.04 gcat/hgEtOH, en la que se han representado los valores de conversión de etanol y
rendimiento de H2 para diferentes temperaturas y para dos valores de la relación S/E.
Como se observa, la conversión de etanol disminuye al aumentar la relación S/E,
siendo las diferencias más notables a baja temperatura, esto es, al operar más alejados
del equilibrio termodinámico. Este resultado es una evidencia de la contribución del
H2O a la atenuación de la velocidad de las reacciones del mecanismo global de
conversión de etanol, aspecto que deberá ser tenido en cuenta en el modelado cinético
del proceso.
Figura 4.10. Efecto de la relación vapor/etanol (S/E) en los valores iniciales de
conversión de etanol y rendimiento de H2 con el catalizador 10Ni/LaAl-
E, para diferentes temperaturas de reformado. Condiciones de reacción:
W/F0=0.04 gcat/hgEtOH, PEtOH=0.083 bar.
0
0.2
0.4
0.6
0.8
1
450 500 550 600 650 700
XE
tOH, R
H2
Temperatura, ºC
XEtOH H2 S/E
3
6

132
Capítulo 4
El rendimiento de H2 aumenta apreciablemente con la relación S/E para altas
temperaturas, dado que al tener el agua poco efecto sobre la conversión en este caso
(por ser ésta muy elevada) predomina el efecto beneficioso del exceso de agua sobre la
selectividad de H2, al potenciar las reacciones que lo producen, como se ha
mencionado anteriormente. Sin embargo, a baja temperatura (lejos del equilibrio), el
aumento del rendimiento de H2 con la relación S/E es muy pequeño porque en este
caso es más importante el efecto atenuante del agua sobre la conversión de etanol.
Carrera-Cerritos y cols. (2010) observaron un efecto similar de la relación S/E
para el catalizador 10Ni/12La2O3-Al2O3 preparado por el método sol-gel, operando en
lecho fijo a 600 ºC y tiempo espacial de ~0.10 gcat/hgEtOH, dado que obtuvieron
conversión completa con S/E=3, mientras que con S/E=6 ésta disminuyó hasta 0.89 al
cabo de 1.5 h de reacción, y el rendimiento de H2 no aumentó con la relación S/E.
4.2.2. Estabilidad del catalizador
4.2.2.1. Efecto de la temperatura
El efecto de la temperatura en la estabilidad del catalizador 10Ni/LaAl-E se
muestra en las Figuras 4.11 y 4.12, en las que se ha representado la evolución con el
tiempo de la conversión y el rendimiento de H2 (Figura 4.11) y de los productos
carbonados (Figura 4.12), para tres temperatura de operación, y operando con relación
S/E=3 y elevado tiempo espacial (0.18 gcath/gEtOH). Debe indicarse que no se muestran
los rendimientos de etileno, dado que no se ha detectado este compuesto en estas
condiciones de operación, con elevado valor del tiempo espacial.
Los resultados mostrados indican una más rápida desactivación cuanto menor es
la temperatura de reacción, dado que a 500 ºC la conversión disminuye muy poco
durante las primeras 8-10 h, pero cae de forma pronunciada y casi linealmente con el
tiempo a partir de 10 h de reacción. A 600 ºC la disminución de la conversión es solo
ligeramente apreciable a partir de 15 h de reacción, mientras que a 650 ºC no se
observa aparentemente desactivación, obteniéndose conversión completa durante toda
la reacción.
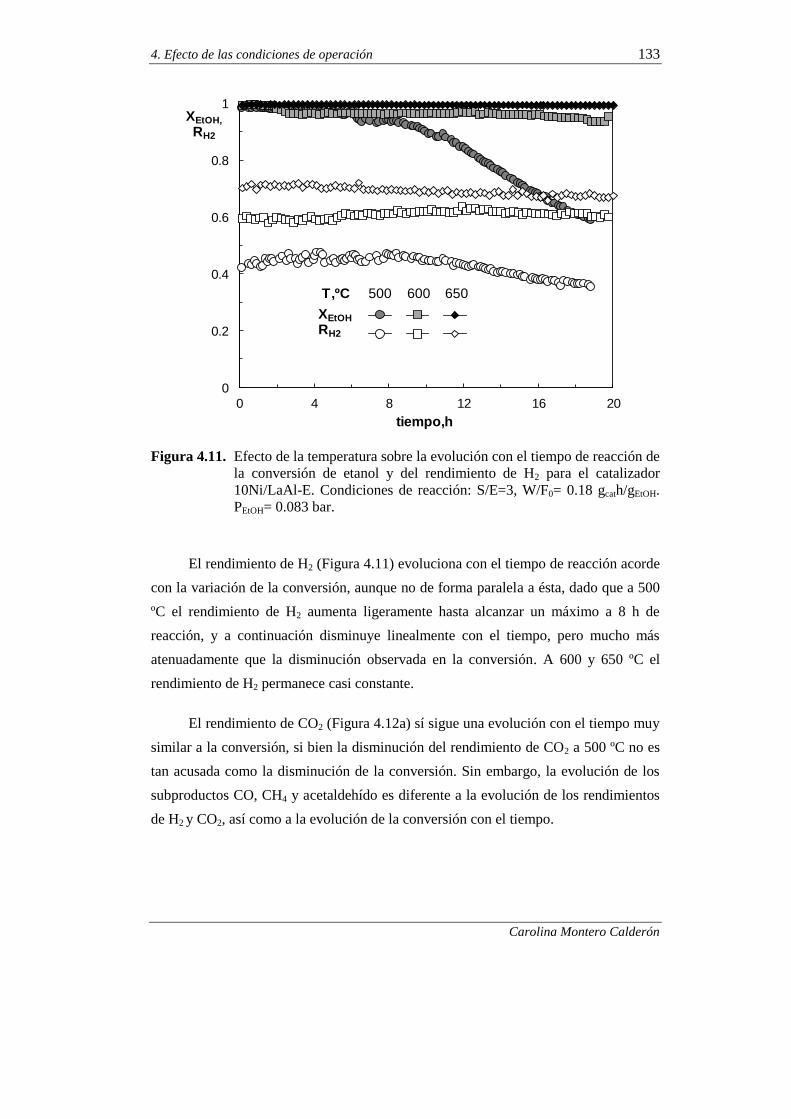
4. Efecto de las condiciones de operación 133
Carolina Montero Calderón
Figura 4.11. Efecto de la temperatura sobre la evolución con el tiempo de reacción de
la conversión de etanol y del rendimiento de H2 para el catalizador
10Ni/LaAl-E. Condiciones de reacción: S/E=3, W/F0= 0.18 gcath/gEtOH.
PEtOH= 0.083 bar.
El rendimiento de H2 (Figura 4.11) evoluciona con el tiempo de reacción acorde
con la variación de la conversión, aunque no de forma paralela a ésta, dado que a 500
ºC el rendimiento de H2 aumenta ligeramente hasta alcanzar un máximo a 8 h de
reacción, y a continuación disminuye linealmente con el tiempo, pero mucho más
atenuadamente que la disminución observada en la conversión. A 600 y 650 ºC el
rendimiento de H2 permanece casi constante.
El rendimiento de CO2 (Figura 4.12a) sí sigue una evolución con el tiempo muy
similar a la conversión, si bien la disminución del rendimiento de CO2 a 500 ºC no es
tan acusada como la disminución de la conversión. Sin embargo, la evolución de los
subproductos CO, CH4 y acetaldehído es diferente a la evolución de los rendimientos
de H2 y CO2, así como a la evolución de la conversión con el tiempo.
0
0.2
0.4
0.6
0.8
1
0 4 8 12 16 20
XEtOH,
RH2
tiempo,h
T,ºC 500 600 650
XEtOH
RH2
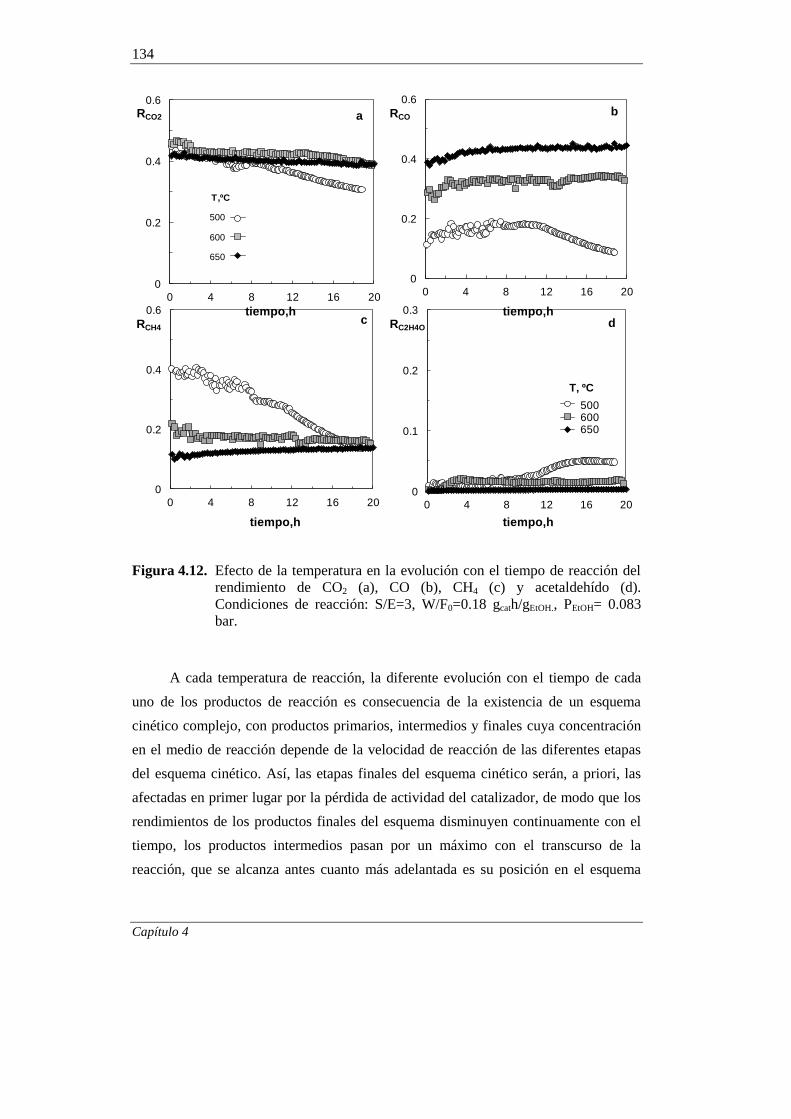
134
Capítulo 4
Figura 4.12. Efecto de la temperatura en la evolución con el tiempo de reacción del
rendimiento de CO2 (a), CO (b), CH4 (c) y acetaldehído (d).
Condiciones de reacción: S/E=3, W/F0=0.18 gcath/gEtOH., PEtOH= 0.083
bar.
A cada temperatura de reacción, la diferente evolución con el tiempo de cada
uno de los productos de reacción es consecuencia de la existencia de un esquema
cinético complejo, con productos primarios, intermedios y finales cuya concentración
en el medio de reacción depende de la velocidad de reacción de las diferentes etapas
del esquema cinético. Así, las etapas finales del esquema cinético serán, a priori, las
afectadas en primer lugar por la pérdida de actividad del catalizador, de modo que los
rendimientos de los productos finales del esquema disminuyen continuamente con el
tiempo, los productos intermedios pasan por un máximo con el transcurso de la
reacción, que se alcanza antes cuanto más adelantada es su posición en el esquema
0
0.2
0.4
0.6
0 4 8 12 16 20
RCO2
tiempo,h
a
T,ºC
500
600
650
0
0.2
0.4
0.6
0 4 8 12 16 20
RCO
tiempo,h
b
0
0.2
0.4
0.6
0 4 8 12 16 20
RCH4
tiempo,h
c
0
0.1
0.2
0.3
0 4 8 12 16 20
RC2H4O
tiempo,h
d
500600650
T, ºC

4. Efecto de las condiciones de operación 135
Carolina Montero Calderón
cinético, y el rendimiento de reactantes aumenta con el tiempo (Aguayo y cols., 2002;
Gayubo y cols., 2004a,b, 2010a,b, 2011, 2012; Valle y cols., 2010; Mier y cols., 2011;
Epelde y cols., 2014a,b). Este es el comportamiento que cabe esperar para un proceso
con desactivación no selectiva del catalizador (una misma actividad para todas las
etapas del esquema cinético), si bien puede ser alterado en el caso de desactivación
selectiva, en el que unas etapas pueden sufrir una más rápida desactivación que el
resto, lo que podría originar una rápida disminución de algunos productos intermedios
en el esquema cinético. Estos aspectos deben ser tenidos en cuenta en el modelado
cinético del proceso, que se aborda en el último apartado de esta Tesis.
Analizando la Figura 4.12, a 500 ºC se observa que el rendimiento de CO
(Figura 4.12b) pasa por un máximo con el transcurso de la reacción, empezando con
un valor de 0.11 a t=0h, llegando a 0.18 en el intervalo 10-12 h, y disminuyendo
posteriormente de forma lineal con el tiempo. Este comportamiento es
cualitativamente similar para las otras temperaturas estudiadas, si bien el máximo se
alcanza a mayor tiempo de reacción a medida que aumenta la temperatura, de modo
que a 600 ºC el máximo en el rendimiento de CO se obtiene prácticamente al final del
periodo de 20 h estudiado, y a 650 ºC se observa un aumento continuado del
rendimiento de CO (de forma asintótica) durante las 20 h de reacción.
El máximo obtenido a 500 ºC se explica por el carácter de producto intermedio
del CO a esta temperatura, de modo que al inicio de la reacción, cuando la conversión
de etanol es completa, pero empieza a ser manifiesta la desactivación de las etapas
finales de transformación de CO a esta temperatura (WGS y metanación), se produce
un aumento del rendimiento de CO. Cuando la desactivación es tan notable que afecta
ya a las etapas primeras del esquema cinético (deshidrogenación para formar
acetaldehído y descomposición de éste y de etanol para formar CO y CH4), entonces
disminuye el rendimiento de CO.
El rendimiento de CH4 (Figura 4.12c) a 500 ºC disminuye continuamente con el
tiempo, lentamente al comienzo de la reacción y más rápidamente, y de forma casi
lineal, a partir de 8-10 h de reacción, cuando la disminución de la conversión es ya
notable. Este resultado es indicativo del carácter de producto final de CH4 en estas
condiciones de reacción y puede explicarse porque a esta temperatura en el sistema de

136
Capítulo 4
reacciones de metanación y de reformado de metano, está favorecida la reacción de
metanación, y ésta es una reacción relativamente lenta, por lo que será rápidamente
afectada por la desactivación del catalizador, con lo que disminuye el rendimiento de
metano desde el inicio de la reacción.
A alta temperatura (650 ºC), para la cual son muy rápidas las reacciones de
descomposición para formar CO y CH4, hay un ligero y continuado aumento del
rendimiento de ambos compuestos con el tiempo, que parece indicar que estas
reacciones de descomposición sufren una desactivación más lenta que las reacciones
que conllevan transformación de CO y CH4, como la WGS o el reformado de metano.
Por encima de 600 ºC no hay prácticamente formación de acetaldehído (Figura
4.12d), lo que se explica porque es un producto primario en el esquema de reacción,
que desaparece por descomposición para formar CO y CH4, y puede también
contemplarse su reacción de reformado, siendo estas reacciones irreversibles y muy
rápidas a estas altas temperaturas. Sin embargo, a 500 ºC si comienza a observarse la
formación de acetaldehído a partir de 8 h de reacción, cuando la desactivación del
catalizador afecta de forma evidente a las reacciones de su transformación (debido a
que, al ser baja la temperatura, el tiempo espacial es menor del necesario para
conversión completa), y el rendimiento de este producto aumenta tendiendo
asintóticamente a un máximo, correspondiente al momento en que la reacción de
deshidrogenación se vea afectada por la desactivación.
Estas tendencias observadas son cualitativamente similares para las relaciones
S/E =6 y 9, aunque las variaciones con el tiempo de la conversión y de los
rendimientos son más lentas al aumentar la relación S/E, como se muestra en el
siguiente apartado.
No obstante, para extraer conclusiones sobre la mayor o menor desactivación
observada a cada temperatura, debe tenerse en cuenta que la velocidad de disminución
de la conversión con el tiempo es altamente dependiente del nivel de conversión
alcanzado. Si se opera con exceso de catalizador respecto del necesario para alcanzar
las conversiones de equilibrio de las diferentes etapas del esquema cinético, tanto la
conversión a la salida del reactor como los rendimientos de productos permanecerán

4. Efecto de las condiciones de operación 137
Carolina Montero Calderón
constantes durante un cierto tiempo (el necesario para que el exceso de centros activos
haya sido desactivado), a partir del cual comenzarán a disminuir la conversión o a
observarse variaciones en la distribución de productos de reacción. Para un tiempo
espacial dado, el exceso de catalizador respecto del necesario para alcanzar la
conversión completa es mayor a medida que aumenta la temperatura, dado que
aumenta la velocidad de las reacciones, por lo que es esperable que la conversión se
mantenga constante durante un mayor tiempo de reacción. En consecuencia, para
apreciar bien el efecto de la temperatura en la estabilidad sería necesario comparar
ensayos realizados en condiciones de igual extensión de la reacción para cada
temperatura, lo que es difícil de conseguir, dado que deberán utilizarse diferentes
valores (desconocidos) de tiempo espacial a cada temperatura.
Para tener una visión más amplia del efecto de la temperatura en la estabilidad
del catalizador, considerando también condiciones de baja conversión, se ha
comparado la evolución con el tiempo de los índices de reacción obtenidos a diferente
temperatura para valores muy bajos del tiempo espacial, de 0.02 gcath/gEtOH, (esto es,
suficientemente alejado del régimen termodinámico), y los resultados se muestran en
la Figura 4.13.
Como se observa en la Figura 4.13, para un valor muy bajo del tiempo espacial
se produce una muy rápida desactivación del catalizador en las 2 primeras horas de
reacción, que es más rápida a medida que aumenta la temperatura, de modo que a 650
ºC (Figura 4.13b) el catalizador está totalmente desactivado tras 4 h de reacción, y a
partir de ese momento la conversión de etanol y los rendimientos de productos se
mantienen casi constantes e iguales a los valores correspondientes a la conversión por
rutas térmicas. Sin embargo, a menor temperatura (Figura 4.13a) la desactivación
continúa a lo largo del tiempo, si bien de forma mucho más atenuada a partir de 4 h de
reacción.
Dada la rápida desactivación, y la poca extensión de todas las reacciones del
esquema cinético en estas condiciones, el rendimiento de los productos de reacción
disminuye continuamente con el tiempo, con excepción del rendimiento de
acetaldehído (primer producto de reacción), que pasa por un ligero máximo al inicio
de la reacción para la mayor temperatura estudiada.
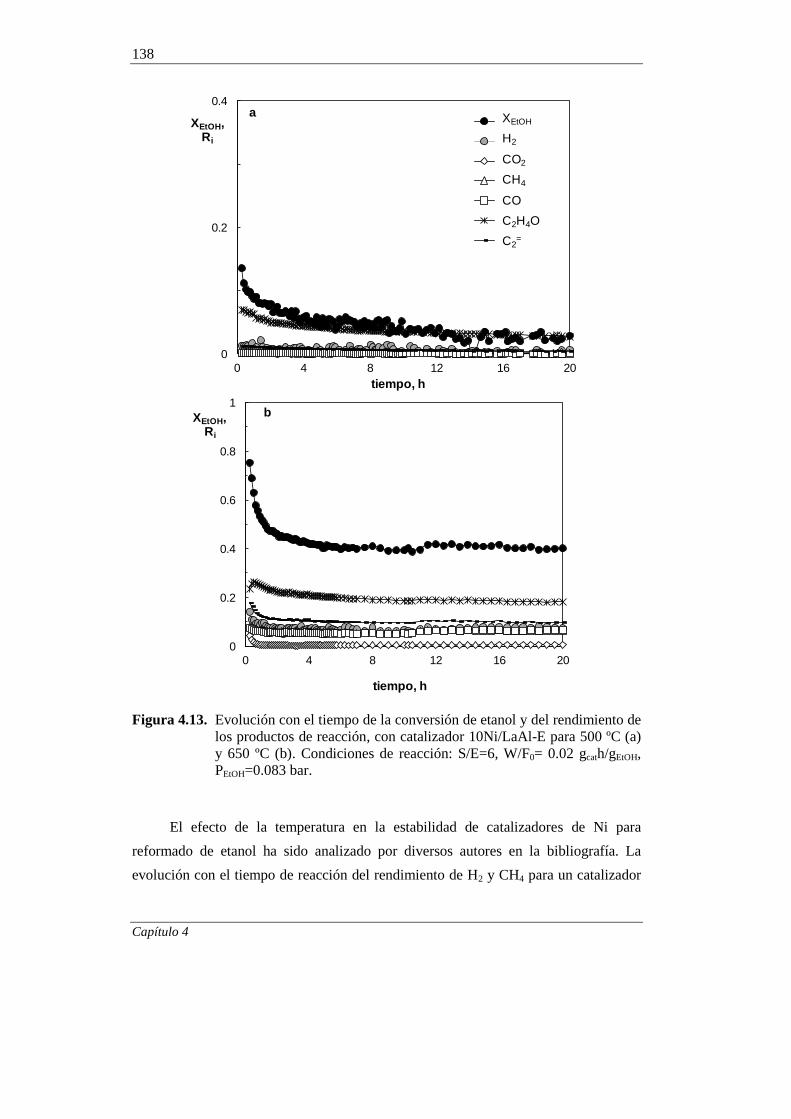
138
Capítulo 4
Figura 4.13. Evolución con el tiempo de la conversión de etanol y del rendimiento de
los productos de reacción, con catalizador 10Ni/LaAl-E para 500 ºC (a)
y 650 ºC (b). Condiciones de reacción: S/E=6, W/F0= 0.02 gcath/gEtOH,
PEtOH=0.083 bar.
El efecto de la temperatura en la estabilidad de catalizadores de Ni para
reformado de etanol ha sido analizado por diversos autores en la bibliografía. La
evolución con el tiempo de reacción del rendimiento de H2 y CH4 para un catalizador
0
0.2
0.4
0 4 8 12 16 20
XEtOH,Ri
tiempo, h
XEtOH
H2
CO2
CH4
CO
C2H4O
C2=
a
0
0.2
0.4
0.6
0.8
1
0 4 8 12 16 20
XEtOH,Ri
tiempo, h
b

4. Efecto de las condiciones de operación 139
Carolina Montero Calderón
de NiLaZr y relación S/E=9 ha sido analizada por Bussi y cols. (2013) para una
velocidad espacial (GHSV) de 41000 h−1
, quienes señalan que el rendimiento de H2
tiende a ser constante durante las 8 primeras horas de reacción a 650 ºC, pero
observando que a 500 ºC disminuye ligeramente desde ~0.42 hasta ~0.33. El
rendimiento de metano disminuye de manera continua con el tiempo de reacción, lo
que según los autores es debido a cambios en las propiedades superficiales por
desactivación del catalizador. Para el catalizador de Ni/Ce2O3/Zr2O3 y S/E=9, Patel y
cols. (2013) observan que a las temperaturas estudiadas (600-700 ºC) no hay
variaciones apreciables del rendimiento de H2 y de la conversión de etanol con el
tiempo de reacción (6 h). Señalan que esta estabilidad del catalizar puede deberse a la
capacidad de almacenamiento del oxígeno por el Ce presente en el soporte, que
ayudaría a la gasificación del carbón depositado en los centros activos. Además,
señalan que la combinación Ce-Zr brinda resistencia térmica y mecánica al catalizador
y promueve la reacción WGS.
4.2.2.2. Efecto de la relación molar S/E
En la Figura 4.14 se muestra la evolución con el tiempo de reacción de la
conversión de etanol y del rendimiento de productos, para dos valores de la relación
molar vapor/etanol (S/E). Los resultados ponen de manifiesto la gran atenuación de la
desactivación que conlleva el aumento de la relación molar S/E. Si bien no se muestra,
cabe indicar que la diferencia es especialmente en el intervalo entre S/E= 3 y 6,
mientras que las diferencias son menos notables entre S/E = 6 y 9. Como resultado,
para S/E=9 tanto la conversión como los rendimientos de productos permanecen
prácticamente constantes durante las 20 h de reacción. Como se mostrará en el
Capítulo 5, esta atenuación de la desactivación está asociada a una atenuación de la
deposición de coque, que es la única causa de la desactivación del catalizador a esta
relativamente baja temperatura de reformado. Similar efecto de la relación S/E sobre
la desactivación se ha observado a otras temperaturas y tiempos espaciales.
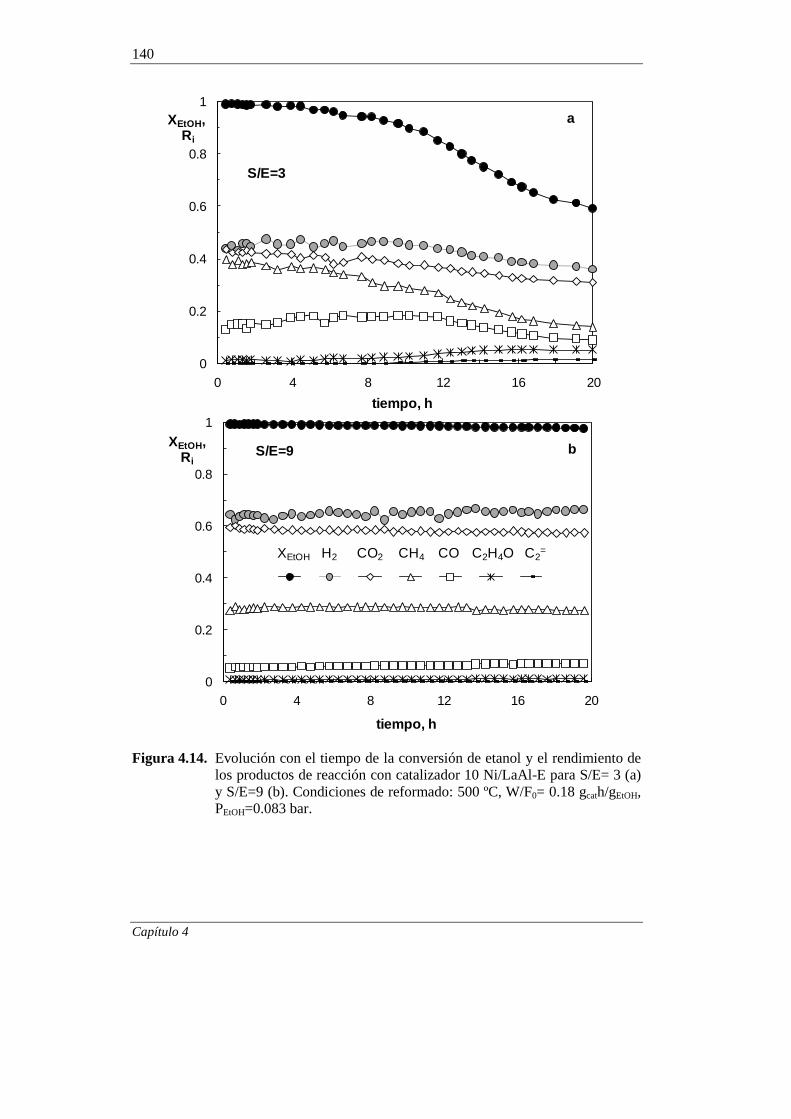
140
Capítulo 4
Figura 4.14. Evolución con el tiempo de la conversión de etanol y el rendimiento de
los productos de reacción con catalizador 10 Ni/LaAl-E para S/E= 3 (a)
y S/E=9 (b). Condiciones de reformado: 500 ºC, W/F0= 0.18 gcath/gEtOH,
PEtOH=0.083 bar.
0
0.2
0.4
0.6
0.8
1
0 4 8 12 16 20
XEtOH,Ri
tiempo, h
S/E=3
a
0
0.2
0.4
0.6
0.8
1
0 4 8 12 16 20
XEtOH,Ri
tiempo, h
S/E=9
XEtOH H2 CO2 CH4 CO C2H4O C2=
b

4. Efecto de las condiciones de operación 141
Carolina Montero Calderón
4.2.2.3. Efecto del tiempo espacial
El efecto del tiempo espacial sobre la velocidad de desactivación se muestra en
las Figuras 4.15-4-17, correspondientes cada una a una temperatura de operación y
para S/E=6. La Figura 4.15 muestra la evolución con el tiempo de la conversión de
etanol y del rendimiento de H2 (Figura 4.15a) y los rendimientos de CO, CH4 y
acetaldehído como subproductos de reacción (Figura 4.15b) a 500 ºC y para dos
valores diferentes del tiempo espacial. Las Figuras 4.16 y 4.17 muestran la evolución
con el tiempo, para diferentes valores del tiempo espacial, de la conversión del etanol
(a), rendimiento de H2 (b), CO (c), CH4 (d), acetaldehído (e) y etileno (f) para 550 y
600 ºC, respectivamente. El rendimiento de CO2 no ha sido representado debido a que
tiene similar tendencia que el rendimiento de H2.
Para todas las temperaturas se observa un rápido descenso de la conversión de
etanol y de los rendimientos de H2 y de subproductos carbonados a bajo tiempo
espacial, que indica que en tales condiciones se produce una muy rápida desactivación
del catalizador. Los valores de conversión y rendimientos descienden bruscamente en
las primeras 2-4 h de reacción, más rápido cuanto menor es la conversión inicial, y
posteriormente más atenuadamente, alcanzando los niveles correspondientes a la
conversión debida al efecto térmico. Por tanto, para el catalizador totalmente
desactivado el único subproducto que se obtiene en cantidades apreciables a 500 ºC es
acetaldehído. Se obtienen también rendimientos apreciables de etileno a 550 ºC y a
600 ºC se observan un pequeño rendimiento residual de H2 obtenido por reacciones
térmicas.
El descenso de la conversión y las variaciones de rendimientos de productos se
van atenuando a medida que aumenta el tiempo espacial, y por encima de 0.18
gcath/gEtOH la desactivación es sumamente lenta, especialmente a mayor temperatura,
debido al anteriormente comentado efecto de la temperatura sobre la desactivación.
La ausencia de acetaldehído y etileno para alto tiempo espacial a 600 ºC, se
debe a que tales condiciones corresponden a un gran exceso de catalizador respecto al
necesario para conversión completa, por lo que la extensión de las reacciones de
transformación de ambos hidrocarburos (descomposición y reformado) es completa.
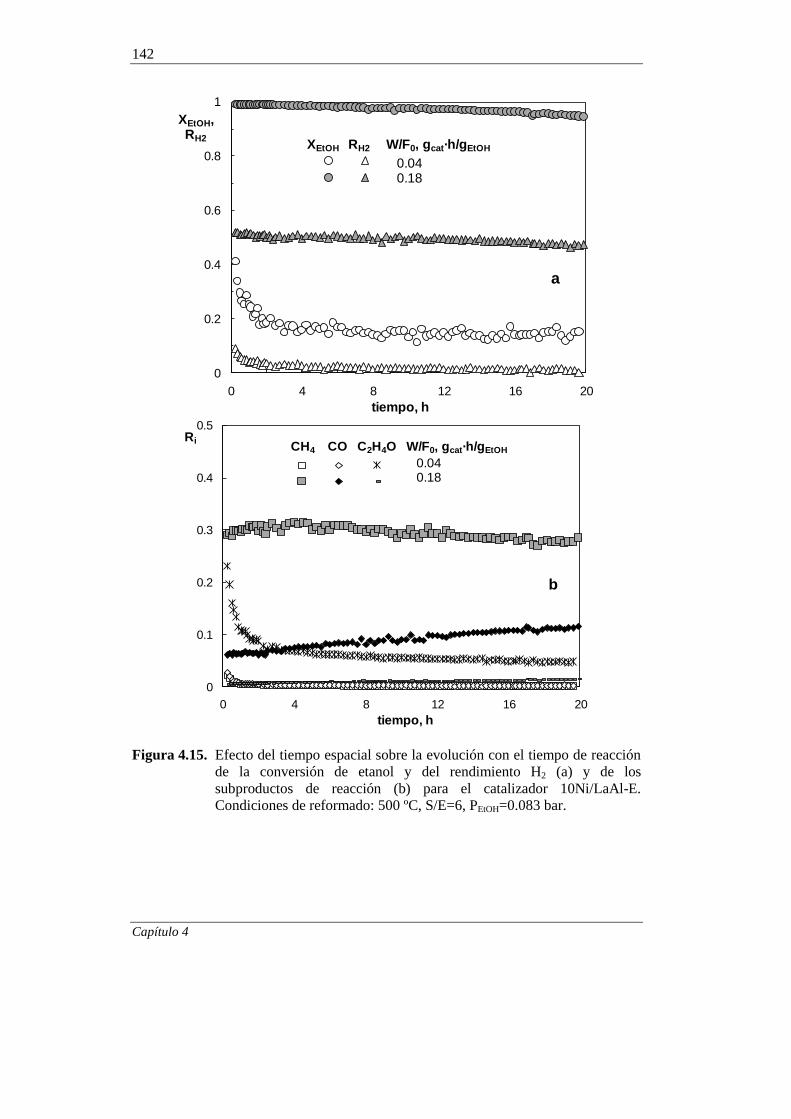
142
Capítulo 4
Figura 4.15. Efecto del tiempo espacial sobre la evolución con el tiempo de reacción
de la conversión de etanol y del rendimiento H2 (a) y de los
subproductos de reacción (b) para el catalizador 10Ni/LaAl-E.
Condiciones de reformado: 500 ºC, S/E=6, PEtOH=0.083 bar.
0
0.2
0.4
0.6
0.8
1
0 4 8 12 16 20
XEtOH,RH2
tiempo, h
XEtOH RH2 W/F0, gcat·h/gEtOH
0.040.18
a
0
0.1
0.2
0.3
0.4
0.5
0 4 8 12 16 20
Ri
tiempo, h
CH4 CO C2H4O W/F0, gcat·h/gEtOH
0.040.18
b
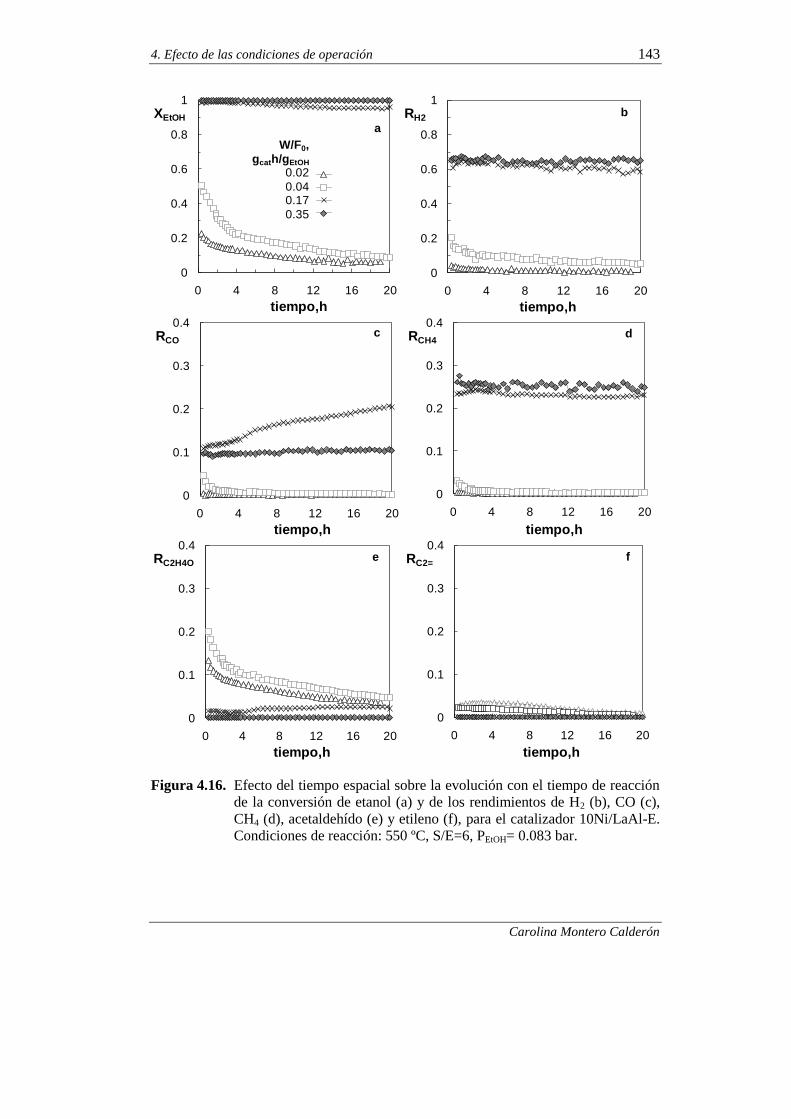
4. Efecto de las condiciones de operación 143
Carolina Montero Calderón
Figura 4.16. Efecto del tiempo espacial sobre la evolución con el tiempo de reacción
de la conversión de etanol (a) y de los rendimientos de H2 (b), CO (c),
CH4 (d), acetaldehído (e) y etileno (f), para el catalizador 10Ni/LaAl-E.
Condiciones de reacción: 550 ºC, S/E=6, PEtOH= 0.083 bar.
0
0.2
0.4
0.6
0.8
1
0 4 8 12 16 20
XEtOH
tiempo,h
a
W/F0,
gcath/gEtOH
0.02
0.040.17
0.35
0
0.2
0.4
0.6
0.8
1
0 4 8 12 16 20
RH2
tiempo,h
b
0
0.1
0.2
0.3
0.4
0 4 8 12 16 20
RCO
tiempo,h
c
0
0.1
0.2
0.3
0.4
0 4 8 12 16 20
RCH4
tiempo,h
d
0
0.1
0.2
0.3
0.4
0 4 8 12 16 20
RC2H4O
tiempo,h
e
0
0.1
0.2
0.3
0.4
0 4 8 12 16 20
RC2=
tiempo,h
f
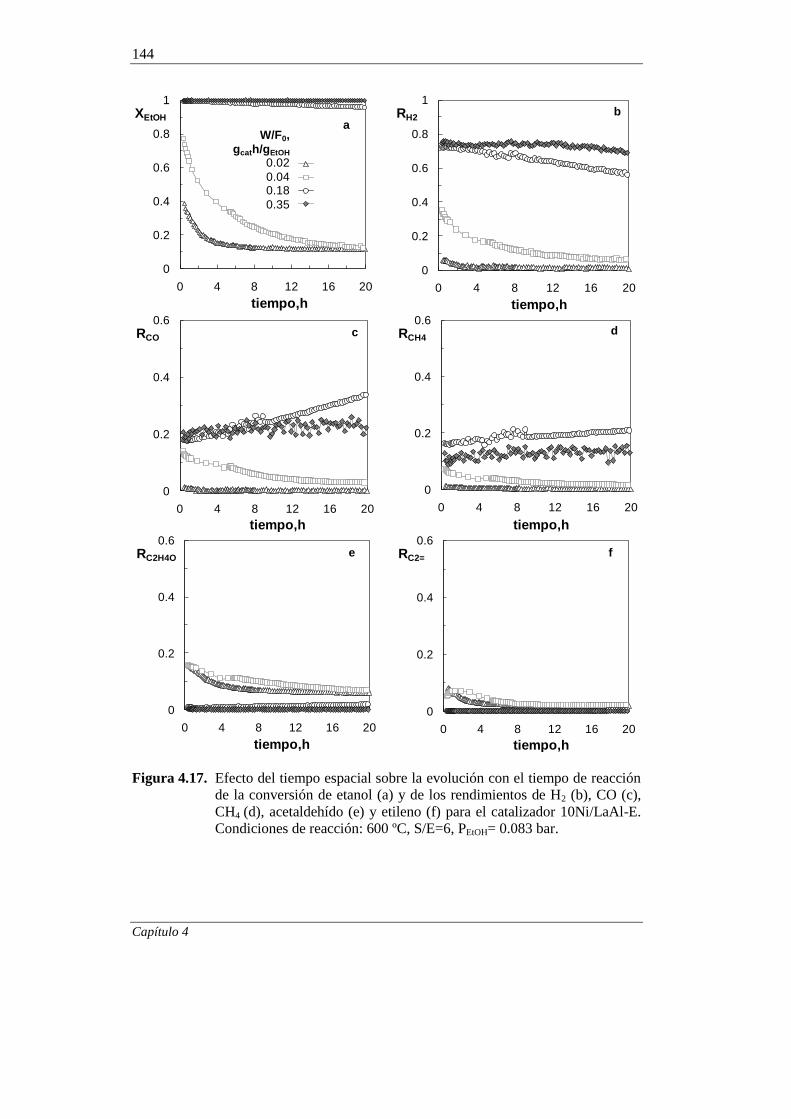
144
Capítulo 4
Figura 4.17. Efecto del tiempo espacial sobre la evolución con el tiempo de reacción
de la conversión de etanol (a) y de los rendimientos de H2 (b), CO (c),
CH4 (d), acetaldehído (e) y etileno (f) para el catalizador 10Ni/LaAl-E.
Condiciones de reacción: 600 ºC, S/E=6, PEtOH= 0.083 bar.
0
0.2
0.4
0.6
0.8
1
0 4 8 12 16 20
XEtOH
tiempo,h
aW/F0,
gcath/gEtOH
0.02
0.040.18
0.35
0
0.2
0.4
0.6
0.8
1
0 4 8 12 16 20
RH2
tiempo,h
b
0
0.2
0.4
0.6
0 4 8 12 16 20
RCO
tiempo,h
c
0
0.2
0.4
0.6
0 4 8 12 16 20
RCH4
tiempo,h
d
0
0.2
0.4
0.6
0 4 8 12 16 20
RC2H4O
tiempo,h
e
0
0.2
0.4
0.6
0 4 8 12 16 20
RC2=
tiempo,h
f

4. Efecto de las condiciones de operación 145
Carolina Montero Calderón
Estos resultados ponen de manifiesto la importante contribución a la
desactivación del catalizador de los productos intermedios de reacción (acetaldehído y
etileno, especialmente el primero, por su mayor concentración en el medio de
reacción), así como del propio reactante (el etanol), dada la rápida desactivación
observada cuando la concentración de todos ellos es elevada. Por el contrario, los
subproductos CO y CH4 tienen escasa contribución a la desactivación del catalizador,
dado que ésta se atenúa cuando aumentan las concentraciones de ambos productos
carbonosos.
Para corroborar el papel del acetaldehído en la desactivación se realizaron dos
ensayos de reformado con vapor de acetaldehído (SRA), en las siguientes condiciones:
600 ºC; relación molar S/Ac= 12.3; W/F0 = 0.04 gcat y 0.21gcat (b); PAc= 0.083 bar. Los
resultados de conversión y rendimientos de productos de reacción obtenidos en ambos
ensayos se muestran en la Figura 4.18.
A bajo tiempo espacial (Figura 4.18a) el acetaldehído es el componentes
mayoritario en el medio de reacción, y se produce una muy rápida desactivación, que
es total al cabo de unos 40 min, permaneciendo luego constantes la composiciones con
el tiempo en los valores correspondientes a la conversión debida el efecto térmico.
Para elevado tiempo espacial (Figura 4.18b) se promueve las reacciones de
transformación de acetaldehído (descomposición y reformado), así como de los
productos derivados de la descomposición del acetaldehído (CO y CH4), de modo que
la conversión es prácticamente completa y permanece casi constante durante el ensayo
realizado a elevado tiempo espacial. No obstante, los rendimientos de productos sí
varían apreciablemente durante la reacción para este elevado valor del tiempo
espacial, resultado que pone de manifiesto la desactivación del catalizador, y que
dicha desactivación afecta de diferente forma a las diferentes etapas de reacción, de
modo que los productos finales (H2 y CO2) disminuyen, mientras que los productos
intermedios (CO y CH4) aumentan con el tiempo, resultado que indica una mucho más
rápida desactivación para las reacciones WGS y de reformado de metano que para la
descomposición de acetaldehído.
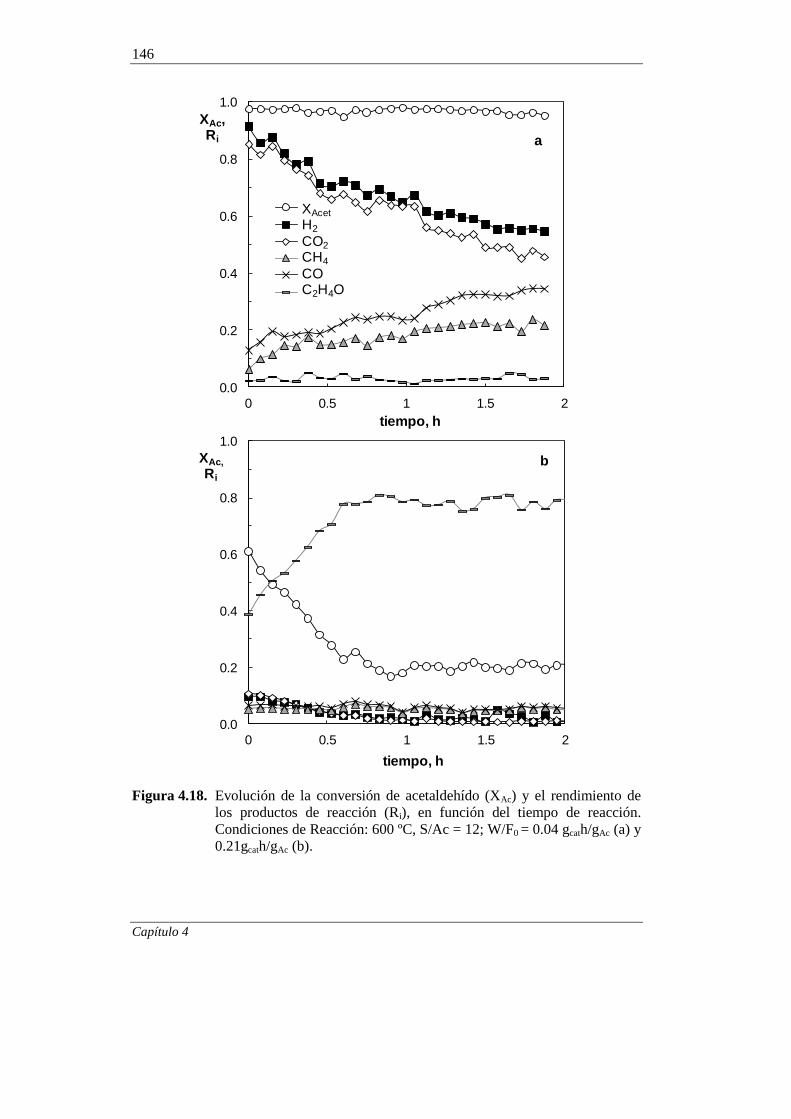
146
Capítulo 4
Figura 4.18. Evolución de la conversión de acetaldehído (XAc) y el rendimiento de
los productos de reacción (Ri), en función del tiempo de reacción.
Condiciones de Reacción: 600 ºC, S/Ac = 12; W/F0 = 0.04 gcath/gAc (a) y
0.21gcath/gAc (b).
0.0
0.2
0.4
0.6
0.8
1.0
0 0.5 1 1.5 2
XAc,Ri
tiempo, h
XAcet
H2
CO2
CH4
COC2H4O
a
0.0
0.2
0.4
0.6
0.8
1.0
0 0.5 1 1.5 2
XAc,
Ri
tiempo, h
b

4. Efecto de las condiciones de operación 147
Carolina Montero Calderón
4.2.2.4. Ensayos de muy larga duración
Como se ha señalado previamente, a altos tiempos espaciales (> 0.35 gcath/gEtOH)
se opera en condiciones cercanas al equilibrio termodinámico y tras 20 h de reacción
no se ha observado desactivación del catalizador para ninguna de las relaciones S/E y
temperaturas estudiadas.
Con objeto de analizar con mayor detalle la estabilidad del catalizador, se han
realizado experimentos de muy larga duración (hasta 200 h), para diferentes relaciones
S/E y a elevada temperatura (600 y 650 ºC), para poder determinar si la sinterización
es una causa de la desactivación. Los resultados de estos ensayos se muestran en las
Figuras 4.19 y 4.20, que corresponden a 600 y 650 ºC, respectivamente. Se muestra la
evolución con el tiempo de la conversión de etanol y de los rendimientos de H2, CO2,
CO y CH4, que son los únicos productos de reacción detectados en estas condiciones.
La Figura 4.19 pone de manifiesto el ya comentado beneficioso efecto del agua
sobre la reacción de reformado, dado que una mayor relación S/E favorece la
formación de H2, y disminuye la formación de CO y CH4. Analizando la evolución
con el tiempo de los índices de reacción, en ambas figuras se observa que el
catalizador se comporta de forma muy estable, de modo que la conversión permanece
muy elevada (> 0.97) al cabo de 200 h de reacción y es muy pequeña también la
variación de los rendimientos de los productos de reacción, con muy pequeño aumento
o disminución de los mismos, según el carácter de cada componente como producto
intermedio o final en el esquema de reacción en las diferentes condiciones estudiadas.
Se ha cuantificado mediante TPO el contenido de coque depositado en estos
ensayos, y los resultados se presentan en la Tabla 4.1. Analizando estos resultados, se
comprueba que a pesar del elevado contenido de coque el catalizador mantiene su
actividad porque en estas condiciones el coque es de tipo fibrilar y no desactivante
(aspecto que se analiza en detalle en el capítulo siguiente). Se observa igualmente una
atenuación del contenido de coque al aumentar la relación S/E, así como al aumentar
la temperatura. El efecto de las variables de operación sobre la deposición de coque se
estudia con mayor detalle en el capítulo siguiente.
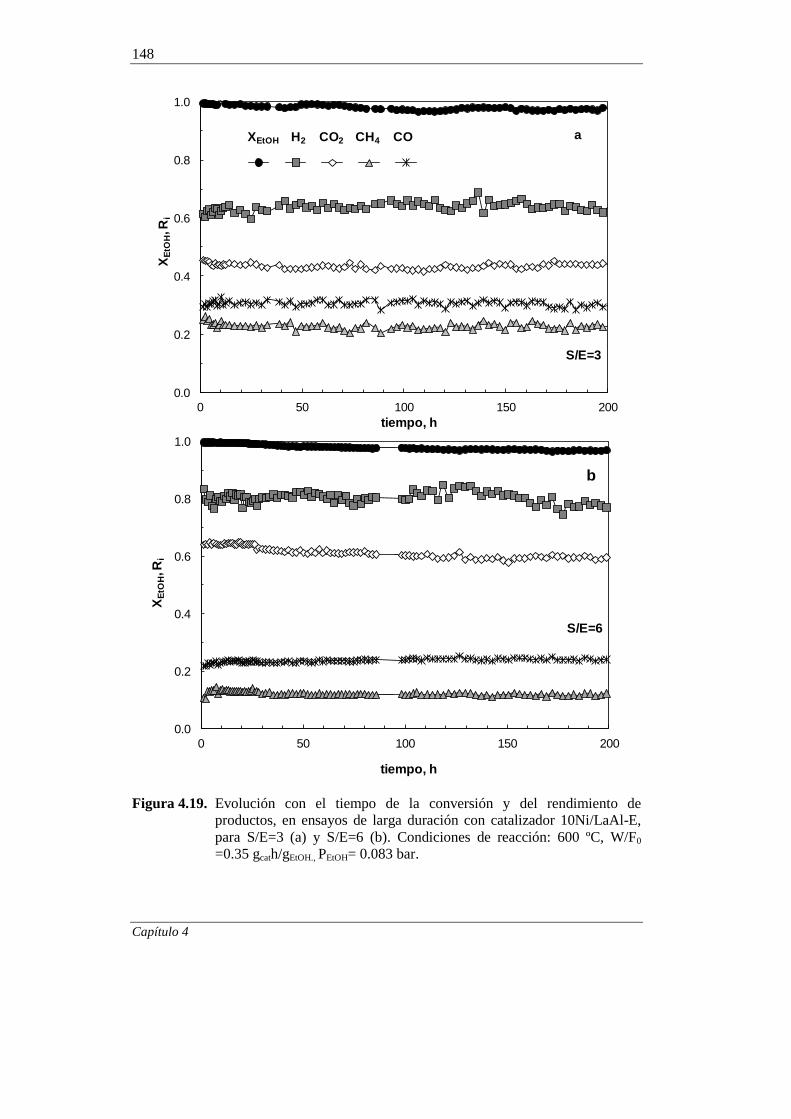
148
Capítulo 4
Figura 4.19. Evolución con el tiempo de la conversión y del rendimiento de
productos, en ensayos de larga duración con catalizador 10Ni/LaAl-E,
para S/E=3 (a) y S/E=6 (b). Condiciones de reacción: 600 ºC, W/F0
=0.35 gcath/gEtOH., PEtOH= 0.083 bar.
0.0
0.2
0.4
0.6
0.8
1.0
0 50 100 150 200
XE
tOH, R
i
tiempo, h
S/E=3
XEtOH H2 CO2 CH4 CO a
0.0
0.2
0.4
0.6
0.8
1.0
0 50 100 150 200
XE
tOH, R
i
tiempo, h
S/E=6
b
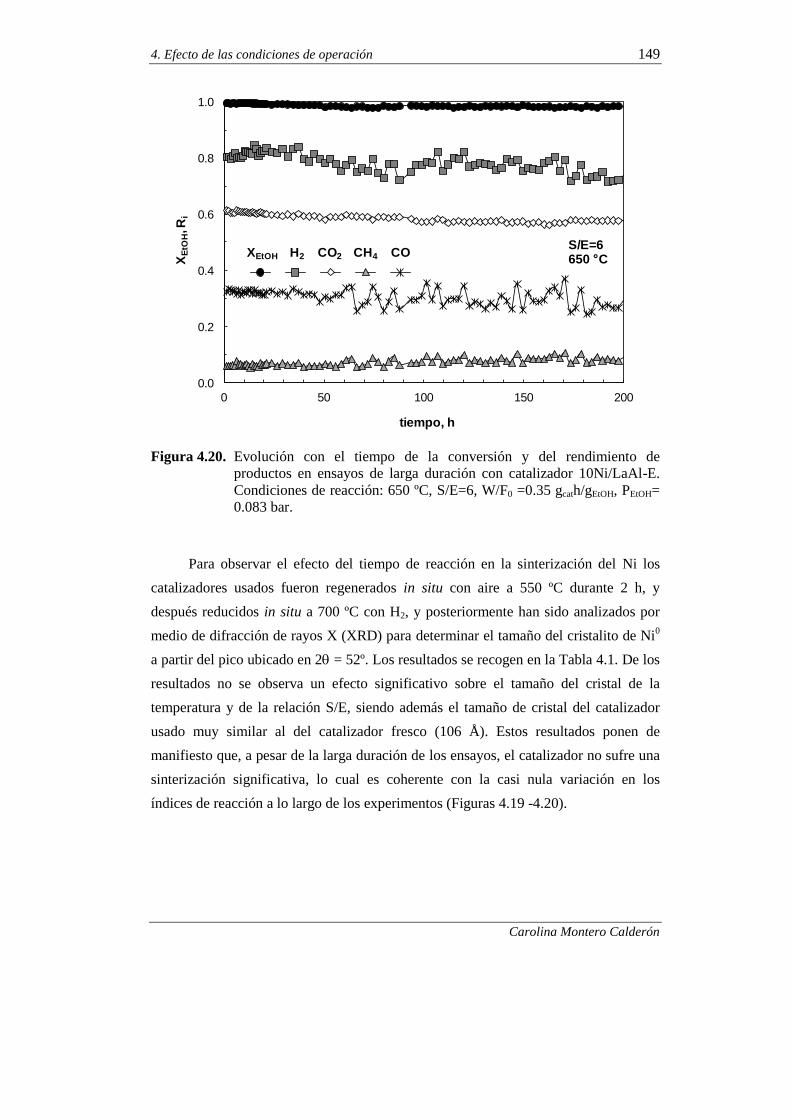
4. Efecto de las condiciones de operación 149
Carolina Montero Calderón
Figura 4.20. Evolución con el tiempo de la conversión y del rendimiento de
productos en ensayos de larga duración con catalizador 10Ni/LaAl-E.
Condiciones de reacción: 650 ºC, S/E=6, W/F0 =0.35 gcath/gEtOH, PEtOH=
0.083 bar.
Para observar el efecto del tiempo de reacción en la sinterización del Ni los
catalizadores usados fueron regenerados in situ con aire a 550 ºC durante 2 h, y
después reducidos in situ a 700 ºC con H2, y posteriormente han sido analizados por
medio de difracción de rayos X (XRD) para determinar el tamaño del cristalito de Ni0
a partir del pico ubicado en 2 = 52º. Los resultados se recogen en la Tabla 4.1. De los
resultados no se observa un efecto significativo sobre el tamaño del cristal de la
temperatura y de la relación S/E, siendo además el tamaño de cristal del catalizador
usado muy similar al del catalizador fresco (106 Å). Estos resultados ponen de
manifiesto que, a pesar de la larga duración de los ensayos, el catalizador no sufre una
sinterización significativa, lo cual es coherente con la casi nula variación en los
índices de reacción a lo largo de los experimentos (Figuras 4.19 -4.20).
0.0
0.2
0.4
0.6
0.8
1.0
0 50 100 150 200
XE
tOH, R
i
tiempo, h
S/E=6650 C
XEtOH H2 CO2 CH4 CO
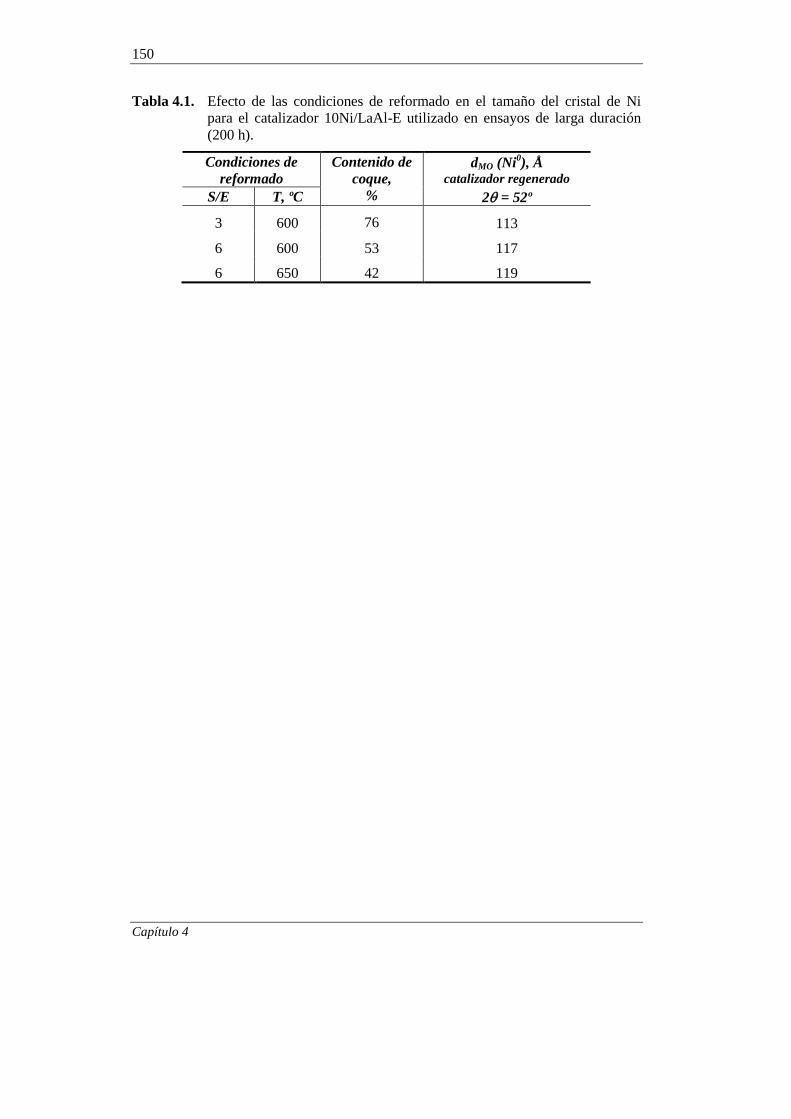
150
Capítulo 4
Tabla 4.1. Efecto de las condiciones de reformado en el tamaño del cristal de Ni
para el catalizador 10Ni/LaAl-E utilizado en ensayos de larga duración
(200 h).
Condiciones de
reformado
Contenido de
coque,
%
dMO (Ni0), Å
catalizador regenerado
S/E T, ºC 2 = 52º
3 600 76 113
6 600 53 117
6 650 42 119

5. ESTUDIO DEL CATALIZADOR
10Ni/LaAl-E DESACTIVADO


153
Carolina Montero Calderón
5. ESTUDIO DEL CATALIZADOR 10Ni/αLaAl-E DESACTIVADO
En este Capítulo se presentan los resultados del estudio del catalizador
10Ni/LaAl-E tras ser utilizado, que se ha centrado principalmente en el análisis del
contenido y naturaleza del coque depositado, al ser ésta es la única causa de la
desactivación del catalizador, dado que, como se ha mostrado en el capítulo anterior,
no tiene lugar una significante sinterización del Ni en las condiciones estudiadas.
La formación de coque en el proceso SRE tiene lugar mediante reacciones
secundarias a las de formación de H2 y del resto de subproductos gaseosos, tal como
se ha descrito en el Apartado 1.5.1, en el que se han identificado las siguientes
reacciones que pueden dar lugar a la formación de coque: i) la reacción de Boudouard,
Ec. (1.24), o disociación del CO, que es exotérmica, por lo que su constante de
equilibrio disminuye al aumentar la temperatura (Trimm, 1997; Alberton y cols.,
2007; Koo y cols., 2014); ii) la descomposición del CH4, Ec. (1.25), que es
endotérmica, por lo que se favorece al aumentar la temperatura de reformado y,
además, da lugar a la producción de nanotubos o nanofibras de carbono (Chen y cols.,
2005; Lobo y cols., 2011; Nasir Udim y cols., 2014); iii) la polimerización de etileno,
Ec. (1.23). También se ha descrito la formación de coque mediante otras reacciones
como la hidrogenación del CO (Ec. 5.1) y la descomposición del etanol (Ec. 5.2) (Li y
cols., 2008; Wang y cols., 2011).
CO + H2 ↔ H2O + C (5.1)
CH3CH2OH → C + CO + 3H2 (5.2)
Recientemente, Vicente y cols. (2014a) han estudiado la desactivación por
coque de catalizadores de Ni y Co, concluyendo que la naturaleza del coque y las
propiedades físicas del soporte son factores determinantes de la desactivación a
temperaturas moderadas (500 ºC), en ausencia de sinterización. Se observó que el
coque está constituido por dos fracciones de diferente naturaleza: a) coque de tipo
monoatómico amorfo y polimérico, con picos en el perfil TPO a temperaturas menores
a 450 ºC, que se denomina “encapsulante” y es el principal causante del bloqueo de
los centros metálicos. Sus precursores son los productos intermedios de reacción

154
Capítulo 5
(acetaldehído y etileno), así como el etanol reactante, por la formación de iones etoxi;
b) coque fibrilar, con picos en el perfil TPO a temperatura superior a 500 ºC, que no
origina desactivación y cuyos precursores son CO y CH4 mediante las reacciones de
Boudouard y la descomposición, respectivamente.
En este capítulo se ha estudiado el efecto de las condiciones de reformado
(temperatura, tiempo espacial, relación molar S/E) sobre la cantidad y naturaleza del
coque depositado en el catalizador 10Ni/LaAl-E y se han contrastado los resultados
con la composición del medio de reacción (mostrada en el Capítulo 4), para
determinar los precursores en la formación de coque. Además, se ha analizado los
cambios de las propiedades del catalizador y en las características del coque con el
transcurso del tiempo de reacción (para igualdad del resto de condiciones de
operación), con el objeto de determinar posibles cambios en la morfología de las
partículas de Ni como consecuencia de la evolución de la deposición de coque. Para
ello, se han empleado diversas técnicas de análisis, que incluyen: i) oxidación a
temperatura programada (TPO), para determinar el contenido de coque, así como su
naturaleza (en base a la posición de los picos de combustión); ii) técnicas
espectroscópicas (espectroscopía FTIR, espectroscopía Raman), análisis XPS e
imágenes SEM y TEM. Las propiedades físicas de los catalizadores desactivados
(superficie específica y distribución de volumen de poros) se han caracterizado
mediante adsorción-desorción de N2, y su estructura cristalina mediante análisis de
XRD. Todas estas técnicas analíticas se han descrito en el Capítulo 2.
Al final del capítulo se recogen los resultados obtenidos en el estudio de la
regeneración del catalizador mediante gasificación, ensayo que se ha llevado a cabo
con la finalidad de analizar alternativas a la combustión con aire para la eliminación
del coque.
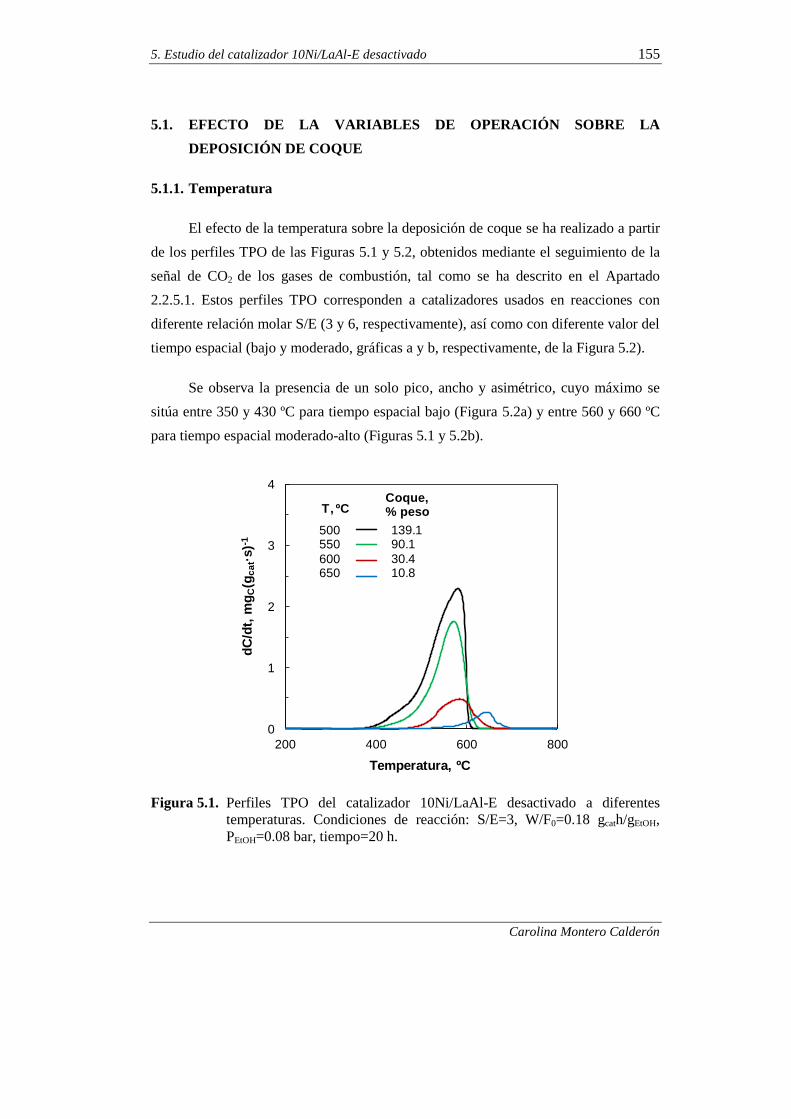
5. Estudio del catalizador 10Ni/LaAl-E desactivado 155
Carolina Montero Calderón
5.1. EFECTO DE LA VARIABLES DE OPERACIÓN SOBRE LA
DEPOSICIÓN DE COQUE
5.1.1. Temperatura
El efecto de la temperatura sobre la deposición de coque se ha realizado a partir
de los perfiles TPO de las Figuras 5.1 y 5.2, obtenidos mediante el seguimiento de la
señal de CO2 de los gases de combustión, tal como se ha descrito en el Apartado
2.2.5.1. Estos perfiles TPO corresponden a catalizadores usados en reacciones con
diferente relación molar S/E (3 y 6, respectivamente), así como con diferente valor del
tiempo espacial (bajo y moderado, gráficas a y b, respectivamente, de la Figura 5.2).
Se observa la presencia de un solo pico, ancho y asimétrico, cuyo máximo se
sitúa entre 350 y 430 ºC para tiempo espacial bajo (Figura 5.2a) y entre 560 y 660 ºC
para tiempo espacial moderado-alto (Figuras 5.1 y 5.2b).
Figura 5.1. Perfiles TPO del catalizador 10Ni/LaAl-E desactivado a diferentes
temperaturas. Condiciones de reacción: S/E=3, W/F0=0.18 gcath/gEtOH,
PEtOH=0.08 bar, tiempo=20 h.
0
1
2
3
4
200 400 600 800
dC
/dt,
mg
C(g
ca
t·s)-
1
Temperatura, ºC
500550
600650
T, ºCCoque,% peso
139.190.1
30.410.8
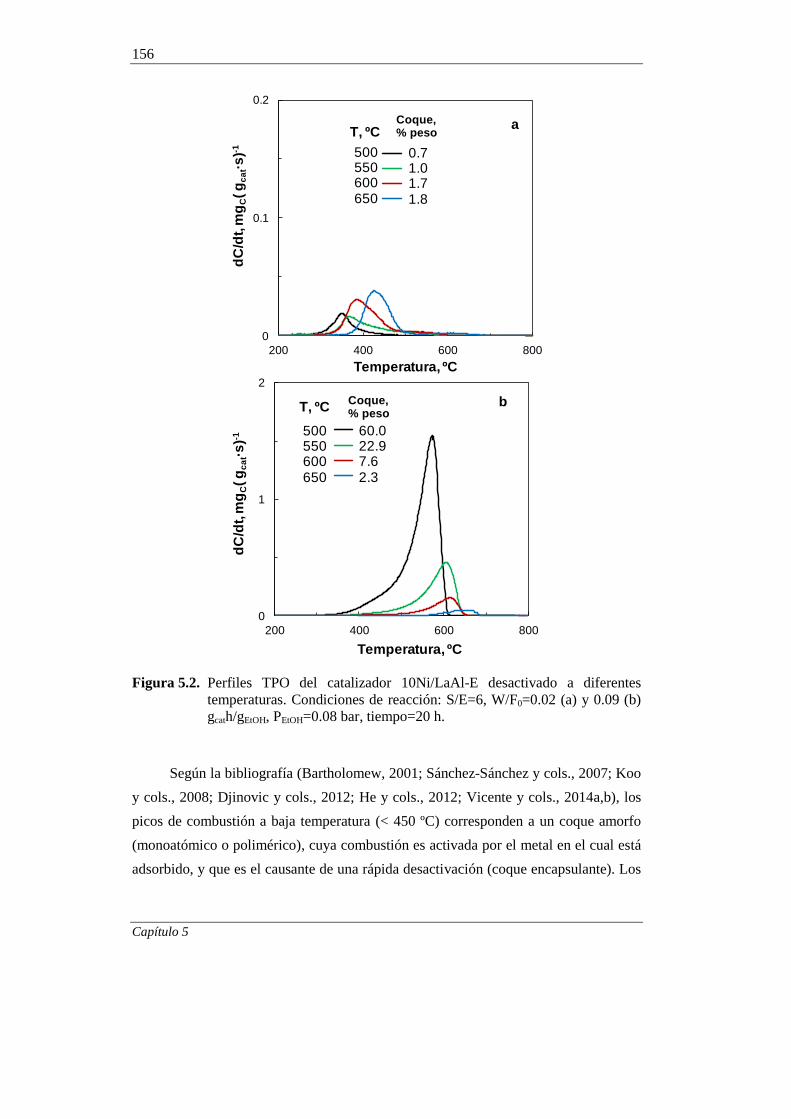
156
Capítulo 5
Figura 5.2. Perfiles TPO del catalizador 10Ni/LaAl-E desactivado a diferentes
temperaturas. Condiciones de reacción: S/E=6, W/F0=0.02 (a) y 0.09 (b)
gcath/gEtOH, PEtOH=0.08 bar, tiempo=20 h.
Según la bibliografía (Bartholomew, 2001; Sánchez-Sánchez y cols., 2007; Koo
y cols., 2008; Djinovic y cols., 2012; He y cols., 2012; Vicente y cols., 2014a,b), los
picos de combustión a baja temperatura (< 450 ºC) corresponden a un coque amorfo
(monoatómico o polimérico), cuya combustión es activada por el metal en el cual está
adsorbido, y que es el causante de una rápida desactivación (coque encapsulante). Los
0
0.1
0.2
200 400 600 800
dC
/dt,
mg
C( g
ca
t·s
)-1
Temperatura, ºC
500550600650
T, ºCCoque,% peso
0.71.01.71.8
a
0
1
2
200 400 600 800
dC
/dt,
mg
C( g
ca
t·s
)-1
Temperatura, ºC
500550600650
T, ºCCoque,% peso
60.022.97.62.3
b

5. Estudio del catalizador 10Ni/LaAl-E desactivado 157
Carolina Montero Calderón
picos a mayores temperaturas de combustión corresponden a coques con diferente
grado de grafitización, entre los que se incluyen los filamentos de carbón. Este
segundo tipo de coque no bloquea los centros metálicos y, en consecuencia, tiene
menor incidencia en la desactivación del catalizador.
Como se aprecia en las Figuras 5.1 y 5.2, la temperatura de reformado tiene un
efecto notable en el contenido de coque depositado, siendo el efecto diferente en
función del tiempo espacial utilizado. Para un tiempo espacial muy bajo, de 0.02
gcath/gEtOH (Figura 5.2a), el contenido de coque, que es muy pequeño, aumenta al
aumentar la temperatura de reformado, si bien el aumento es moderado (de 0.7 y 1.8
% en peso para 500 y 650 ºC, respectivamente). Sin embargo, para un tiempo espacial
moderado-alto (Figura 5.1 y 5.2b) el aumento de la temperatura de reformado atenúa
notablemente la deposición de coque, que en ambos casos es muy elevada
(especialmente para tiempo espacial de 0.09 gcath/gEtOH). En todos los casos se observa
que al aumentar la temperatura de reformado los picos se desplazan a mayor
temperatura de combustión, lo que indica que el coque es más grafítico, en
consecuencia es más difícil de eliminar mediante combustión con aire.
Las diferencias en el contenido y naturaleza del coque se explican en base a la
diferente composición del medio de reacción en las diferentes condiciones
experimentales, dado que ambos tipos de coque (encapsulante y fibrilar) tienen su
origen en la evolución de diferentes compuestos del medio de reacción que son los
precursores del coque (Vicente y cols., 2014a,b). Estas composiciones del medio de
reacción se han mostrado en las Figuras 4.12-4.15 del Capítulo 4 donde se observa que
a tiempo espacial bajo (Figura 4.13) la concentración de todos los productos de
reacción es muy baja, exceptuando al acetaldehído y en menor medida al etileno (cuya
formación es más importante al aumentar la temperatura).
Por tanto, en concordancia con estudios previos (Vicente y cols., 2014a,b), cabe
considerar que a bajo tiempo espacial el coque proviene de la descomposición del
acetaldehído y etileno, y de la participación del etanol a través de los iones etoxi , y es
un coque de tipo encapsulante, que se adsorbe en los sitios metálicos, bloquea la
entrada de los reactivos y desactiva rápidamente todas las reacciones involucradas en
el proceso de reformado. En estas condiciones, al aumentar la temperatura es mayor la

158
Capítulo 5
concentración de los precursores de este coque de tipo encapsulante (acetaldehído y
etileno), lo que resulta una mayor deposición de este tipo de coque. Esta interpretación
explica que la velocidad de desactivación sea mayor a 650 ºC que a 550 ºC (Figura
4.13).
Por otro lado, el hecho de que el pico de combustión se desplace a mayor
temperatura de combustión al aumentar la temperatura de reformado, indica que se
trata de un coque más evolucionado hacia estructuras grafíticas. Además, el mayor
contenido de CO en el medio de reacción favorecerá la formación de cierta cantidad
de coque de naturaleza fibrilar, mediante la reacción de Boudouard.
Para un tiempo espacial moderado (Figura 4.14) o elevado (Figuras 4.11 y 4.12)
son notables los rendimientos de CO y de CH4 y son muy bajos o insignificantes los
de acetaldehído y etileno. En estas condiciones el CO y CH4 son presumiblemente los
precursores de la formación del coque de naturaleza fibrilar observado en las Figuras
5.1 y 5.2b.
Es destacable que a pesar del elevado contenido de coque depositado
(especialmente a baja temperatura), la velocidad de desactivación observada en las
Figuras 4.12, y 4.14 es notablemente más lenta que la observada a bajo tiempo
espacial (Figura 4.13), lo que corrobora que el coque de naturaleza fibrilar contribuye
poco a la desactivación del catalizador. La Figura 4.12 mostraba que el aumento de
temperatura favorece la formación de CO y atenúa la formación de metano, lo que
ratifica la contribución al coque total en estas condiciones del coque obtenido por
reacción de Boudouard a partir de CO, que sería de más difícil combustión que el
coque formado por descomposición del metano.
Para estudiar el efecto de la temperatura sobre la estructura del coque, los
catalizadores desactivados se han analizado mediante espectroscopia Raman, técnica
que permite obtener información sobre la estructura de los carbones amorfos,
incluyendo el coque. En la Figura 5.3 se muestran los espectros Raman
correspondientes a catalizadores utilizados en reacciones de reformado con S/E=3 y
tiempo espacial de 0.04 gcath/gEtOH a 550 y 650 ºC. Los perfiles TPO para estos
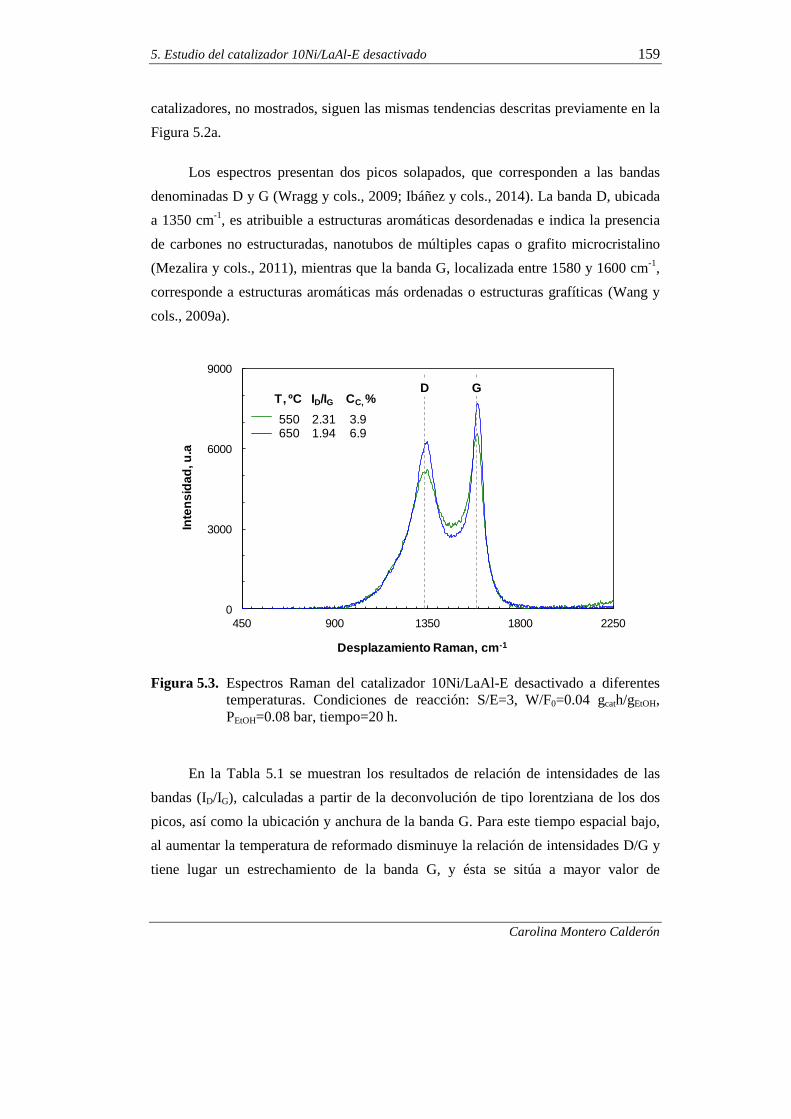
5. Estudio del catalizador 10Ni/LaAl-E desactivado 159
Carolina Montero Calderón
catalizadores, no mostrados, siguen las mismas tendencias descritas previamente en la
Figura 5.2a.
Los espectros presentan dos picos solapados, que corresponden a las bandas
denominadas D y G (Wragg y cols., 2009; Ibáñez y cols., 2014). La banda D, ubicada
a 1350 cm-1
, es atribuible a estructuras aromáticas desordenadas e indica la presencia
de carbones no estructuradas, nanotubos de múltiples capas o grafito microcristalino
(Mezalira y cols., 2011), mientras que la banda G, localizada entre 1580 y 1600 cm-1
,
corresponde a estructuras aromáticas más ordenadas o estructuras grafíticas (Wang y
cols., 2009a).
Figura 5.3. Espectros Raman del catalizador 10Ni/LaAl-E desactivado a diferentes
temperaturas. Condiciones de reacción: S/E=3, W/F0=0.04 gcath/gEtOH,
PEtOH=0.08 bar, tiempo=20 h.
En la Tabla 5.1 se muestran los resultados de relación de intensidades de las
bandas (ID/IG), calculadas a partir de la deconvolución de tipo lorentziana de los dos
picos, así como la ubicación y anchura de la banda G. Para este tiempo espacial bajo,
al aumentar la temperatura de reformado disminuye la relación de intensidades D/G y
tiene lugar un estrechamiento de la banda G, y ésta se sitúa a mayor valor de
0
3000
6000
9000
450 900 1350 1800 2250
Inte
ns
ida
d, u
.a
Desplazamiento Raman, cm-1
550 2.31 3.9650 1.94 6.9
D GT, ºC ID/IG CC, %

160
Capítulo 5
desplazamiento Raman, lo que indica que se han generado estructuras de mayor
cristalinidad. Este resultado es coherente con el desplazamiento a mayor temperatura
de los picos de combustión observado en los perfiles TPO (Figuras 5.1 y 5.2).
Tabla 5.1. Efecto de la temperatura en las características del coque depositado.
Condiciones de reacción:S/E=3, W/F0= 0.04 gcath/gEtOH, PEtOH=0.08 bar.
T, ºC ID/IG Banda G
Posición,cm-1
Anchura, cm-1
550 2.31 1590.4 65.2
650 1.94 1594.3 56.9
5.1.2. Relación molar S/E
En la Figura 5.4 se muestra el efecto de la relación S/E sobre el contenido y
perfil TPO del coque depositado en diferentes reacciones, para un valor dado del
tiempo espacial y dos temperaturas diferentes, 500 y 600 ºC. En los perfiles TPO del
coque depositado a 500 ºC (Figura 5.4a) se observa un pico asimétrico con máximo
ubicado a unos 580 ºC, cuya posición disminuye ligeramente a menor temperatura al
aumentar la relación S/E, desde 584 ºC para S/E=3 hasta 577 ºC para S/E=9. Similar
comportamiento se observa para el coque depositado a 600 ºC (Figura 5.4b), cuyo
máximo de combustión se sitúa en 625, 623 y 616 ºC, para relaciones S/E=3, 6 y 9,
respectivamente. Se trata, por tanto, de un coque de naturaleza mayoritariamente
fibrilar, lo que es coherente con la composición del medio de reacción, mayoritario en
CO y CH4 (Figuras 4.12 y 4.15) para este elevado valor del tiempo espacial. Estos
compuestos son precursores de coque fibrilar mediante reacciones de Boudouard y
metanación, respectivamente.
Como se observa, el aumento de la relación S/E atenúa notablemente la
deposición de coque, lo cual puede explicarse porque: i) el alto contenido de agua
atenúa las reacciones de formación de los precursores de coque (CO y CH4), cuya
concentración en el medio de reacción disminuye apreciablemente al aumentar la
relación S/E, y; ii) el elevado contenido de agua promueve las reacciones de
gasificación del coque, Ec. (1.26), siendo este efecto mucho más importante a elevada
temperatura (Chiou y cols., 2014). Esta notable atenuación del contenido de coque al
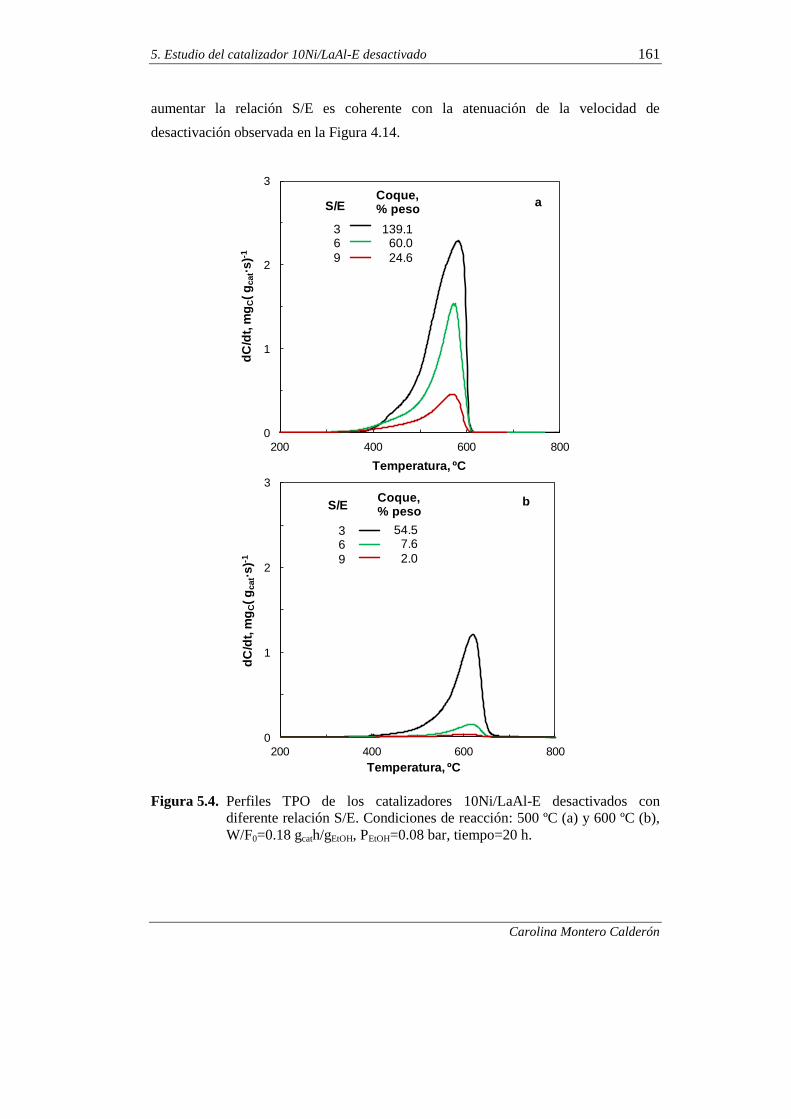
5. Estudio del catalizador 10Ni/LaAl-E desactivado 161
Carolina Montero Calderón
aumentar la relación S/E es coherente con la atenuación de la velocidad de
desactivación observada en la Figura 4.14.
Figura 5.4. Perfiles TPO de los catalizadores 10Ni/LaAl-E desactivados con
diferente relación S/E. Condiciones de reacción: 500 ºC (a) y 600 ºC (b),
W/F0=0.18 gcath/gEtOH, PEtOH=0.08 bar, tiempo=20 h.
0
1
2
3
200 400 600 800
dC
/dt,
mg
C(
gcat·
s)-1
Temperatura, ºC
36
9
S/ECoque,% peso
139.160.0
24.6
a
0
1
2
3
200 400 600 800
dC
/dt,
mg
C(
gcat·
s)-1
Temperatura, ºC
36
9
S/ECoque,% peso
54.57.6
2.0
b
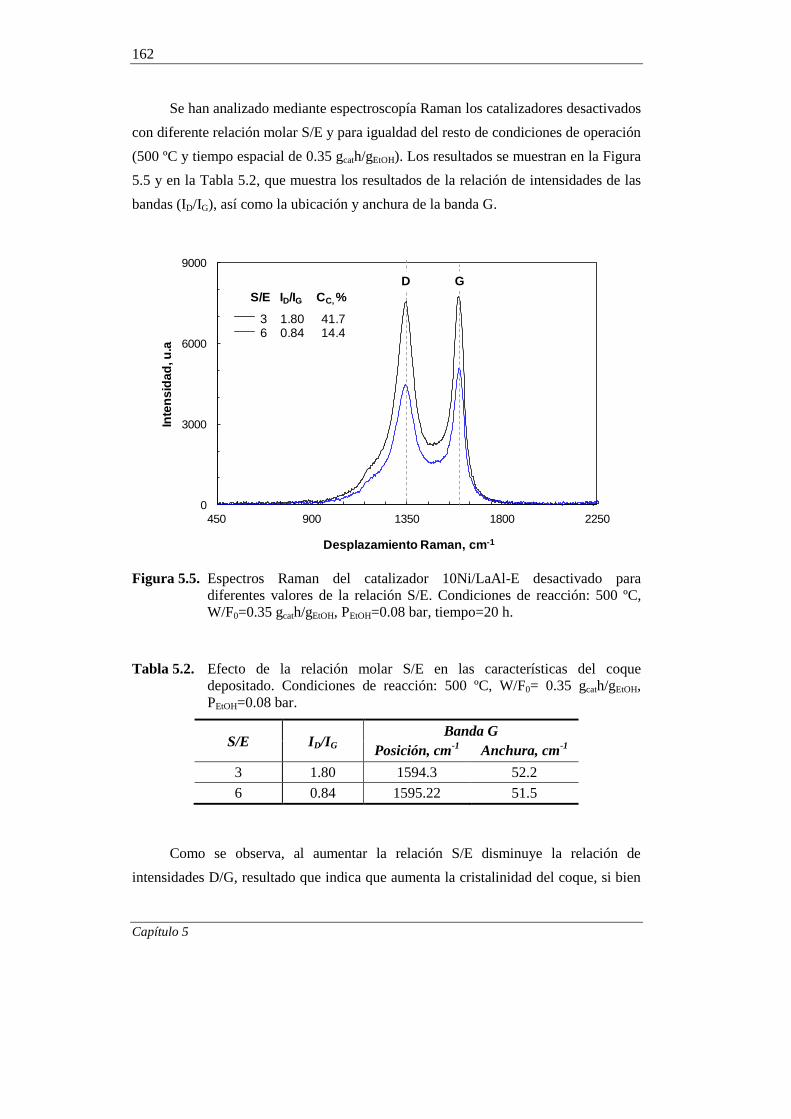
162
Capítulo 5
Se han analizado mediante espectroscopía Raman los catalizadores desactivados
con diferente relación molar S/E y para igualdad del resto de condiciones de operación
(500 ºC y tiempo espacial de 0.35 gcath/gEtOH). Los resultados se muestran en la Figura
5.5 y en la Tabla 5.2, que muestra los resultados de la relación de intensidades de las
bandas (ID/IG), así como la ubicación y anchura de la banda G.
Figura 5.5. Espectros Raman del catalizador 10Ni/LaAl-E desactivado para
diferentes valores de la relación S/E. Condiciones de reacción: 500 ºC,
W/F0=0.35 gcath/gEtOH, PEtOH=0.08 bar, tiempo=20 h.
Tabla 5.2. Efecto de la relación molar S/E en las características del coque
depositado. Condiciones de reacción: 500 ºC, W/F0= 0.35 gcath/gEtOH,
PEtOH=0.08 bar.
S/E ID/IG Banda G
Posición, cm-1
Anchura, cm-1
3 1.80 1594.3 52.2
6 0.84 1595.22 51.5
Como se observa, al aumentar la relación S/E disminuye la relación de
intensidades D/G, resultado que indica que aumenta la cristalinidad del coque, si bien
0
3000
6000
9000
450 900 1350 1800 2250
Inte
ns
ida
d, u
.a
Desplazamiento Raman, cm-1
D G
3 1.80 41.76 0.84 14.4
S/E ID/IG CC, %

5. Estudio del catalizador 10Ni/LaAl-E desactivado 163
Carolina Montero Calderón
la posición y anchura de la banda G están muy poco afectadas. Este resultado es
coherente con la obtención de un pico de combustión más asimétrico (con mayor
proporción de coque que quema a baja temperatura) para una baja relación molar S/E,
tal como se observa en la Figura 5.4.
5.1.3. Tiempo espacial
El efecto del tiempo espacial sobre el contenido de coque depositado y sobre los
perfiles TPO se muestra en las Figuras 5.6 y 5.7, respectivamente, correspondiendo
cada gráfica de estas figuras a una temperatura de reformado diferente.
El contenido de coque (Figura 5.6) pasa por un pronunciado máximo, que se
alcanza a menor valor del tiempo espacial cuanto mayor es la temperatura. Para baja
temperatura (Figura 5.6a), la posición del máximo depende de la relación molar S/E,
alcanzándose para un mayor valor del tiempo espacial al aumentar la relación S/E. En
concordancia con lo mostrado en los apartados anteriores, el contenido de coque
disminuye notablemente al aumentar la relación S/E y al aumentar la temperatura de
reformado.
Los contenidos de coque de la Figura 5.6 no guardan relación con la velocidad
de desactivación observada, que es muy elevada a bajo tiempo espacial (Figura 4.13 y
4.15), condiciones en las que el contenido de coque es bajo. Sin embargo, la
desactivación está muy atenuada para tiempos espaciales superiores a 0.09 gcath/gEtOH
(Figuras 4.11, 4.12, 4.16, 4.17), para los que contenido de coque es muy elevado. Este
resultado corrobora el hecho ya comentado de que la velocidad de desactivación
depende principalmente de la naturaleza del coque, en mucha mayor medida que del
contenido total de coque.
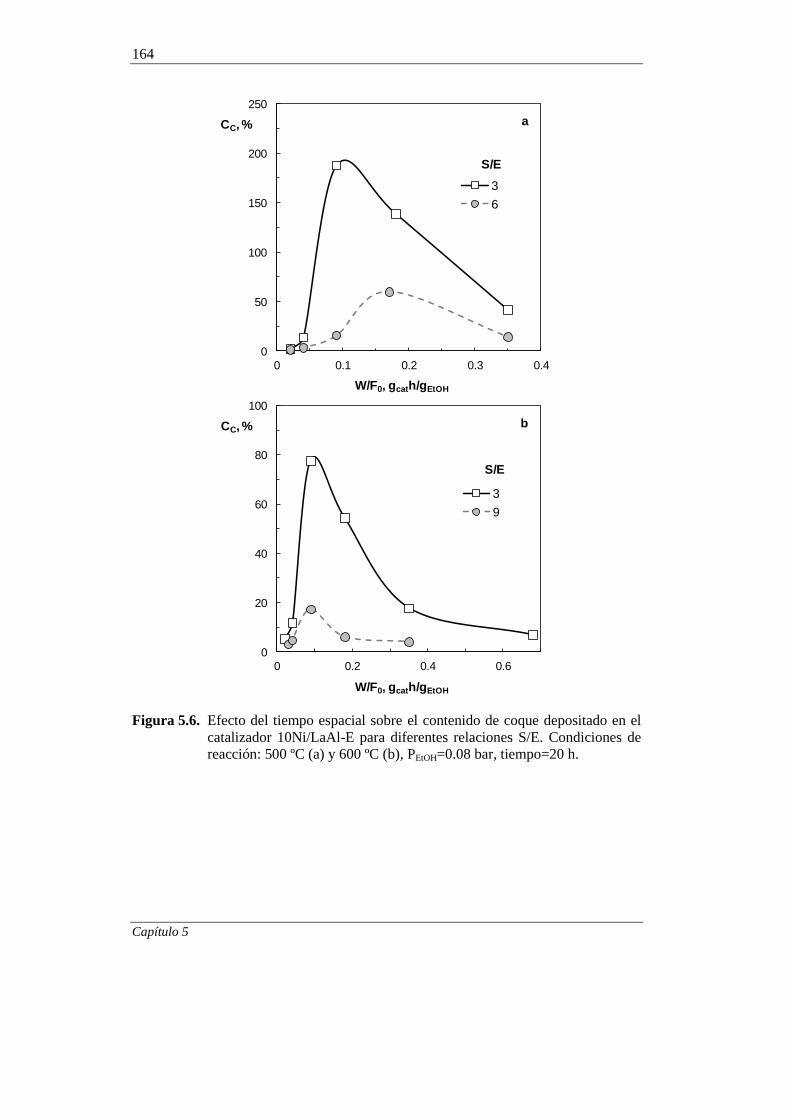
164
Capítulo 5
Figura 5.6. Efecto del tiempo espacial sobre el contenido de coque depositado en el
catalizador 10Ni/LaAl-E para diferentes relaciones S/E. Condiciones de
reacción: 500 ºC (a) y 600 ºC (b), PEtOH=0.08 bar, tiempo=20 h.
0
50
100
150
200
250
0 0.1 0.2 0.3 0.4
CC, %
W/F0, gcath/gEtOH
3
6
S/E
a
0
20
40
60
80
100
0 0.2 0.4 0.6
CC, %
W/F0, gcath/gEtOH
3
9
S/E
b
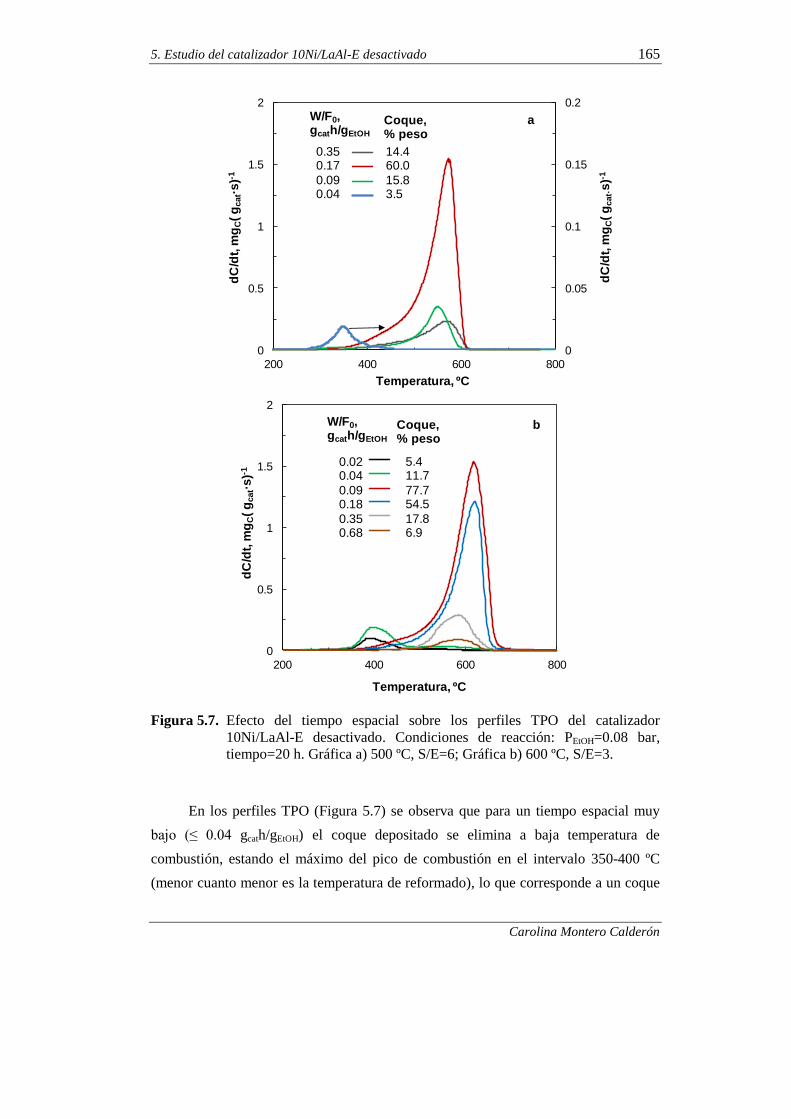
5. Estudio del catalizador 10Ni/LaAl-E desactivado 165
Carolina Montero Calderón
Figura 5.7. Efecto del tiempo espacial sobre los perfiles TPO del catalizador
10Ni/LaAl-E desactivado. Condiciones de reacción: PEtOH=0.08 bar,
tiempo=20 h. Gráfica a) 500 ºC, S/E=6; Gráfica b) 600 ºC, S/E=3.
En los perfiles TPO (Figura 5.7) se observa que para un tiempo espacial muy
bajo (≤ 0.04 gcath/gEtOH) el coque depositado se elimina a baja temperatura de
combustión, estando el máximo del pico de combustión en el intervalo 350-400 ºC
(menor cuanto menor es la temperatura de reformado), lo que corresponde a un coque
0
0.05
0.1
0.15
0.2
0
0.5
1
1.5
2
200 400 600 800
dC
/dt,
mg
C(
gcat·s
)-1
dC
/dt,
mg
C(
gcat·
s)-1
Temperatura, ºC
0.350.17
0.090.04
W/F0,gcath/gEtOH
14.4 60.0
15.83.5
Coque,% peso
a
0
0.5
1
1.5
2
200 400 600 800
dC
/dt,
mg
C(
gcat·
s)-1
Temperatura, ºC
0.020.04
0.090.18
0.350.68
W/F0,gcath/gEtOH
Coque,% peso
5.411.7
77.754.5
17.86.9
b

166
Capítulo 5
de naturaleza encapsulante y con gran capacidad de desactivación del catalizador,
como se ha puesto de manifiesto en la Figura 4.13.
A tiempos espaciales elevados (≥ 0.35 gcath/gEtOH) hay un pico mayoritario a
elevada temperatura de combustión, que corresponde a un coque de naturaleza fibrilar,
y, por tanto, con poca capacidad de desactivación del catalizador. El máximo se sitúa a
mayor temperatura de combustión al aumentar la temperatura de reformado, como se
ha comentado anteriormente..
Para un tiempo espacial intermedio (0.09-0.18) se observa un pico mayoritario a
alta temperatura de combustión, que corresponde a un coque de naturaleza fibrilar, lo
que explica que la desactivación observada en tales condiciones (Figuras 4.11, 4.12,
4.14-4.17) no sea excesivamente rápida, a pesar de que el contenido de coque es muy
elevado. Para estos tiempos espaciales intermedios se puede observar, no obstante, que
una pequeña fracción de coque comienza a quemar a baja temperatura (inferior a 400
ºC), e incluso se observa que para el tiempo espacial de 0.09 gcath/gEtOH a 500 ºC
(Figura 5.7a) se forma un muy pequeño pico de coque encapsulante.
Como ya se ha discutido, estas diferencias en el contenido y naturaleza del
coque depositado para los diferentes tiempos espaciales se explican en base a la
composición del medio de reacción, en el que existen diferentes concentraciones de
los precursores de ambos tipos de coque (encapsulante y fibrilar) según el avance de la
reacción, lo que depende del tiempo espacial. Los elevados rendimientos de
acetaldehído y etileno, así como la elevada concentración de etanol, para muy bajo
tiempo espacial (Figura 4.13, 4.16 y 4.17), justifican la formación de coque de
naturaleza encapsulante, mientras que las elevadas concentraciones de CO y CH4 para
tiempos espaciales moderados y altos (Figuras 4.11, 4.12, 4.14-4.17) son responsables
de la formación de coque de naturaleza fibrilar. Entre estos dos últimos potenciales
precursores de coque, se puede concluir que el CO contribuye en mayor medida a la
formación de coque, dado que el máximo contenido de coque (Figura 5.6) se obtiene
en general para el tiempo espacial para el cual es máximo el rendimiento de CO
(Figura 4.7).
Estas conclusiones son corroboradas por los resultados de la Figura 5.8, que
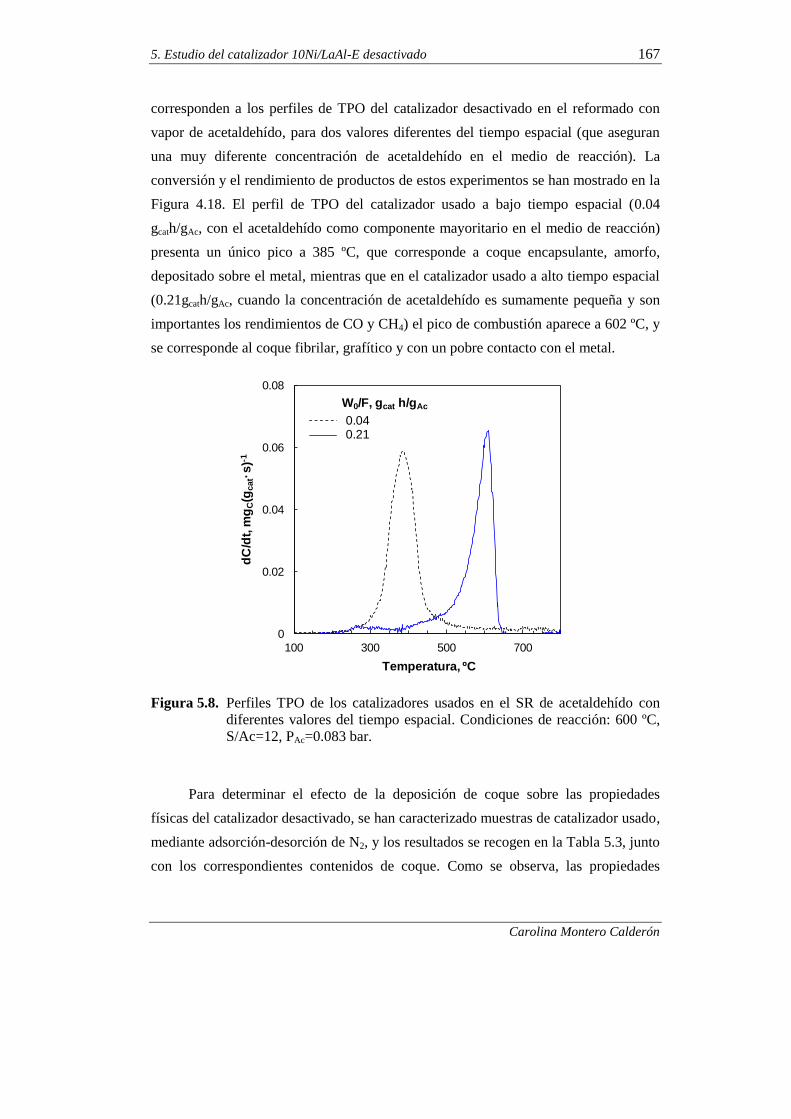
5. Estudio del catalizador 10Ni/LaAl-E desactivado 167
Carolina Montero Calderón
corresponden a los perfiles de TPO del catalizador desactivado en el reformado con
vapor de acetaldehído, para dos valores diferentes del tiempo espacial (que aseguran
una muy diferente concentración de acetaldehído en el medio de reacción). La
conversión y el rendimiento de productos de estos experimentos se han mostrado en la
Figura 4.18. El perfil de TPO del catalizador usado a bajo tiempo espacial (0.04
gcath/gAc, con el acetaldehído como componente mayoritario en el medio de reacción)
presenta un único pico a 385 ºC, que corresponde a coque encapsulante, amorfo,
depositado sobre el metal, mientras que en el catalizador usado a alto tiempo espacial
(0.21gcath/gAc, cuando la concentración de acetaldehído es sumamente pequeña y son
importantes los rendimientos de CO y CH4) el pico de combustión aparece a 602 ºC, y
se corresponde al coque fibrilar, grafítico y con un pobre contacto con el metal.
Figura 5.8. Perfiles TPO de los catalizadores usados en el SR de acetaldehído con
diferentes valores del tiempo espacial. Condiciones de reacción: 600 ºC,
S/Ac=12, PAc=0.083 bar.
Para determinar el efecto de la deposición de coque sobre las propiedades
físicas del catalizador desactivado, se han caracterizado muestras de catalizador usado,
mediante adsorción-desorción de N2, y los resultados se recogen en la Tabla 5.3, junto
con los correspondientes contenidos de coque. Como se observa, las propiedades
0
0.02
0.04
0.06
0.08
100 300 500 700
dC
/dt,
mg
C(g
cat·
s)-1
Temperatura, ºC
W0/F, gcat h/gAc
0.040.21
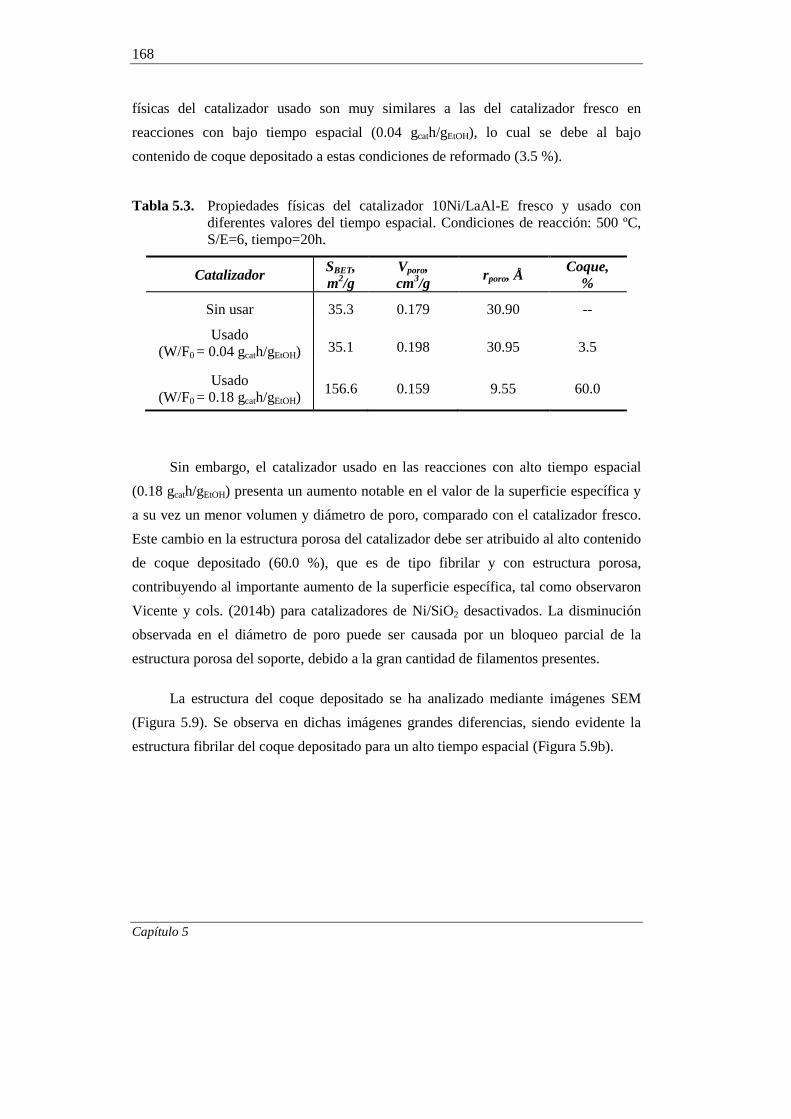
168
Capítulo 5
físicas del catalizador usado son muy similares a las del catalizador fresco en
reacciones con bajo tiempo espacial (0.04 gcath/gEtOH), lo cual se debe al bajo
contenido de coque depositado a estas condiciones de reformado (3.5 %).
Tabla 5.3. Propiedades físicas del catalizador 10Ni/LaAl-E fresco y usado con
diferentes valores del tiempo espacial. Condiciones de reacción: 500 ºC,
S/E=6, tiempo=20h.
Catalizador SBET,
m2/g
Vporo,
cm3/g
rporo, Å Coque,
%
Sin usar 35.3 0.179 30.90 --
Usado
(W/F0 = 0.04 gcath/gEtOH) 35.1 0.198 30.95 3.5
Usado
(W/F0 = 0.18 gcath/gEtOH) 156.6 0.159 9.55 60.0
Sin embargo, el catalizador usado en las reacciones con alto tiempo espacial
(0.18 gcath/gEtOH) presenta un aumento notable en el valor de la superficie específica y
a su vez un menor volumen y diámetro de poro, comparado con el catalizador fresco.
Este cambio en la estructura porosa del catalizador debe ser atribuido al alto contenido
de coque depositado (60.0 %), que es de tipo fibrilar y con estructura porosa,
contribuyendo al importante aumento de la superficie específica, tal como observaron
Vicente y cols. (2014b) para catalizadores de Ni/SiO2 desactivados. La disminución
observada en el diámetro de poro puede ser causada por un bloqueo parcial de la
estructura porosa del soporte, debido a la gran cantidad de filamentos presentes.
La estructura del coque depositado se ha analizado mediante imágenes SEM
(Figura 5.9). Se observa en dichas imágenes grandes diferencias, siendo evidente la
estructura fibrilar del coque depositado para un alto tiempo espacial (Figura 5.9b).

5. Estudio del catalizador 10Ni/LaAl-E desactivado 169
Carolina Montero Calderón
Figura 5.9. Imágenes SEM del coque depositado sobre el catalizador 10Ni/LaAl-E
Condiciones de reacción: 500 ºC, S/E=6.3, W/F0= 0.09 (a) y 0.18
gcath/gEtOH (b), PEtOH=0.08 bar, tiempo=20 h.
El catalizador utilizado en la reacción con alto tiempo espacial (0.18 gcath/gEtOH),
ha sido regenerado mediante combustión con aire, para observar las posibles
variaciones en la superficie tras la regeneración. La regeneración ha sido realizada en
un horno de mufla mediante calentamiento con una rampa de 5 ºC/ min hasta alcanzar
550 ºC (con objeto de no superar la temperatura de calcinación), seguido de una
isoterma a 550 ºC durante 2 h para garantizar la eliminación total del coque. La
muestra de catalizador recuperada fue analizada mediante microscopia electrónica de
barrido de electrones retrodispersados (BSE), técnica que, a pesar de tener poca
resolución, tiene alta sensibilidad a las variaciones en el número atómico de los
elementos en la superficie, por lo que permite identificar los partículas Ni presentes en
la muestra. La Figura 5.10 muestra el resultado, y se pueden apreciar las partículas de
NiOx distribuidas en el soporte.

170
Capítulo 5
Figura 5.10. Imagen BSE de la superficie del catalizador 10Ni/LaAl-E regenerado
por combustión del coque a 550 ºC. Condiciones de reacción: 500 ºC,
S/E=6, W/F0=0.18 gcath/gEtOH, PEtOH=0.08 bar, tiempo=20 h.
De estos resultados, así como de las imágenes TEM mostradas en el siguiente
apartado, se puede concluir que las fibras de coque que se van formado sobre el
catalizador (Figura 5.9b) logran separar las partículas metálicas de Ni que tienen poca
interacción con el soporte, las cuales se ubican en el extremo de las fibras, y cuando
estas fibras de coque son eliminadas por la combustión, los óxidos de Ní vuelven a
integrarse al soporte. Esta evolución de los cristales ya fue observada anteriormente
por Lobo y cols. (2011).

5. Estudio del catalizador 10Ni/LaAl-E desactivado 171
Carolina Montero Calderón
5.2. EVOLUCIÓN DEL Ni y DEL COQUE CON EL TIEMPO DE
REACCIÓN
En este apartado se estudia la evolución morfológica con el tiempo de reacción
de las partículas del Ni y del coque en el catalizador 10Ni/LaAl-E, con el objetivo de
relacionar estas evoluciones entre sí y mejorar la comprensión de la desactivación del
catalizador. El estudio se ha realizado en condiciones en las que la desactivación al
cabo de 20 h de reacción es relativamente severa, para poder analizar el catalizador en
diferentes estados de desactivación, tanto incipiente (al inicio de la reacción), como
prácticamente completa (al cabo de 20 h). Las condiciones utilizadas han sido: 500 ºC;
relación molar S/E= 3; W/F0 = 0.09 gcath/gEtOH; PEtOH=0.083 bar. En todos los casos las
condiciones fluidodinámicas se han mantenido constantes de acuerdo a lo especificado
en el Apartado 2.3.3.
Los resultados de evolución con el tiempo de reacción de la conversión de
etanol (XEtOH) y de los rendimientos de los productos de reacción se muestran en la
Figura 5.11. No se incluye el rendimiento de etileno, dado que fue despreciable en
estas condiciones. La evolución de la conversión y los rendimientos permite establecer
3 etapas diferenciadas:
Etapa 1 (t < 4 h). La conversión de etanol es elevada (en torno a 0.90) y
permanece aproximadamente constante, con un rendimiento de H2 también
constante de 0.40. El rendimiento de acetaldehído es bajo, porque la extensión
de su descomposición a CO y CH4 es elevada, permaneciendo también casi
constantes los rendimientos de estos productos, en este intervalo.
Etapa 2 (4 h < t < 8 h): Tiene lugar una acusada disminución (del 49 %) de la
conversión de etanol y una aún mayor disminución de los rendimientos de CO y
CH4 (que disminuye cerca del 73 %). Sin embargo, el rendimiento de H2 solo
disminuye un 37 %, resultado que parece indicar que la desactivación afecta de
diferente forma a las etapas del esquema cinético, o bien puede ser debido a la
contribución de la reacción de descomposición de CH4 a la formación de H2, a la
vez que de coque, a medida que transcurre la reacción.
Etapa 3 (t > 8 h): La conversión de etanol y los rendimientos de todos los
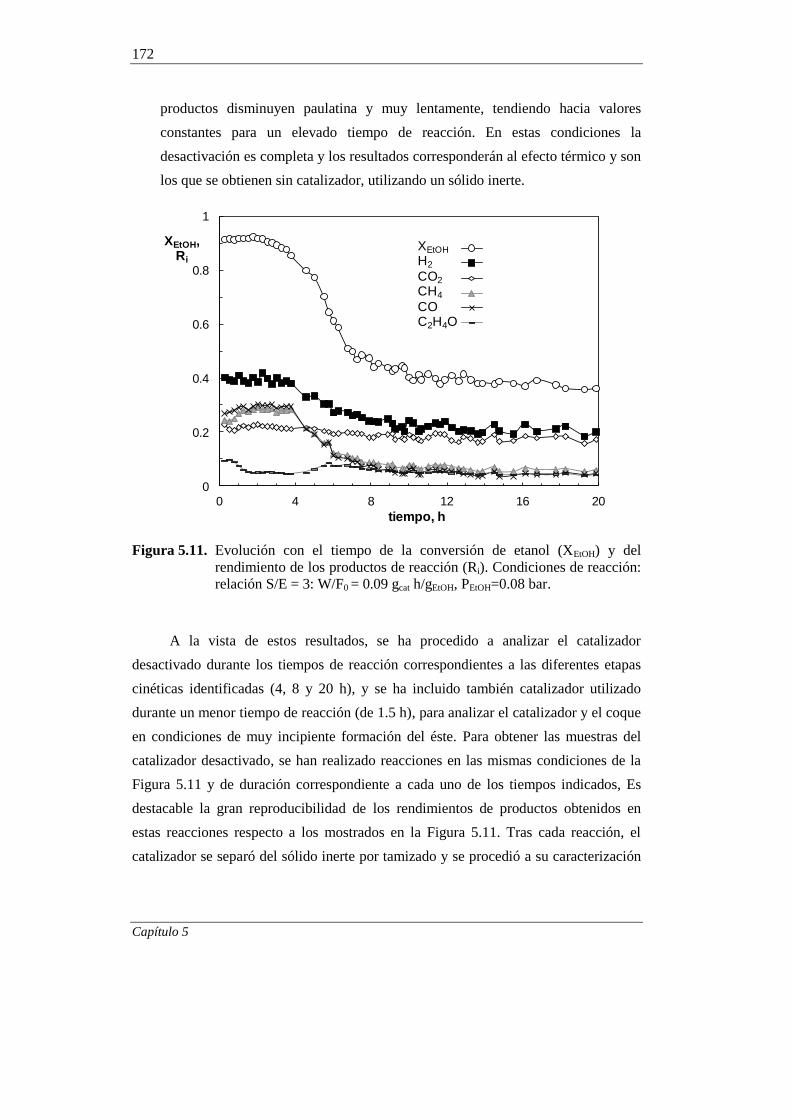
172
Capítulo 5
productos disminuyen paulatina y muy lentamente, tendiendo hacia valores
constantes para un elevado tiempo de reacción. En estas condiciones la
desactivación es completa y los resultados corresponderán al efecto térmico y son
los que se obtienen sin catalizador, utilizando un sólido inerte.
Figura 5.11. Evolución con el tiempo de la conversión de etanol (XEtOH) y del
rendimiento de los productos de reacción (Ri). Condiciones de reacción:
relación S/E = 3: W/F0 = 0.09 gcat h/gEtOH, PEtOH=0.08 bar.
A la vista de estos resultados, se ha procedido a analizar el catalizador
desactivado durante los tiempos de reacción correspondientes a las diferentes etapas
cinéticas identificadas (4, 8 y 20 h), y se ha incluido también catalizador utilizado
durante un menor tiempo de reacción (de 1.5 h), para analizar el catalizador y el coque
en condiciones de muy incipiente formación del éste. Para obtener las muestras del
catalizador desactivado, se han realizado reacciones en las mismas condiciones de la
Figura 5.11 y de duración correspondiente a cada uno de los tiempos indicados, Es
destacable la gran reproducibilidad de los rendimientos de productos obtenidos en
estas reacciones respecto a los mostrados en la Figura 5.11. Tras cada reacción, el
catalizador se separó del sólido inerte por tamizado y se procedió a su caracterización
0
0.2
0.4
0.6
0.8
1
0 4 8 12 16 20
XEtOH,Ri
tiempo, h
XEtOH
H2
CO2
CH4
COC2H4O
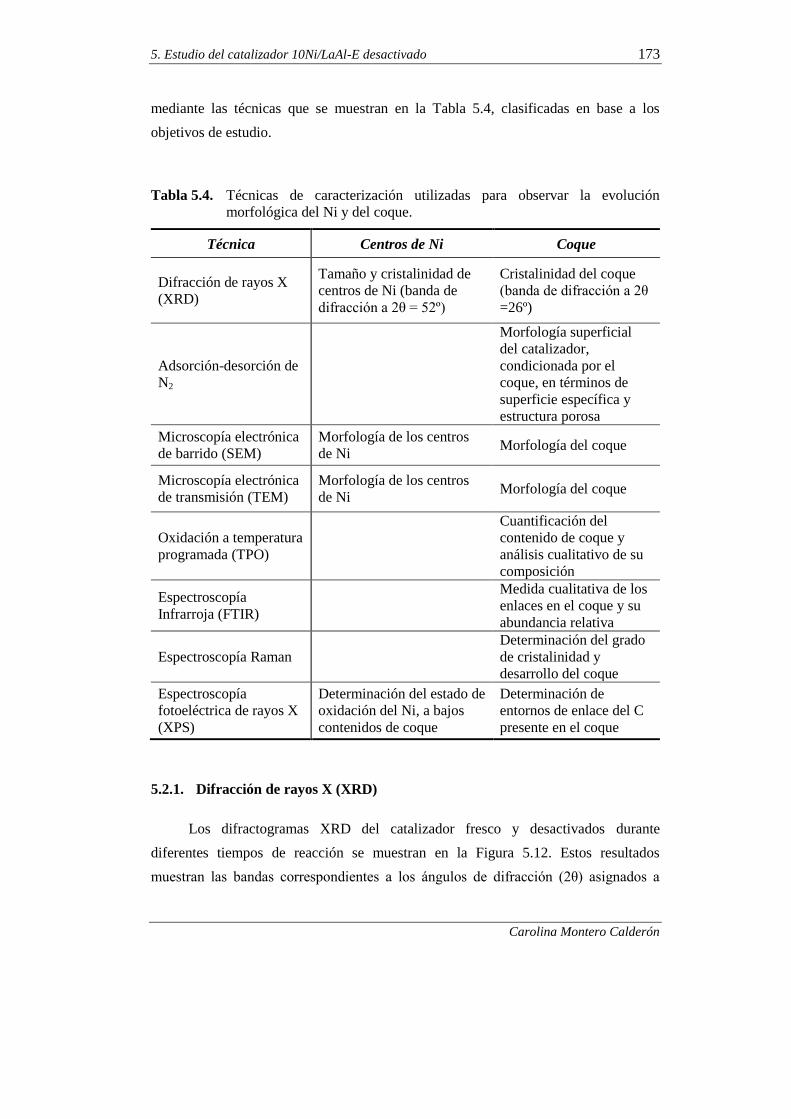
5. Estudio del catalizador 10Ni/LaAl-E desactivado 173
Carolina Montero Calderón
mediante las técnicas que se muestran en la Tabla 5.4, clasificadas en base a los
objetivos de estudio.
Tabla 5.4. Técnicas de caracterización utilizadas para observar la evolución
morfológica del Ni y del coque.
Técnica Centros de Ni Coque
Difracción de rayos X
(XRD)
Tamaño y cristalinidad de
centros de Ni (banda de
difracción a 2θ = 52º)
Cristalinidad del coque
(banda de difracción a 2θ
=26º)
Adsorción-desorción de
N2
Morfología superficial
del catalizador,
condicionada por el
coque, en términos de
superficie específica y
estructura porosa
Microscopía electrónica
de barrido (SEM)
Morfología de los centros
de Ni Morfología del coque
Microscopía electrónica
de transmisión (TEM)
Morfología de los centros
de Ni Morfología del coque
Oxidación a temperatura
programada (TPO)
Cuantificación del
contenido de coque y
análisis cualitativo de su
composición
Espectroscopía
Infrarroja (FTIR)
Medida cualitativa de los
enlaces en el coque y su
abundancia relativa
Espectroscopía Raman
Determinación del grado
de cristalinidad y
desarrollo del coque
Espectroscopía
fotoeléctrica de rayos X
(XPS)
Determinación del estado de
oxidación del Ni, a bajos
contenidos de coque
Determinación de
entornos de enlace del C
presente en el coque
5.2.1. Difracción de rayos X (XRD)
Los difractogramas XRD del catalizador fresco y desactivados durante
diferentes tiempos de reacción se muestran en la Figura 5.12. Estos resultados
muestran las bandas correspondientes a los ángulos de difracción (2θ) asignados a
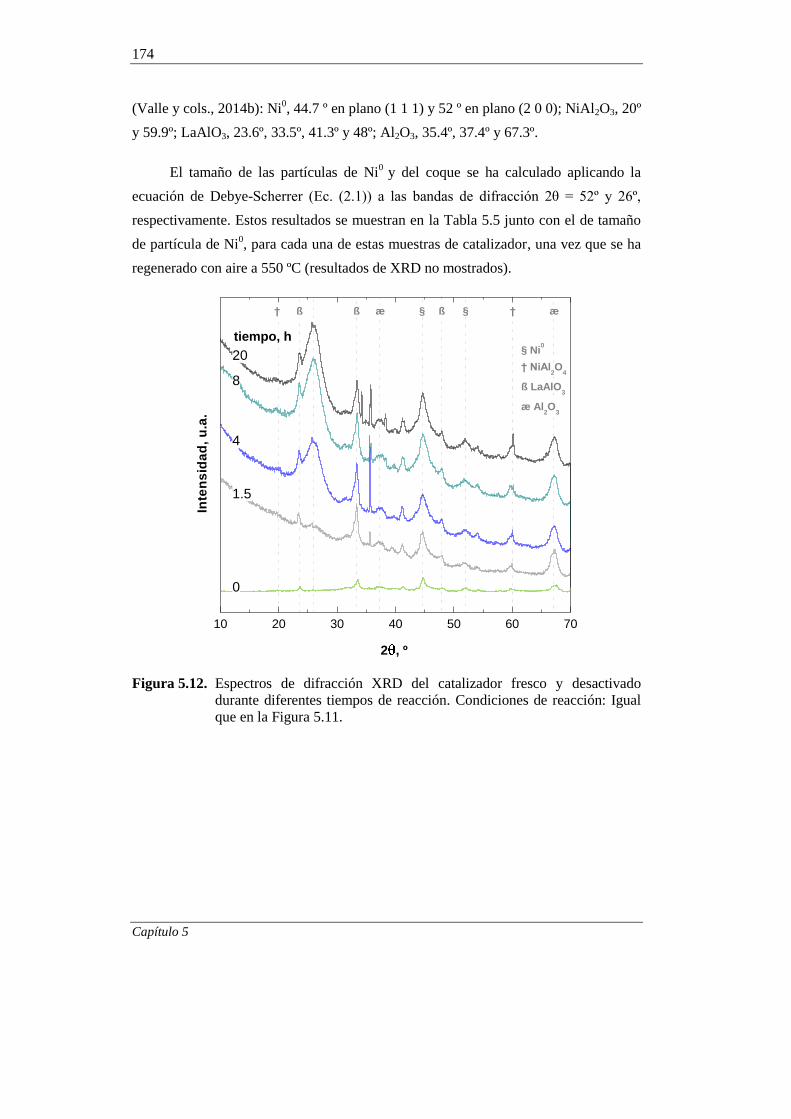
174
Capítulo 5
(Valle y cols., 2014b): Ni0, 44.7 º en plano (1 1 1) y 52 º en plano (2 0 0); NiAl2O3, 20º
y 59.9º; LaAlO3, 23.6º, 33.5º, 41.3º y 48º; Al2O3, 35.4º, 37.4º y 67.3º.
El tamaño de las partículas de Ni0
y del coque se ha calculado aplicando la
ecuación de Debye-Scherrer (Ec. (2.1)) a las bandas de difracción 2θ = 52º y 26º,
respectivamente. Estos resultados se muestran en la Tabla 5.5 junto con el de tamaño
de partícula de Ni0, para cada una de estas muestras de catalizador, una vez que se ha
regenerado con aire a 550 ºC (resultados de XRD no mostrados).
Figura 5.12. Espectros de difracción XRD del catalizador fresco y desactivado
durante diferentes tiempos de reacción. Condiciones de reacción: Igual
que en la Figura 5.11.
ß߆ †§§
§ Ni0
† NiAl2O
4
ß LaAlO3
æ Al2O
3
10 20 30 40 50 60 70
ææ
tiempo, h
20
8
4
1.5
Inte
ns
ida
d,
u.a
.
2 , º
0
ß
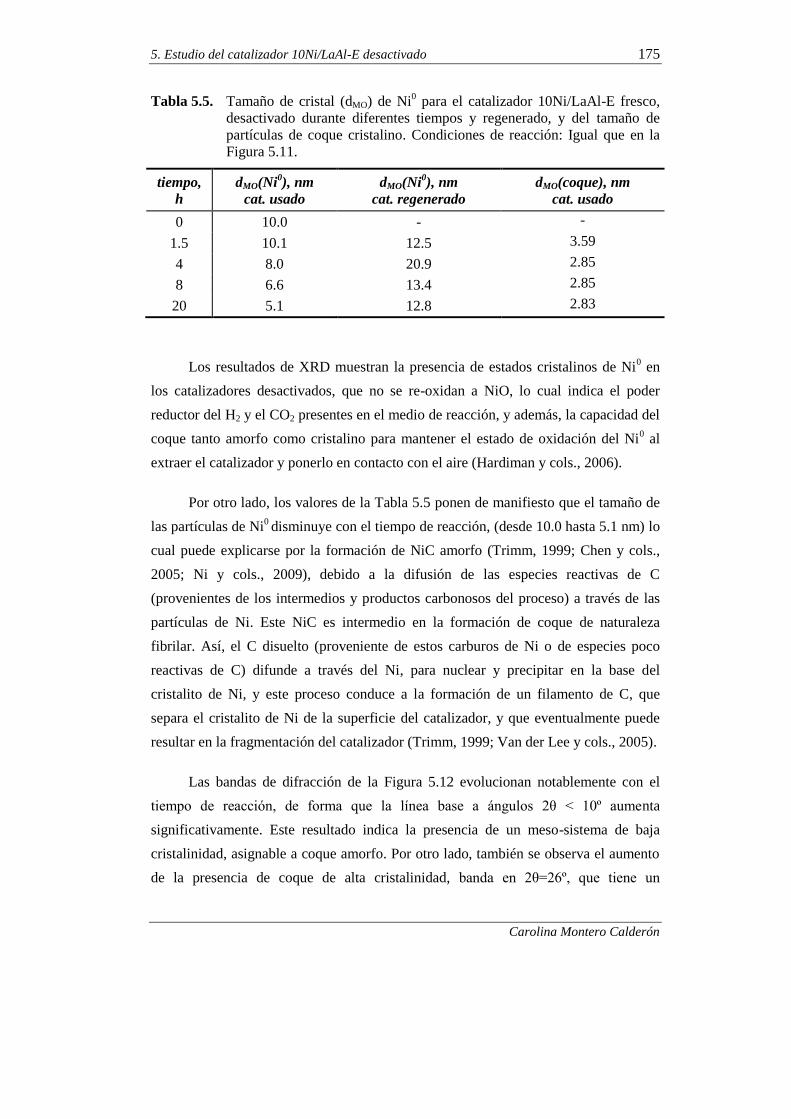
5. Estudio del catalizador 10Ni/LaAl-E desactivado 175
Carolina Montero Calderón
Tabla 5.5. Tamaño de cristal (dMO) de Ni0 para el catalizador 10Ni/LaAl-E fresco,
desactivado durante diferentes tiempos y regenerado, y del tamaño de
partículas de coque cristalino. Condiciones de reacción: Igual que en la
Figura 5.11.
tiempo,
h
dMO(Ni0), nm
cat. usado
dMO(Ni0), nm
cat. regenerado
dMO(coque), nm
cat. usado
0 10.0 - -
1.5 10.1 12.5 3.59
4 8.0 20.9 2.85
8 6.6 13.4 2.85
20 5.1 12.8 2.83
Los resultados de XRD muestran la presencia de estados cristalinos de Ni0 en
los catalizadores desactivados, que no se re-oxidan a NiO, lo cual indica el poder
reductor del H2 y el CO2 presentes en el medio de reacción, y además, la capacidad del
coque tanto amorfo como cristalino para mantener el estado de oxidación del Ni0 al
extraer el catalizador y ponerlo en contacto con el aire (Hardiman y cols., 2006).
Por otro lado, los valores de la Tabla 5.5 ponen de manifiesto que el tamaño de
las partículas de Ni0 disminuye con el tiempo de reacción, (desde 10.0 hasta 5.1 nm) lo
cual puede explicarse por la formación de NiC amorfo (Trimm, 1999; Chen y cols.,
2005; Ni y cols., 2009), debido a la difusión de las especies reactivas de C
(provenientes de los intermedios y productos carbonosos del proceso) a través de las
partículas de Ni. Este NiC es intermedio en la formación de coque de naturaleza
fibrilar. Así, el C disuelto (proveniente de estos carburos de Ni o de especies poco
reactivas de C) difunde a través del Ni, para nuclear y precipitar en la base del
cristalito de Ni, y este proceso conduce a la formación de un filamento de C, que
separa el cristalito de Ni de la superficie del catalizador, y que eventualmente puede
resultar en la fragmentación del catalizador (Trimm, 1999; Van der Lee y cols., 2005).
Las bandas de difracción de la Figura 5.12 evolucionan notablemente con el
tiempo de reacción, de forma que la línea base a ángulos 2θ < 10º aumenta
significativamente. Este resultado indica la presencia de un meso-sistema de baja
cristalinidad, asignable a coque amorfo. Por otro lado, también se observa el aumento
de la presencia de coque de alta cristalinidad, banda en 2θ=26º, que tiene un

176
Capítulo 5
crecimiento muy significativo entre 1.5-8 h. En la Tabla 5.5 se muestran los resultados
del tamaño de coque grafítico y cristalino calculado para esta banda de difracción,
observándose que el tamaño de partículas del coque tiende a un valor estable en torno
a 2.85 nm.
Por otro lado, cabe señalar que la regeneración del catalizador por combustión
de coque con aire produce una recuperación del tamaño de las partículas de Ni, que
son readsorbidas en el soporte.
5.2.2. Adsorción-desorción de N2
En la Figura 5.13 se muestran las isotermas de adsorción-desorción de N2
correspondientes al catalizador fresco y desactivado durante diferentes tiempos de
reacción. Se observa que la isoterma del catalizador fresco corresponde a la de un
material mesoporoso. En los catalizadores usados se aprecia un progresivo aumento
del volumen total adsorbido con el tiempo de reacción, a la vez que la curva de
histéresis cambia desde el tipo H3 (catalizador fresco) al H4 (t ≥4 h). Es decir, la
superficie del catalizador evoluciona desde la correspondiente a materiales
compuestos por partículas laminares y poros de tipo rendija hasta la correspondiente a
poros más estrechos. A medida que el catalizador se desactiva, aumenta el volumen de
N2 adsorbido (P/P0 < 0.4), como consecuencia de la generación de micro y
mesoporosidad en el catalizador. Paralelamente, el volumen de N2 adsorbido en la
región de altas presiones parciales (P/P0 > 0.4) es menor, lo que indica el bloqueo de
los macroporos del catalizador. A tiempos elevados (t = 20 h), el volumen de N2
adsorbido vuelve a aumentar, lo que indica una recuperación de macroporos.
La Tabla 5.6 resume los resultados de superficie BET, volumen de poros y radio
medio de los mismos. La superficie BET aumenta con el tiempo de reacción, de forma
que se genera superficie accesible, a la vez que disminuyen el volumen y el radio
medio de poro. Estos resultados evidencian el bloqueo parcial que sufren los
macroporos del catalizador (Wang y Lu, 1999; Vicente y cols., 2014a), a la vez que se
crea una nueva micro y mesoporosidad que hacen que aumente el área superficial del
catalizador (Roy y cols., 2012). Estos resultados difieren de la disminución de la
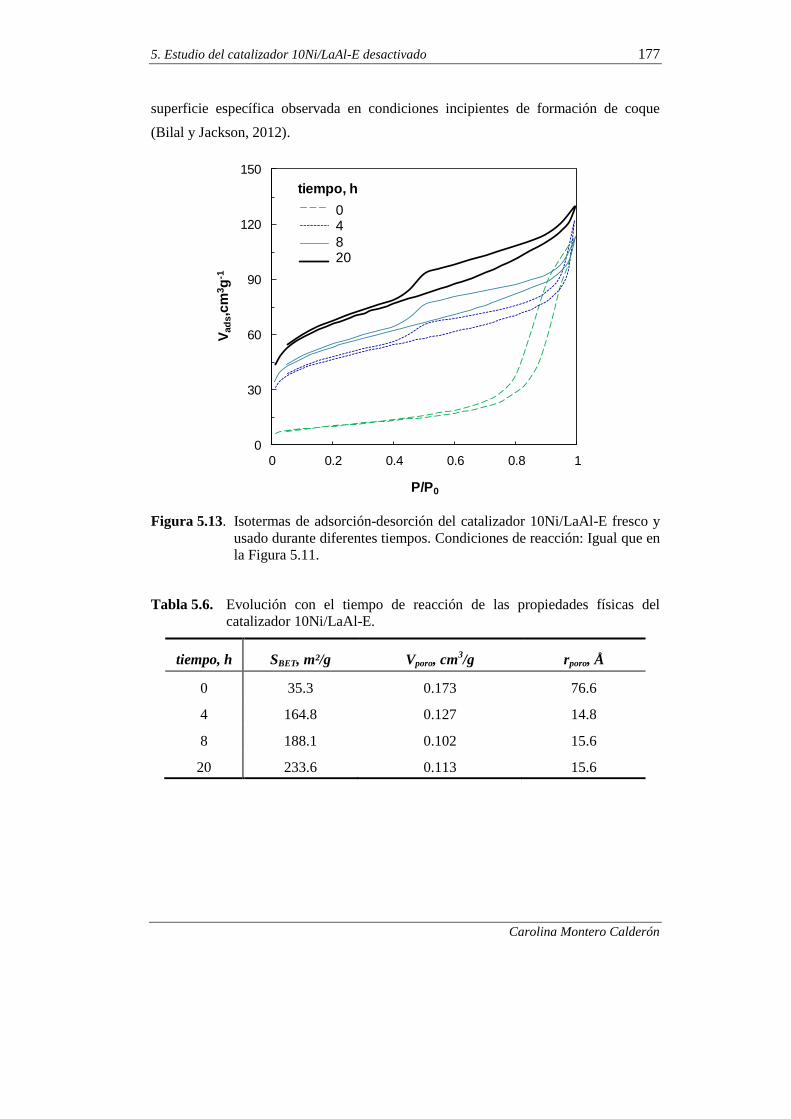
5. Estudio del catalizador 10Ni/LaAl-E desactivado 177
Carolina Montero Calderón
superficie específica observada en condiciones incipientes de formación de coque
(Bilal y Jackson, 2012).
Figura 5.13. Isotermas de adsorción-desorción del catalizador 10Ni/LaAl-E fresco y
usado durante diferentes tiempos. Condiciones de reacción: Igual que en
la Figura 5.11.
Tabla 5.6. Evolución con el tiempo de reacción de las propiedades físicas del
catalizador 10Ni/LaAl-E.
tiempo, h SBET, m²/g Vporo, cm3/g rporo, Å
0 35.3 0.173 76.6
4 164.8 0.127 14.8
8 188.1 0.102 15.6
20 233.6 0.113 15.6
0
30
60
90
120
150
0 0.2 0.4 0.6 0.8 1
Vad
s,c
m3g
-1
P/P0
tiempo, h
04
820

178
Capítulo 5
5.2.3. Microscopía electrónica de barrido (SEM)
Los resultados de microscopía electrónica de barrido (SEM) y de electrón
retrodispersado (BSE) se muestran en la Figura 5.14. En estas imágenes se han
colorear en amarillo (color no real) las regiones más brillantes de BSE para las
partículas de Ni0 sobre la imagen SEM.
Figura 5.14. Imágenes SEM y BSE superpuestas con regiones amarillas para
identificar el Ni en el catalizador usado durante diferentes tiempos de
reacción. Condiciones de reacción: Igual que en la Figura 5.11.
A t = 1.5 h (Figura 5.14a) la superficie presenta estructuras fibrilares de coque
incipientes, en cuyo extremo se deposita una partícula de Ni0. Esta formación de coque
fibrilar ha sido ampliamente descrito cualitativamente y cuantitativamente para el
reformado de etanol (Bilal y Jackson, 2013). También en este periodo, se puede
distinguir parte del soporte del catalizador por la presencia de zonas planas y porosas.

5. Estudio del catalizador 10Ni/LaAl-E desactivado 179
Carolina Montero Calderón
A t = 4 h (Figura 5.14b) y t = 8 h (Figura 5.14c) se observan un crecimiento de
la longitud del coque fibrilar en todas las direcciones, a la vez que deja de ser visible
el soporte.
A partir de 8 h (Figura 5.14c) pero sobre todo para 20 h (Figura 5.14d) se
observa la aparición de un coque que crece desde la superficie del soporte llenando los
espacios creados entre las fibras, que se correspondería con el denominado coque
pirolítico (Bartholomew, 2001). Según este autor, este coque se forma en estados
avanzados de desactivación del catalizador y contribuye con el coque encapsulante en
el bloqueo del acceso del reactante a los centros metálicos del catalizador. Estos
coques se diferencian en su carácter más desactivante respecto al coque fibrilar.
Los resultados de las Figuras 5.12 y 5.14 demuestran que las partículas de Ni0
presentes a aproximadamente 4 h de reacción (de 8 nm, Tabla 5.5), promueven el
crecimiento de coque fibrilar. Sin embargo, las partículas de Ni0 con tamaños
inferiores, para mayores tiempos de reacción, favorecen el crecimiento del coque
pirolítico. Este resultado es concordante con la hipótesis del cambio de mecanismo de
formación de coque (Alberton y cols., 2007; Gohier y cols., 2008) por reducción del
tamaño de Ni0, desde crecimiento de coque fibrilar (para grandes tamaños de Ni) y
hasta crecimiento del coque pirolítico en la superficie (a pequeños tamaños de Ni), lo
cual ha sido descrito a nivel mecanístico (Gómez-Gualdrón y Balbuena, 2013).
Se ha establecido que las partículas pequeñas de Ni son menos activas en la
formación de coque que las grandes (Chen y cols., 2005; Rossetti y cols., 2014), así
como que la formación y crecimiento de fibras de coque provoca una reestructuración
de la capa monoatómica del Ni (Parizotto y cols., 2007). El coque fibrilar no desactiva
significantemente el catalizador (Wang y cols., 2008), sino que provoca la separación
física de las partículas metálicas de Ni0 ubicadas en los extremos de las fibras,
evitando la aglomeración y sinterización de las partículas metálicas (Takenaka y cols.,
2003). Para corroborar que la cantidad y el tamaño de las partículas de Ni0 disminuye
con el tiempo de reacción, se ha calculado la fracción de electrones retrodispersados
por las partículas de Ni0 y se han cuantificado respecto al área total de la imagen. Los
resultados de porcentaje de electrones retrodispersados se muestran en la Figura 5.15,
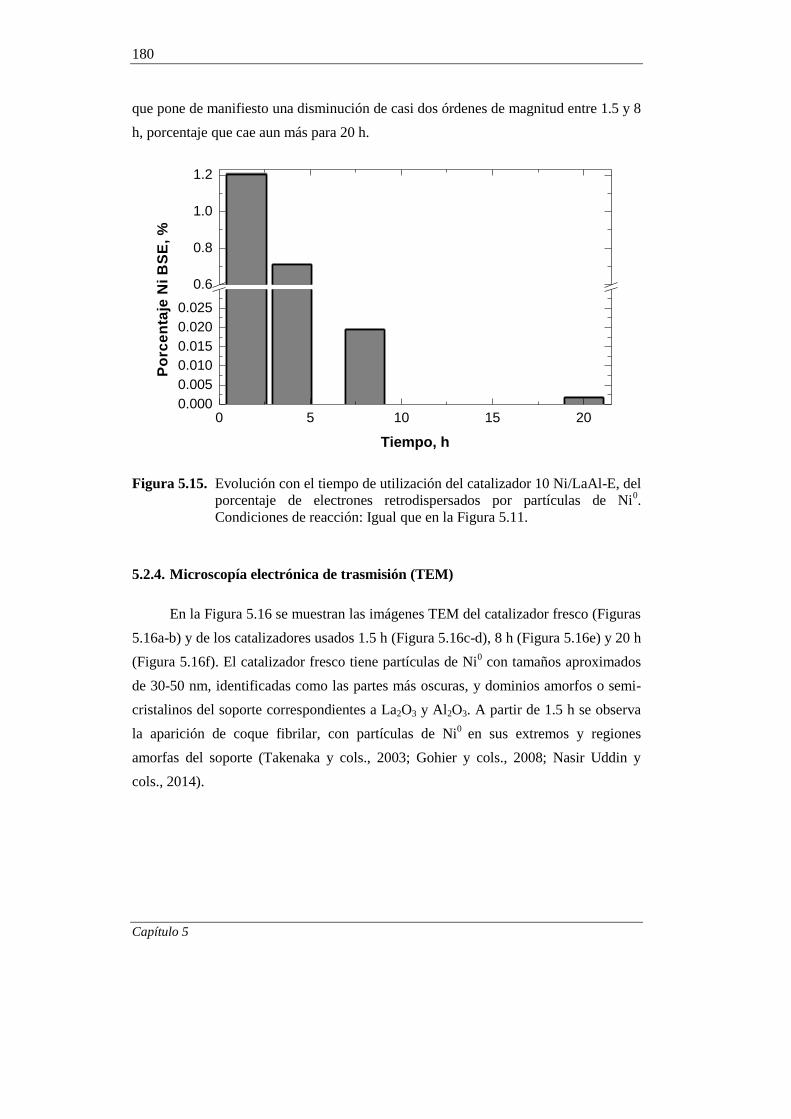
180
Capítulo 5
que pone de manifiesto una disminución de casi dos órdenes de magnitud entre 1.5 y 8
h, porcentaje que cae aun más para 20 h.
Figura 5.15. Evolución con el tiempo de utilización del catalizador 10 Ni/LaAl-E, del
porcentaje de electrones retrodispersados por partículas de Ni0.
Condiciones de reacción: Igual que en la Figura 5.11.
5.2.4. Microscopía electrónica de trasmisión (TEM)
En la Figura 5.16 se muestran las imágenes TEM del catalizador fresco (Figuras
5.16a-b) y de los catalizadores usados 1.5 h (Figura 5.16c-d), 8 h (Figura 5.16e) y 20 h
(Figura 5.16f). El catalizador fresco tiene partículas de Ni0 con tamaños aproximados
de 30-50 nm, identificadas como las partes más oscuras, y dominios amorfos o semi-
cristalinos del soporte correspondientes a La2O3 y Al2O3. A partir de 1.5 h se observa
la aparición de coque fibrilar, con partículas de Ni0
en sus extremos y regiones
amorfas del soporte (Takenaka y cols., 2003; Gohier y cols., 2008; Nasir Uddin y
cols., 2014).
0 5 10 15 200.000
0.005
0.010
0.015
0.020
0.025
0.6
0.8
1.0
1.2P
orc
en
taje
Ni
BS
E,
%
Tiempo, h
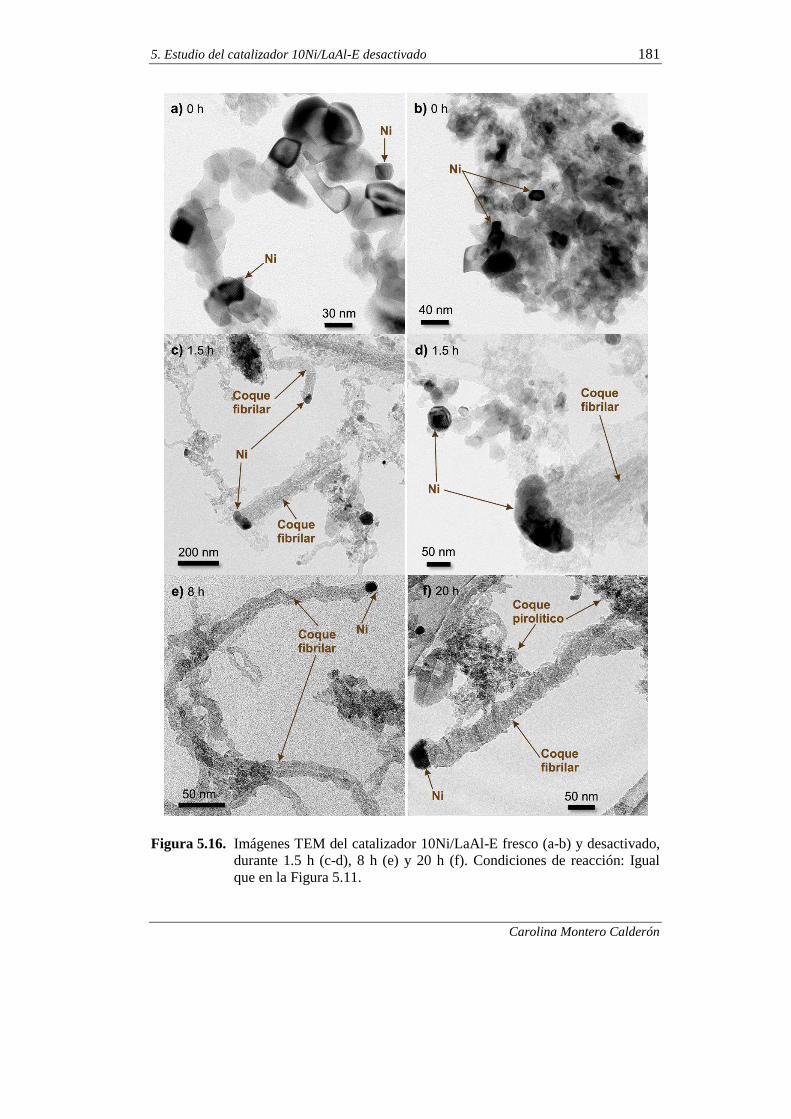
5. Estudio del catalizador 10Ni/LaAl-E desactivado 181
Carolina Montero Calderón
Figura 5.16. Imágenes TEM del catalizador 10Ni/LaAl-E fresco (a-b) y desactivado,
durante 1.5 h (c-d), 8 h (e) y 20 h (f). Condiciones de reacción: Igual
que en la Figura 5.11.

182
Capítulo 5
No obstante, no es fácil observar planos cristalinos en el coque fibrilar, y estos
planos solo se aprecian débilmente a partir de 8 h de reacción. Por otro lado, el coque
pirolítico observado en las imágenes de la Figura 5.14 no se distingue del soporte, lo
que indica el amorfismo de estas dos especies. Por otro lado, los resultados de las
imágenes TEM mostrados en la Figura 5.16 no permiten establecer una correlación
entre el tiempo de reacción y la longitud o achura del coque fibrilar.
5.2.5. Oxidación a temperatura programada (TPO)
En la Figura 5.17a se han representado los perfiles TPO de los catalizadores
usados durante diferentes tiempos. El contenido de coque depositado en los
catalizadores, así como el rendimiento de coque, se muestran en la Figura 5.17b. El
rendimiento de coque (R coque) se ha calculado:
0coque
F
W
h reacción, de tiempo
% Coque,(%)R (5.3)
Los perfiles de TPO presentan un único pico, que es ancho y asimétrico en
todos los casos. Conforme avanza el tiempo de reacción aumenta el área del pico de
combustión (contenido de coque) y su máximo se desplaza a mayor temperatura. El
máximo del pico de combustión del coque depositado tras 1.5 h de reacción se sitúa a
500 ºC con un hombro a 430 ºC. Este hombro crece para el coque depositado tras 4 h
de reacción, pero no evoluciona más para mayor tiempo de reacción. Por otro lado, el
pico de alta temperatura se desplaza a 518, 544 y 552 ºC para tiempos de 4, 8 y 20 h,
respectivamente.
Este resultado indica que conforme avanza el tiempo de reacción el coque
depositado tiene una naturaleza más grafítica, o bien está más alejado espacialmente
del Ni0 (que cataliza su combustión) o una combinación de los dos factores anteriores
(Wang y Lu, 1999). Ahora bien, los espectros XRD mostrados en la Figura 5.12
indican un aumento de la cristalinidad del coque con el tiempo. Además las imágenes
SEM de la Figura 5.14 ponen de manifiesto un crecimiento del contenido de coque
suficiente para que el contacto Ni0-coque no sea suficientemente próximo como para
que la combustión del coque esté catalizada. De esta forma, puede concluirse que el
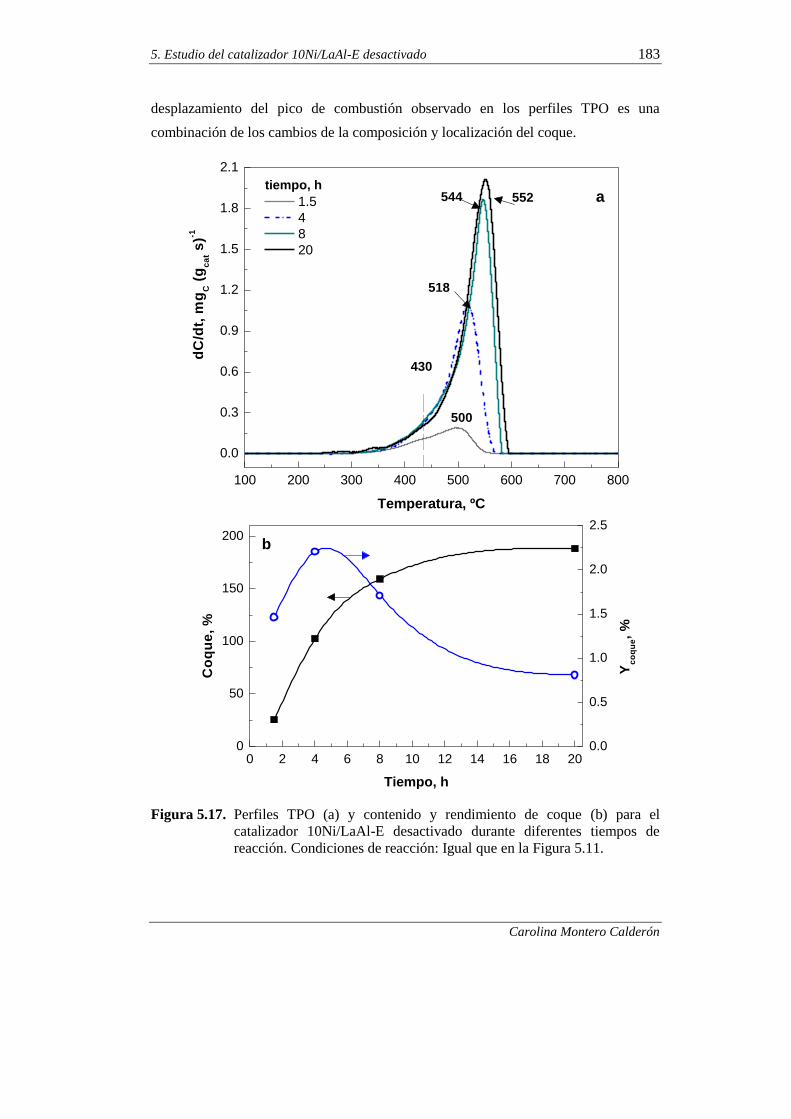
5. Estudio del catalizador 10Ni/LaAl-E desactivado 183
Carolina Montero Calderón
desplazamiento del pico de combustión observado en los perfiles TPO es una
combinación de los cambios de la composición y localización del coque.
Figura 5.17. Perfiles TPO (a) y contenido y rendimiento de coque (b) para el
catalizador 10Ni/LaAl-E desactivado durante diferentes tiempos de
reacción. Condiciones de reacción: Igual que en la Figura 5.11.
100 200 300 400 500 600 700 800
0.0
0.3
0.6
0.9
1.2
1.5
1.8
2.1
a
430
552544
518
dC
/dt,
mg
C (
gc
at s
)-1
Temperatura, ºC
tiempo, h
1.5
4
8
20
500
0 2 4 6 8 10 12 14 16 18 200
50
100
150
200
Co
qu
e,
%
Tiempo, h
0.0
0.5
1.0
1.5
2.0
2.5
b
Yc
oq
ue,
%

184
Capítulo 5
El hombro a 430 ºC corresponde a un coque amorfo, que evoluciona en las
primeras 4 h de reacción, y a partir de ese momento se produce un equilibrio entre su
formación, su transformación en coque más grafítico (que quema a temperaturas
superiores a 500 ºC) y su gasificación para producir CO e H2 (Remiro y cols., 2013b).
Como consecuencia de esta rápida formación de coque amorfo el rendimiento
de coque (Rcoque) tiene un máximo a tiempos de reacción intermedios (Figura 5.17b)
mientras que el contenido de coque aumenta asintóticamente como corresponde a una
evolución que afecta fundamentalmente a su estructura.
Según la bibliografía (Koo y cols., 2008; Vicente y cols., 2014a), la oxidación
de coque fibrilar ocurre a temperaturas mayores a 450 °C, mientras que los picos de
mayor temperatura de combustión (550-600 °C) corresponden a depósitos de coque
con diferentes grados de grafitización, incluyendo filamentos altamente desarrollados.
De esta forma, los resultados de TPO mostrados en la Figura 5.17a indican la
aparición de coque pirolítico para 4 h de reacción, que conlleva el desplazamiento del
máximo del pico de combustión por encima 520 ºC. En cualquier caso, el coque
depositado a partir de este tiempo es una mezcla de coque fibrilar y pirolítico, con una
composición y estructura muy heterogénea.
De la comparación de los perfiles de TPO con los resultados de la concentración
del medio de reacción en estas condiciones (Figura 5.11), se puede concluir que el
coque formado es una combinación de: i) coque encapsulante, proveniente de
oxigenados (particularmente acetaldehído, y etanol, a través de especies etoxi
adsorbidas) y etileno (despreciable en las condiciones experimentales); ii) coque
fibrilar, proveniente de las reacciones de CO y CH4 y; iii) coque pirolítico, promovido
por la alta concentración de oxigenados como el etanol y el acetaldehído (Matas y
cols., 2009). De esta forma, la velocidad de formación de coque en la etapas1 (t < 4 h),
donde esta favorecido el crecimiento del coque fibrilar, es más rápida que en las etapas
2 y 3, donde predomina el crecimiento del coque pirolítico. Se ha establecido que
según disminuye con el tiempo el tamaño de cristal de Ni (Tabla 5.5) el crecimiento de
las fibras pasa por un máximo, que aproximadamente es 4 h en las condiciones
estudiadas, y a partir de ese momento el crecimiento se ralentiza. Esta relación entre la
evolución con el tiempo de la estructura del coque y tamaño de cristal de Ni es acorde

5. Estudio del catalizador 10Ni/LaAl-E desactivado 185
Carolina Montero Calderón
con la bibliografía (Kim y cols., 2000; Chen y cols., 2005).
5.2.6. Espectroscopía FTIR
En la Figura 5.19 se presentan los resultados de las dos regiones de vibración
molecular en el espectro FTIR correspondiente al coque depositado sobre el
catalizador usado durante 4h: 900-1900 cm-1
, Figura 5.19a; y 2700-3100 cm-1
, Figura
5.19b.
Las bandas identificadas han sido asignadas a enlaces presentes en el
catalizador y el coque (Karge y cols., 1996; Robertson, 2002; Epelde y cols., 2014c;
Ochoa y cols., 2014): 1106 cm-1
, óxidos del soporte; 1222 cm-1
, enlaces C-C del
coque; 1444 cm-1
, coque alifático con grupos -CH2 y -CH3; 1575 cm-1
, coque
aromático policondensado; 1631 cm-1
, dienos y dobles enlaces conjugados; 2913 cm
-1,
coque alifático con grupos -CH y -CH2. Estos valores son aproximados ya que pueden
observarse ciertas desviaciones de las bandas.
Como se muestra en la Figura 5.19, cada una de estas bandas se ha
deconvolucionado en picos Lorentzianos, y la evolución del área de las bandas más
representativas se muestra en la Figura 5.20. Las bandas relacionadas con enlaces del
soporte, a 1106 cm-1
, y la de dienos a 1631 cm-1
, disminuyen con el tiempo de
reacción, a la vez que se observa un aumento de las bandas del coque aromático y
policondensado (1575 y 1222 cm-1
) y un aumento menos significativo del coque
alifático (1444 y 2913 cm-1
). Estudios FTIR sobre coque fibrilar señalan como banda
característica de este coque la ubicada a 1450-1460 cm-1
(Teng y Tang, 2008). Estos
resultados permiten correlacionar la composición aromática policondensada del coque
fibrilar y del coque pirolítico, con una diferencia de composición en términos de
enlaces muy baja.
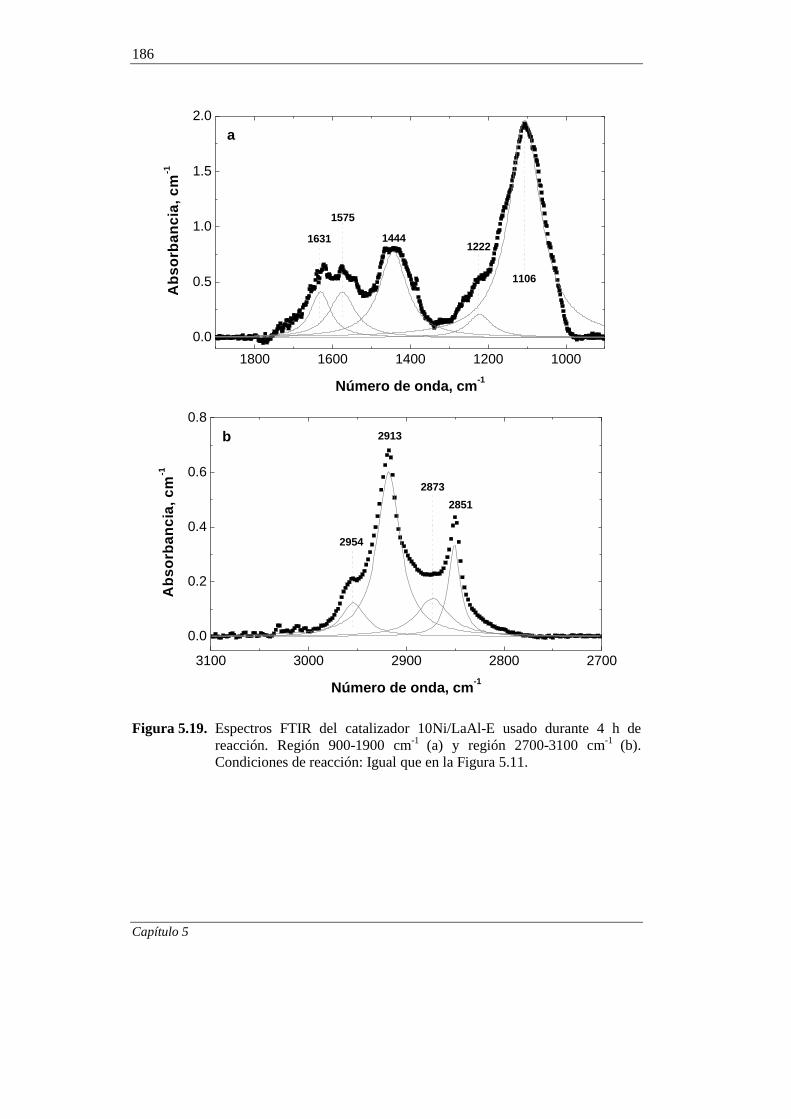
186
Capítulo 5
Figura 5.19. Espectros FTIR del catalizador 10Ni/LaAl-E usado durante 4 h de
reacción. Región 900-1900 cm-1
(a) y región 2700-3100 cm-1
(b).
Condiciones de reacción: Igual que en la Figura 5.11.
1800 1600 1400 1200 1000
0.0
0.5
1.0
1.5
2.0
a
1106
12221444
1575
Ab
so
rba
nc
ia,
cm
-1
Número de onda, cm-1
1631
3100 3000 2900 2800 2700
0.0
0.2
0.4
0.6
0.8
b
2851
2873
2913
2954
Ab
so
rba
nc
ia,
cm
-1
Número de onda, cm-1
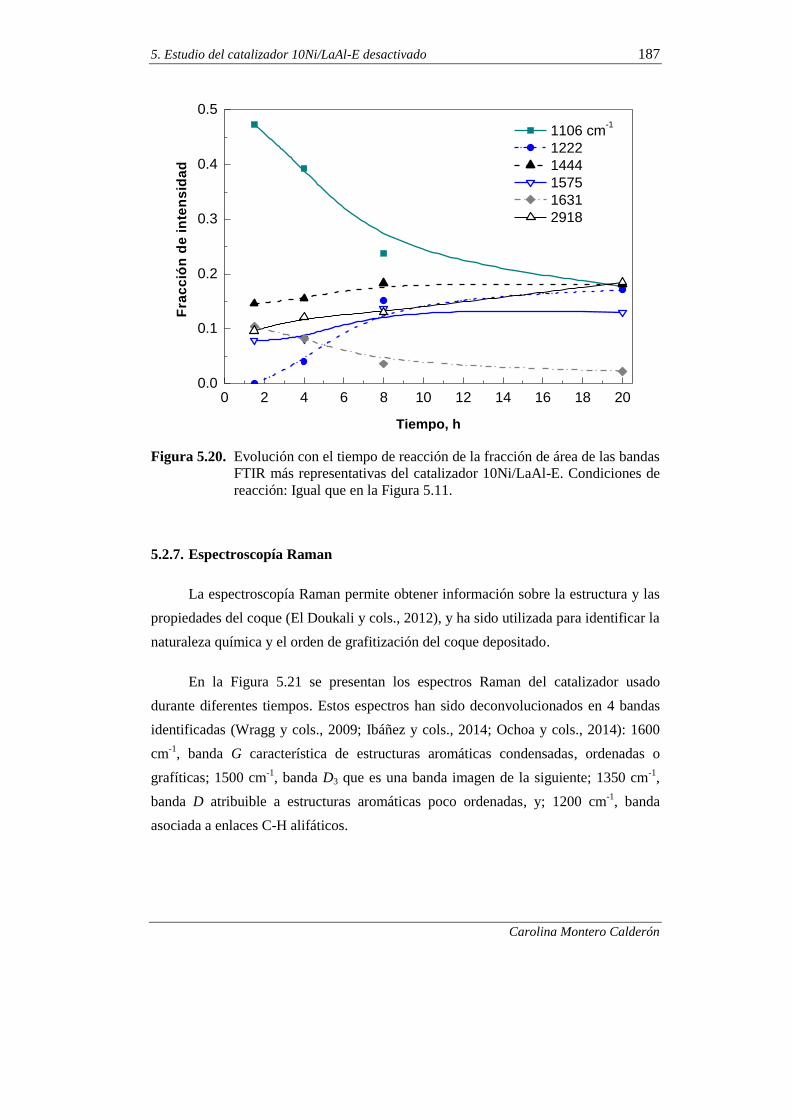
5. Estudio del catalizador 10Ni/LaAl-E desactivado 187
Carolina Montero Calderón
Figura 5.20. Evolución con el tiempo de reacción de la fracción de área de las bandas
FTIR más representativas del catalizador 10Ni/LaAl-E. Condiciones de
reacción: Igual que en la Figura 5.11.
5.2.7. Espectroscopía Raman
La espectroscopía Raman permite obtener información sobre la estructura y las
propiedades del coque (El Doukali y cols., 2012), y ha sido utilizada para identificar la
naturaleza química y el orden de grafitización del coque depositado.
En la Figura 5.21 se presentan los espectros Raman del catalizador usado
durante diferentes tiempos. Estos espectros han sido deconvolucionados en 4 bandas
identificadas (Wragg y cols., 2009; Ibáñez y cols., 2014; Ochoa y cols., 2014): 1600
cm-1
, banda G característica de estructuras aromáticas condensadas, ordenadas o
grafíticas; 1500 cm-1
, banda D3 que es una banda imagen de la siguiente; 1350 cm-1
,
banda D atribuible a estructuras aromáticas poco ordenadas, y; 1200 cm-1
, banda
asociada a enlaces C-H alifáticos.
0 2 4 6 8 10 12 14 16 18 200.0
0.1
0.2
0.3
0.4
0.5
Fra
cc
ión
de
in
ten
sid
ad
Tiempo, h
1106 cm-1
1222
1444
1575
1631
2918
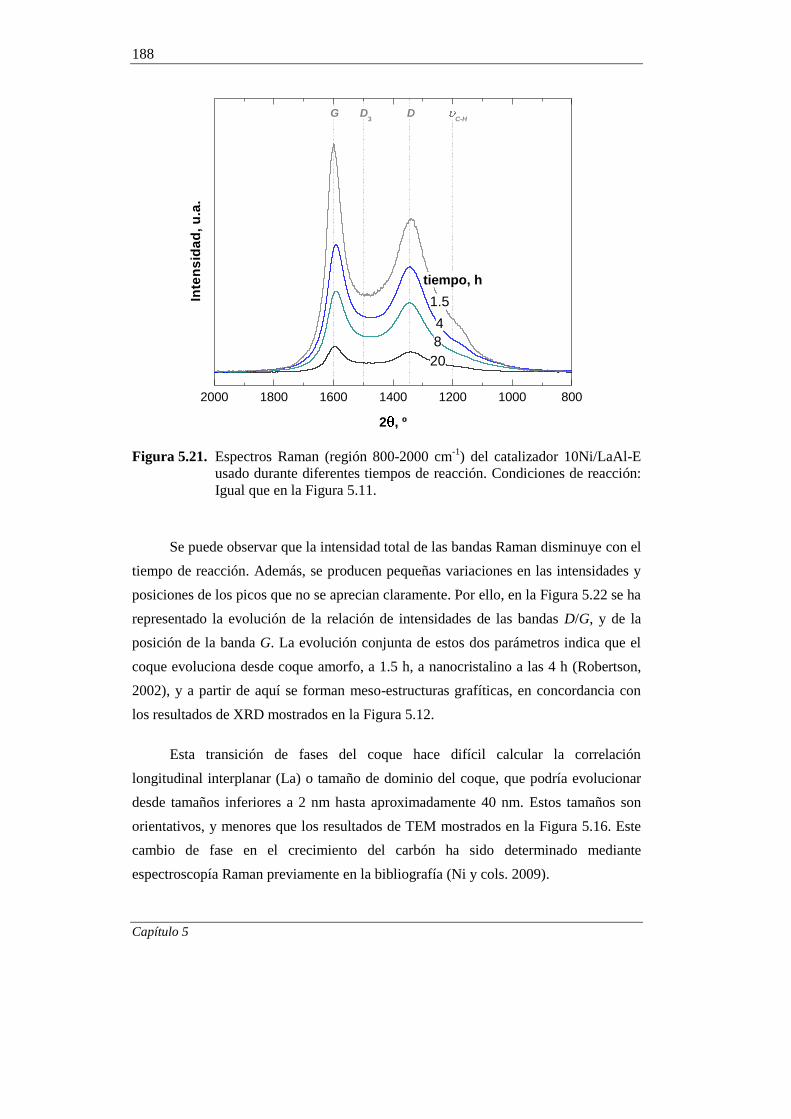
188
Capítulo 5
Figura 5.21. Espectros Raman (región 800-2000 cm-1
) del catalizador 10Ni/LaAl-E
usado durante diferentes tiempos de reacción. Condiciones de reacción:
Igual que en la Figura 5.11.
Se puede observar que la intensidad total de las bandas Raman disminuye con el
tiempo de reacción. Además, se producen pequeñas variaciones en las intensidades y
posiciones de los picos que no se aprecian claramente. Por ello, en la Figura 5.22 se ha
representado la evolución de la relación de intensidades de las bandas D/G, y de la
posición de la banda G. La evolución conjunta de estos dos parámetros indica que el
coque evoluciona desde coque amorfo, a 1.5 h, a nanocristalino a las 4 h (Robertson,
2002), y a partir de aquí se forman meso-estructuras grafíticas, en concordancia con
los resultados de XRD mostrados en la Figura 5.12.
Esta transición de fases del coque hace difícil calcular la correlación
longitudinal interplanar (La) o tamaño de dominio del coque, que podría evolucionar
desde tamaños inferiores a 2 nm hasta aproximadamente 40 nm. Estos tamaños son
orientativos, y menores que los resultados de TEM mostrados en la Figura 5.16. Este
cambio de fase en el crecimiento del carbón ha sido determinado mediante
espectroscopía Raman previamente en la bibliografía (Ni y cols. 2009).
2000 1800 1600 1400 1200 1000 800
D3
G DC-H
tiempo, h
Inte
ns
ida
d,
u.a
.
2 , º
1.5
4
8
20
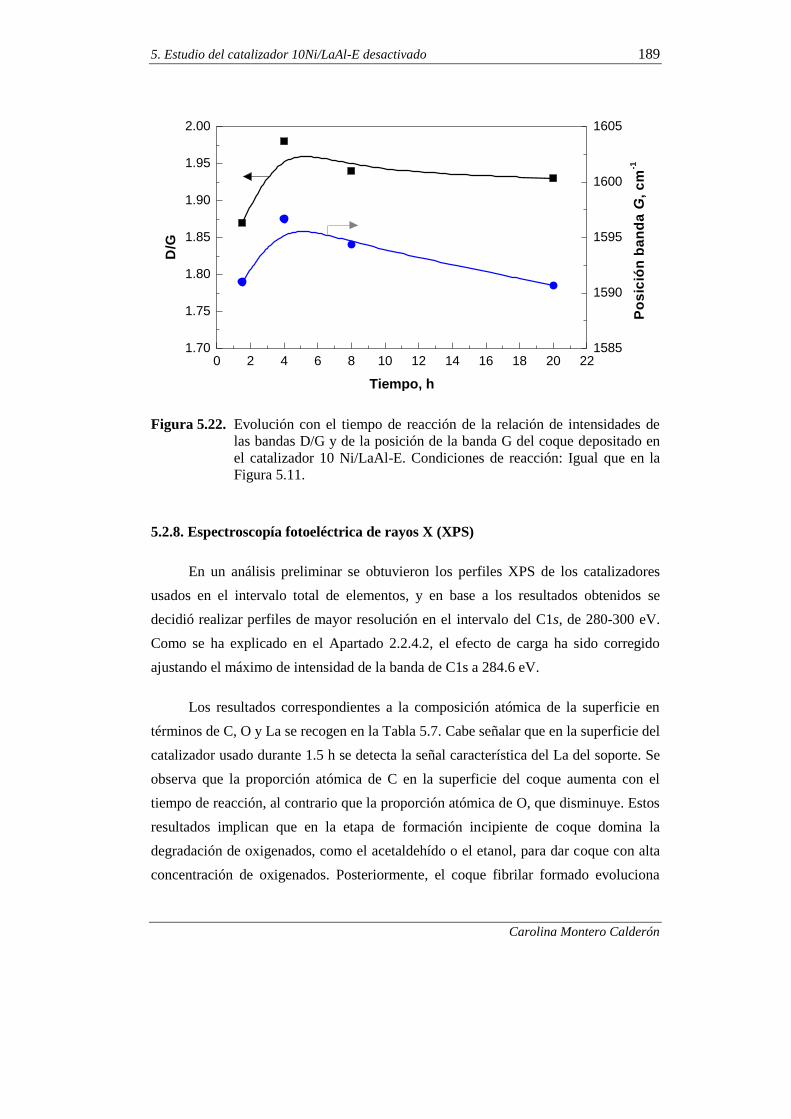
5. Estudio del catalizador 10Ni/LaAl-E desactivado 189
Carolina Montero Calderón
Figura 5.22. Evolución con el tiempo de reacción de la relación de intensidades de
las bandas D/G y de la posición de la banda G del coque depositado en
el catalizador 10 Ni/LaAl-E. Condiciones de reacción: Igual que en la
Figura 5.11.
5.2.8. Espectroscopía fotoeléctrica de rayos X (XPS)
En un análisis preliminar se obtuvieron los perfiles XPS de los catalizadores
usados en el intervalo total de elementos, y en base a los resultados obtenidos se
decidió realizar perfiles de mayor resolución en el intervalo del C1s, de 280-300 eV.
Como se ha explicado en el Apartado 2.2.4.2, el efecto de carga ha sido corregido
ajustando el máximo de intensidad de la banda de C1s a 284.6 eV.
Los resultados correspondientes a la composición atómica de la superficie en
términos de C, O y La se recogen en la Tabla 5.7. Cabe señalar que en la superficie del
catalizador usado durante 1.5 h se detecta la señal característica del La del soporte. Se
observa que la proporción atómica de C en la superficie del coque aumenta con el
tiempo de reacción, al contrario que la proporción atómica de O, que disminuye. Estos
resultados implican que en la etapa de formación incipiente de coque domina la
degradación de oxigenados, como el acetaldehído o el etanol, para dar coque con alta
concentración de oxigenados. Posteriormente, el coque fibrilar formado evoluciona
0 2 4 6 8 10 12 14 16 18 20 221.70
1.75
1.80
1.85
1.90
1.95
2.00
D/G
Tiempo, h
1585
1590
1595
1600
1605
Po
sic
ión
ba
nd
a G
, c
m-1
190
Capítulo 5
hacia estructuras más desoxigenadas y grafíticas, y en la formación de más coque
predomina la descomposición de CH4 y la reacción de Boudouard.
Tabla 5.7. Composición atómica de la superficie del catalizador 10Ni/LaAl-E usado
durante diferente tiempo de reacción. Condiciones de reacción: Igual que
en la Figura 5.11.
tiempo, h % atómico
O/C C O La
1.5 86.18 13.24 0.59 1.701
4 97.92 2.08 - 0.021
8 98.20 1.80 - 0.018
20 98.66 1.34 - 0.014
Los espectros XPS correspondientes a la región del C1s se han representado en
la Figura 5.23. Muestran una banda mayoritaria y varios hombros anexos identificados
como los siguientes entornos electrónicos del C (Estrade-Szwarcockpf, 2004; Zhou y
cols., 2007; Choong y cols., 2011; Coulumbe y cols., 2012; Koo y cols., 2014; Ochoa
y cols., 2014): 284.6 ± 0.0 eV enlaces C-C y C-H; 285.8 ± 0.1 eV enlaces C-O y
defectos; 287.2 ± 0.2 eV enlaces C=O y O-C=O.
La composición de cada entorno electrónico del C ha sido calculada a partir del
área de cada banda y los resultados se muestran en la Tabla 5.8.
Las bandas de 285.8-290.0 eV pueden considerarse transiciones electrónicas
π→π* en defectos o imperfecciones cristalinas del grafito, atribuido a átomos de C
localizados fuera de la configuración grafítica sp2 (Estrada-Szwarcockpf, 2004). La
intensidad de estas bandas aumenta con el tiempo de reacción, conforme disminuye el
contenido de oxígeno en el coque, por lo que se concluye que a partir de 4 h aumentan
los defectos estructurales del carbón. Este resultado, junto con las conclusiones
derivadas de los resultados del análisis TPO (Figura 5.17) indican que la naturaleza
del coque pirolítico (que quema a mayor temperatura) es menos grafítica y más
desordenada que la del coque fibrilar. El hecho de que requiere mayores temperaturas
de combustión para indicar por tanto, que el contacto del Ni (que cataliza la
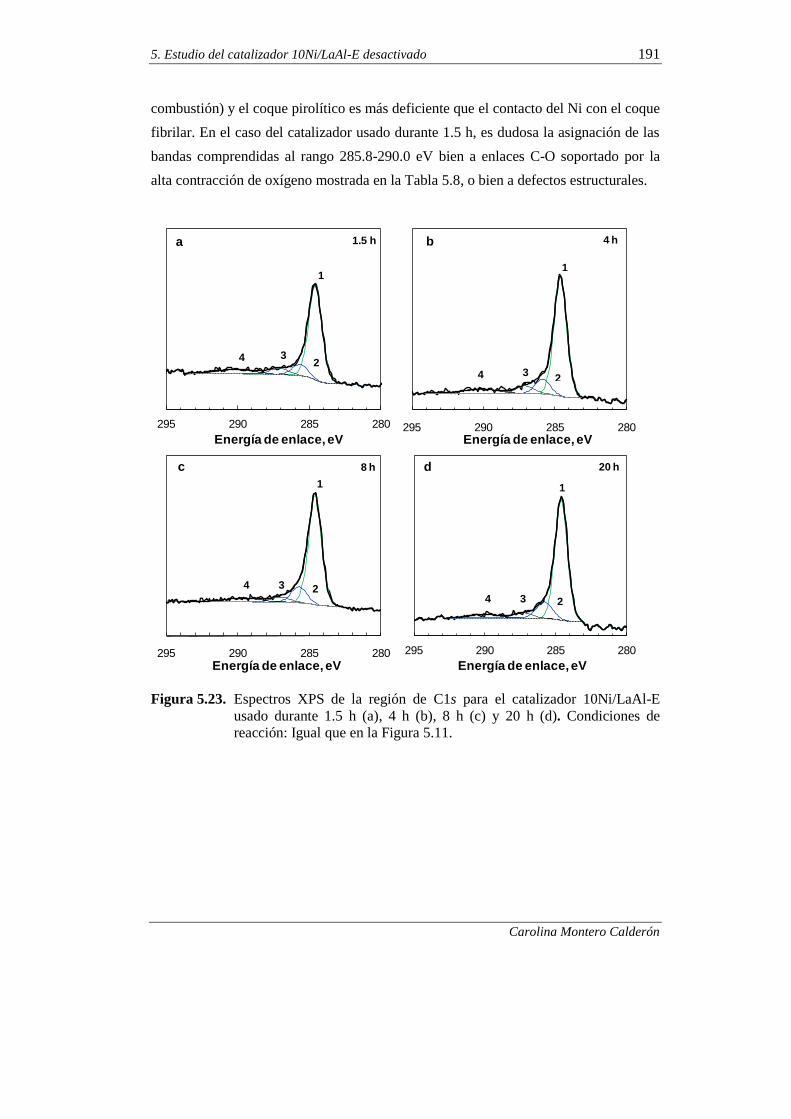
5. Estudio del catalizador 10Ni/LaAl-E desactivado 191
Carolina Montero Calderón
combustión) y el coque pirolítico es más deficiente que el contacto del Ni con el coque
fibrilar. En el caso del catalizador usado durante 1.5 h, es dudosa la asignación de las
bandas comprendidas al rango 285.8-290.0 eV bien a enlaces C-O soportado por la
alta contracción de oxígeno mostrada en la Tabla 5.8, o bien a defectos estructurales.
Figura 5.23. Espectros XPS de la región de C1s para el catalizador 10Ni/LaAl-E
usado durante 1.5 h (a), 4 h (b), 8 h (c) y 20 h (d). Condiciones de
reacción: Igual que en la Figura 5.11.
280285290295
Energía de enlace, eV
1.5 ha
1
234
280285290295Energía de enlace, eV
4 hb
1
234
280285290295Energía de enlace, eV
8 hc
1
234
280285290295
Energía de enlace, eV
20 hd
1
34 2
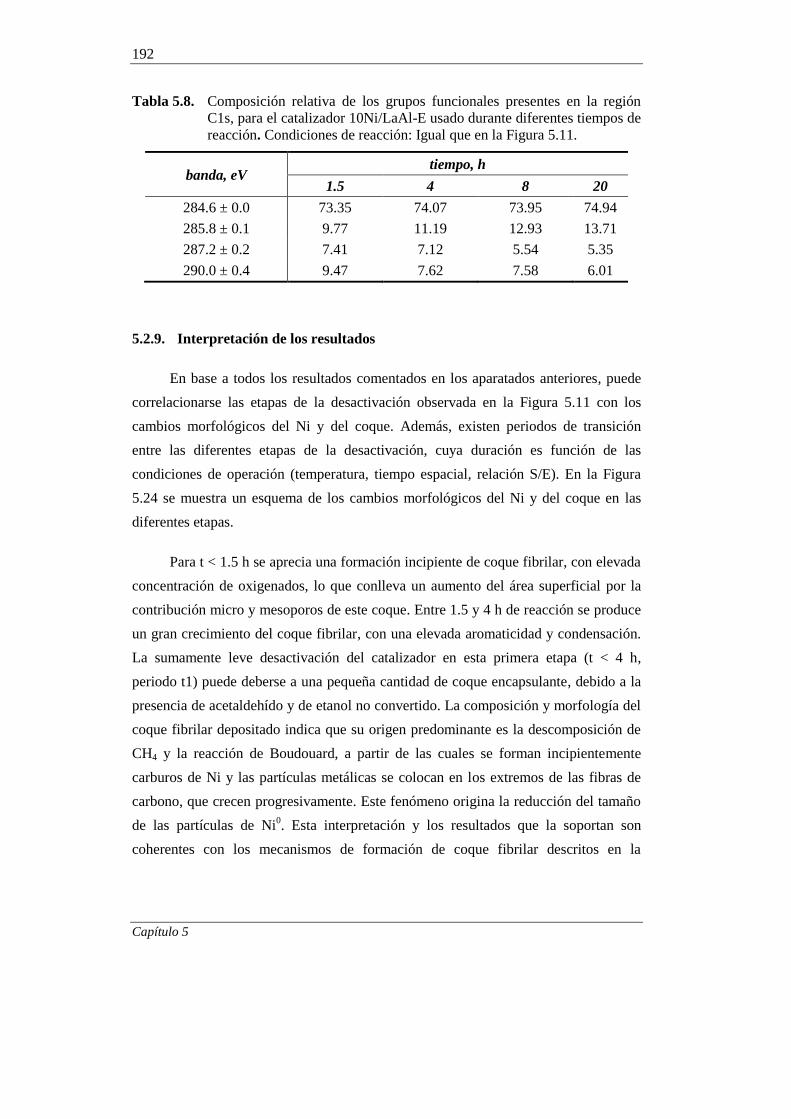
192
Capítulo 5
Tabla 5.8. Composición relativa de los grupos funcionales presentes en la región
C1s, para el catalizador 10Ni/LaAl-E usado durante diferentes tiempos de
reacción. Condiciones de reacción: Igual que en la Figura 5.11.
banda, eV tiempo, h
1.5 4 8 20
284.6 ± 0.0 73.35 74.07 73.95 74.94
285.8 ± 0.1 9.77 11.19 12.93 13.71
287.2 ± 0.2 7.41 7.12 5.54 5.35
290.0 ± 0.4 9.47 7.62 7.58 6.01
5.2.9. Interpretación de los resultados
En base a todos los resultados comentados en los aparatados anteriores, puede
correlacionarse las etapas de la desactivación observada en la Figura 5.11 con los
cambios morfológicos del Ni y del coque. Además, existen periodos de transición
entre las diferentes etapas de la desactivación, cuya duración es función de las
condiciones de operación (temperatura, tiempo espacial, relación S/E). En la Figura
5.24 se muestra un esquema de los cambios morfológicos del Ni y del coque en las
diferentes etapas.
Para t < 1.5 h se aprecia una formación incipiente de coque fibrilar, con elevada
concentración de oxigenados, lo que conlleva un aumento del área superficial por la
contribución micro y mesoporos de este coque. Entre 1.5 y 4 h de reacción se produce
un gran crecimiento del coque fibrilar, con una elevada aromaticidad y condensación.
La sumamente leve desactivación del catalizador en esta primera etapa (t < 4 h,
periodo t1) puede deberse a una pequeña cantidad de coque encapsulante, debido a la
presencia de acetaldehído y de etanol no convertido. La composición y morfología del
coque fibrilar depositado indica que su origen predominante es la descomposición de
CH4 y la reacción de Boudouard, a partir de las cuales se forman incipientemente
carburos de Ni y las partículas metálicas se colocan en los extremos de las fibras de
carbono, que crecen progresivamente. Este fenómeno origina la reducción del tamaño
de las partículas de Ni0. Esta interpretación y los resultados que la soportan son
coherentes con los mecanismos de formación de coque fibrilar descritos en la
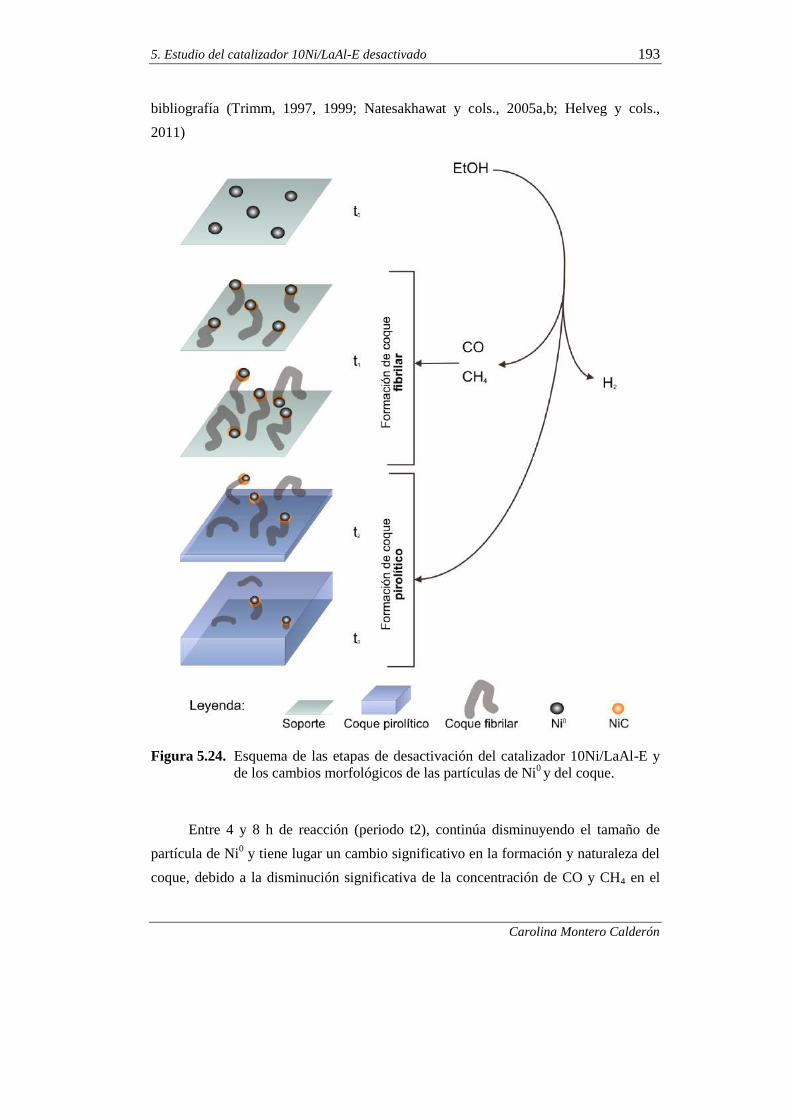
5. Estudio del catalizador 10Ni/LaAl-E desactivado 193
Carolina Montero Calderón
bibliografía (Trimm, 1997, 1999; Natesakhawat y cols., 2005a,b; Helveg y cols.,
2011)
Figura 5.24. Esquema de las etapas de desactivación del catalizador 10Ni/LaAl-E y
de los cambios morfológicos de las partículas de Ni0 y del coque.
Entre 4 y 8 h de reacción (periodo t2), continúa disminuyendo el tamaño de
partícula de Ni0 y tiene lugar un cambio significativo en la formación y naturaleza del
coque, debido a la disminución significativa de la concentración de CO y CH4 en el

194
Capítulo 5
medio de reacción y al aumento de la concentración de acetaldehído y, sobre todo, de
etanol. Es decir, cambian los precursores de formación de coque, lo que conlleva un
cambio en el mecanismo de formación de coque (Shaikjee y Coville, 2012). Así,
aunque continúa aumentando el contenido de coque depositado en el catalizador, el
rendimiento de coque disminuye, y se produce una transición en su crecimiento, con
aumento del coque fibrilar grafítico y aparición del coque pirolítico, identificado como
agente principal de la desactivación.
A partir de 8 h de reacción (periodo t3), se opera en estado de desactivación casi
pseudo-estacionario, con un predominio de la formación de coque pirolítico a partir de
etanol. En consecuencia, tiene lugar la saturación en la formación de coque (el
rendimiento disminuye hasta casi cero), con bloqueo de centros de Ni0 y alta
grafitización del coque.

5. Estudio del catalizador 10Ni/LaAl-E desactivado 195
Carolina Montero Calderón
5.3. REGENERABILIDAD DEL CATALIZADOR 10Ni/LaAl-E MEDIANTE
GASIFICACIÓN DEL COQUE
Como se ha comprobado en el Capítulo 3, el catalizador 10Ni/LaAl equilibrado
permite llevar a cabo de forma reproducible el proceso SRE en ciclos de reacción-
regeneración, cuando la regeneración se realiza mediante combustión con aire a 550
ºC durante 2 h. En estudios previos (Alenazey y cols., 2009; Chen y cols., 2014) se ha
propuesto la gasificación del coque con vapor de agua, Ec. (1.26), como alternativa
para la etapa de regeneración de los catalizadores de Ni desactivados en procesos de
reformado. En esta Tesis se ha querido explorar esta posibilidad, estudiando la
gasificación del coque in situ en el reactor de reformado. El objetivo es comprobar si
la gasificación es eficaz para la completa regeneración del catalizador, de forma que la
operación cíclica sea reproducible.
Para ello, se ha llevado a cabo una reacción de reformado en las siguientes
condiciones de operación: 500 ºC, S/E=3; W/F0= 0.09 gcath/gEtOH, PEtOH=0.08 bar y
tiempo de reacción de 7 h. Tras la reacción, se ha llevado a cabo la etapa de
gasificación in situ manteniendo las mismas condiciones fluidodinámicas en el lecho,
de modo que el caudal de vapor y de inerte se han establecido para obtener una
relación u/umf = 6, siendo los caudales utilizados de 0.1 ml/min de agua y 30 ml
(CN)/min de He. Para analizar el efecto de la temperatura, se ha estudiado la
gasificación en el intervalo 500-650 ºC, con escalones crecientes y duración de unos
40 min a cada temperatura intermedia (500, 550 y 600 ºC), mientras que la duración
del último escalón a 650 ºC fue de 150 min, hasta obtener la ausencia total de los
posibles productos de la gasificación (CO, CO2 e H2) en la corriente de salida. Los
resultados de composición de la corriente de productos durante la etapa de
gasificación se muestran en la Figura 5.25.
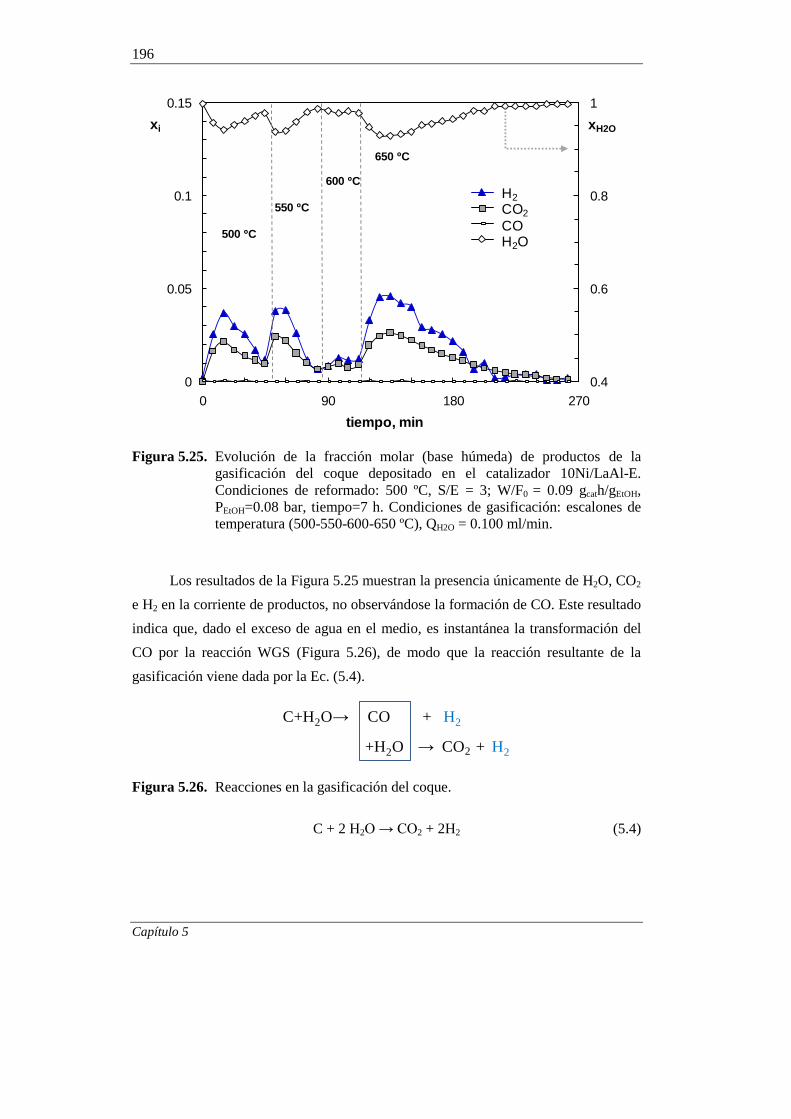
196
Capítulo 5
Figura 5.25. Evolución de la fracción molar (base húmeda) de productos de la
gasificación del coque depositado en el catalizador 10Ni/LaAl-E.
Condiciones de reformado: 500 ºC, S/E = 3; W/F0 = 0.09 gcath/gEtOH,
PEtOH=0.08 bar, tiempo=7 h. Condiciones de gasificación: escalones de
temperatura (500-550-600-650 ºC), QH2O = 0.100 ml/min.
Los resultados de la Figura 5.25 muestran la presencia únicamente de H2O, CO2
e H2 en la corriente de productos, no observándose la formación de CO. Este resultado
indica que, dado el exceso de agua en el medio, es instantánea la transformación del
CO por la reacción WGS (Figura 5.26), de modo que la reacción resultante de la
gasificación viene dada por la Ec. (5.4).
Figura 5.26. Reacciones en la gasificación del coque.
C + 2 H2O → CO2 + 2H2 (5.4)
0.4
0.6
0.8
1
0
0.05
0.1
0.15
0 90 180 270
xH2Oxi
tiempo, min
H2
CO2
COH2O
500 ºC
550 ºC
600 ºC
650 ºC
C+H2O→ CO + H2
+H2O → CO2 + H2

5. Estudio del catalizador 10Ni/LaAl-E desactivado 197
Carolina Montero Calderón
Se observa que a 500 ºC se gasifica una cierta fracción del coque, y al aumentar
la temperatura de 550 ºC se logra gasificar una fracción adicional. Sin embargo, con
un posterior aumento de la temperatura de gasificación hasta 600 ºC el aumento de la
cantidad de coque gasificada es casi insignificante, siendo necesario aumentar la
temperatura a 650 ºC para que continúe gasificándose el resto del coque.
Estos resultados pueden relacionarse con los perfiles de TPO correspondientes
al coque depositado en las mismas condiciones de reacción. Como se observa en las
Figuras 5.4a y 5.7a, y en los resultados para 500 ºC de las Figuras 5.1 y 5.2b, para esta
temperatura de reformado y un tiempo espacial moderado el perfil de TPO del coque
tiene una amplia cola inicial, y el pico mayoritario de coque tiene un máximo por
encima de 550 ºC. Por tanto, si bien el coque depositado es mayoritariamente de tipo
fibrilar, hay una pequeña fracción del coque de diferente naturaleza, que quema antes,
y que, por los resultados de gasificación, es también más fácilmente gasificable,
pudiendo ser gasificado a baja temperatura (en el intervalo 500-500 ºC). Sin embargo,
se requiere una elevada temperatura (650 ºC) para la gasificación del coque fibrilar,
que como se ha indicado quema con facilidad por encima de 550 ºC.
Para verificar si la eliminación del coque por gasificación es eficaz para lograr
la total regeneración del catalizador, se procedió a realizar una segunda etapa de
reacción, en las mismas condiciones de reformado. Los resultados de las dos etapas de
reacción, se muestran en la Figura 5.27. El catalizador regenerado es nuevamente
activo, y tiene una tendencia de evolución con el tiempo de los índices de reacción
similar a la obtenida en el primer ciclo de reacción, aunque se observa una ligera
disminución en la conversión y rendimiento de H2 a tiempo cero respecto de los
valores iniciales con el catalizador fresco. Esta pequeña pérdida de actividad podría
ser debida a que permanece un cierto contenido de coque residual, que no ha podido
ser eliminado por gasificación, dado que no es probable que haya tenido lugar la
sinterización del Ni en las condiciones de gasificación utilizadas, puesto que los
ensayos de larga duración (200 h) a 650 ºC y con una elevada relación S/E (Figura
4.20) mostraron que no tiene lugar la sinterización apreciable del metal.
Cabe indicar que el aumento de temperatura para lograr la total gasificación del
coque no es una estrategia recomendable, dado que una mayor temperatura de
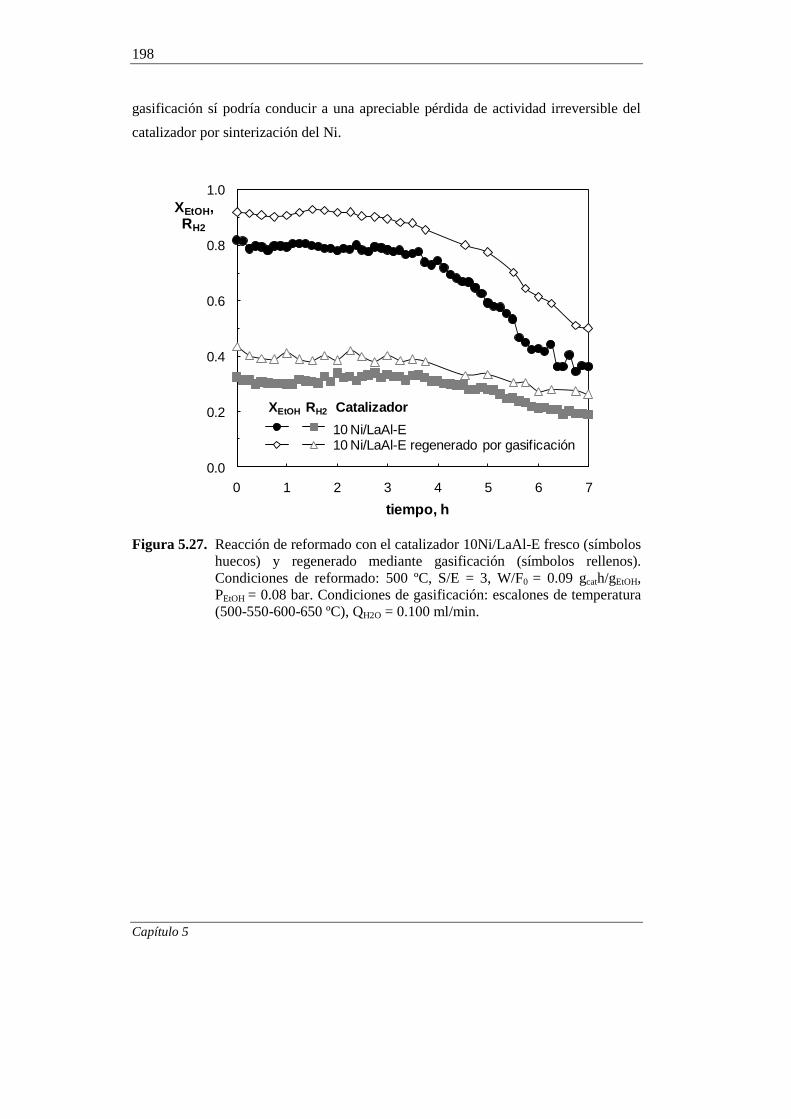
198
Capítulo 5
gasificación sí podría conducir a una apreciable pérdida de actividad irreversible del
catalizador por sinterización del Ni.
Figura 5.27. Reacción de reformado con el catalizador 10Ni/LaAl-E fresco (símbolos
huecos) y regenerado mediante gasificación (símbolos rellenos).
Condiciones de reformado: 500 ºC, S/E = 3, W/F0 = 0.09 gcath/gEtOH,
PEtOH = 0.08 bar. Condiciones de gasificación: escalones de temperatura
(500-550-600-650 ºC), QH2O = 0.100 ml/min.
0.0
0.2
0.4
0.6
0.8
1.0
0 1 2 3 4 5 6 7
XEtOH,RH2
tiempo, h
XEtOH RH2 Catalizador
10 Ni/LaAl-E10 Ni/LaAl-E regenerado por gasificación

6. MODELADO CINÉTICO DEL SRE
SOBRE EL CATALIZADOR 10Ni/LaAl-E


201
Carolina Montero Calderón
6. MODELADO CINÉTICO DEL SRE SOBRE CATALIZADOR
10Ni/LaAl-E
El objetivo de este capítulo es cuantificar, con un modelo cinético, el efecto de
las condiciones de reacción (temperatura, tiempo espacial, relación molar agua y
etanol, tiempo) sobre la distribución de productos en el proceso de obtención de
hidrógeno por reformado con vapor de etanol. En la bibliografía se han propuestos
varios modelos para describir la cinética a tiempo cero (catalizador fresco), si bien en
su mayoría se trata de modelos simplificados, que únicamente tienen en cuenta la
reacción de reformado directo con vapor, Ec. (1.8), sin considerar las reacciones
secundarias involucradas y, por tanto, no consideran ni cuantifican la formación de
subproductos en el proceso. Además, no hay modelos en la bibliografía que
cuantifiquen la desactivación del catalizador.
El estudio se ha realizado en dos etapas:
i) Estableciendo un modelo cinético aplicable a tiempo cero que proporcione un
compromiso adecuado entre sencillez y realismo para ajustar los resultados
experimentales de la composición de los productos de reacción correspondiente a
un amplio intervalo de las variables de operación (temperatura, relación molar
S/E y tiempo espacial). Para ello, se ha seleccionado un esquema cinético con
varias etapas de reacción, y se han determinado las expresiones de la velocidad de
reacción para cada una de las etapas y sus correspondientes parámetros cinéticos.
ii) Incorporando en el modelo una ecuación cinética de desactivación, que cuantifica
la disminución de la actividad del catalizador con el tiempo, considerando el
efecto sobre esta desactivación de la temperatura y concentración de
componentes en el medio de reacción.
La metodología aplicada para el modelado cinético puede resumirse en el
siguiente esquema:

202
Capítulo 6
Figura 6.1. Procedimiento de propuesta y discriminación del modelo cinético.
Cabe recalcar que para el establecimiento del modelo cinético se ha tenido en
cuenta toda la información obtenida en los capítulos anteriores referente al mecanismo
de reacción y comportamiento de desactivación, lo que ha permitido considerar el
papel de todos los componentes del medio de reacción identificados en las diferentes
condiciones de reacción estudiadas: los reactivos (agua y etanol), los productos
mayoritarios (H2, CO2, CO y CH4), así como los subproductos minoritarios
(acetaldehído y etileno).
Establecerdiferentesesquemascinéticos
alternativos
Proponer las expresiones de
velocidad de reacciónpara cada etapa de los
esquemas cinéticos
Cálculo de losparámetros
cinéticos y nivelde ajuste de cada
modelo
Discriminación de los modelos

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 203
Carolina Montero Calderón
6.1. MODELADO CINÉTICO A TIEMPO CERO
6.1.1. Antecedentes bibliográficos
El modelado cinético del proceso de reformado con vapor de etanol ha sido
recientemente estudiado por varios grupos, con diferentes catalizadores y tipos de
reactor. Se han considerado tanto ecuaciones empíricas, de tipo potencial, como
modelos mecanísticos, del tipo de Langmuir-Hinshelwood-Hougen-Watson (LHHW)
y de tipo Eley Rideal (ER), diferenciándose estos modelos mecanísticos entre sí en
que los primeros (LHHW) contemplan la adsorción de todas las especies reactivas en
los centros activos del catalizador, mientras que en los segundos (ER) solo se requiere
la adsorción de una especie de reactante.
6.1.1.1. Modelos cinéticos para catalizadores de Ni
Las Tablas 6.1 y 6.2 recopilan algunas de las ecuaciones cinéticas empíricas y
mecanísticas, respectivamente, planteadas en la bibliografía para la reacción de
reformado con vapor de etanol (Ec. (1.8)) sobre catalizadores de Ni.
Los primeros intentos por determinar el modelo cinético fueron realizados por
Therdthianwong y cols. (2001), quienes analizaron el comportamiento del catalizador
de Ni/Al2O3 en un reactor de lecho fijo. Para aplicar el proceso a celdas de
combustible y atenuar la formación de coque que resulta del craqueo térmico del
etanol a altas temperaturas, plantearon un sistema de reacción con dos etapas en serie:
una primera etapa de pre-reformado de etanol a 400 ºC, para obtener H2 e intermedios
como CH4 y CO2, que posteriormente son transformados en una segunda etapa a
mayor temperatura (650 ºC), mediante reformado de CH4 y WGS inversa. Estos
autores desarrollaron únicamente la ecuación cinética para la primera etapa con una
ecuación potencial, Ec.(6.1), asumiendo que ésta es la etapa controlante del proceso
global.
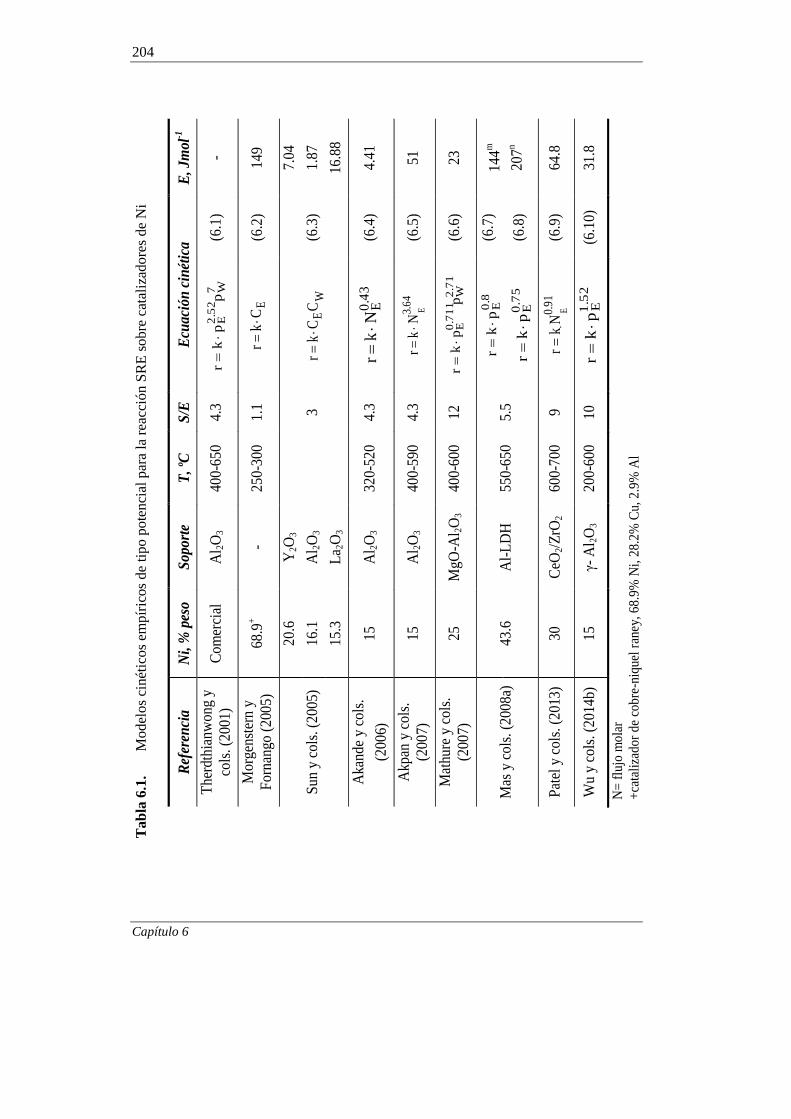
204
Capítulo 6
Ta
bla
6.1
. M
odel
os
cinét
icos
empír
icos
de
tipo p
ote
nci
al p
ara
la r
eacc
ión
SR
E s
ob
re c
atal
izad
ore
s d
e N
i
Ref
eren
cia
Ni,
% p
eso
So
po
rte
T,
ºC
S/E
E
cua
ció
n c
inét
ica
E,
Jmol
-1
The
rdth
ianw
ong
y
cols
. (20
01)
Com
erci
al
Al 2
O3
400-
650
4.
3 7 W
2.5
2E
pp
kr
(6.1
) -
Mor
gens
tern
y
For
nang
o (2
005)
68
.9+
- 25
0-30
0
1.1
EC
kr
(6
.2)
149
Sun
y c
ols.
(20
05)
20.6
Y
2O3
3 W
EC
Ck
r
(6
.3)
7.04
16.1
A
l 2O
3 1.
87
15.3
L
a 2O
3 16
.88
Aka
nde
y co
ls.
(200
6)
15
Al 2
O3
320-
520
4.
3 0
.43
EN
kr
(6.4
) 4.
41
Akp
an y
col
s.
(200
7)
15
Al 2
O3
400-
590
4.
3 6
4.3 E
Nk
r
(6
.5)
51
Mat
hure
y c
ols.
(200
7)
25
MgO
-Al 2
O3
400-
600
12
2
.71
W0
.71
1E
pp
kr
(6.6
) 23
Mas
y c
ols.
(20
08a)
43
.6
Al-
LD
H
550-
650
5.
5
0.8
Ep
kr
0.7
5E
pk
r
(6.7
) 14
4m
207n
(6.8
)
Pat
el y
col
s. (
2013
) 30
C
eO2/
ZrO
2
600-
700
9
91
.0 EN
kr
(6.9
) 64
.8
Wu
y co
ls. (
2014
b)
15
γ- A
l 2O
3
200-
600
10
1
.52
Ep
kr
(6.1
0)
31.8
N=
flu
jo m
ola
r
+ca
tali
zad
or
de
cob
re-n
ique
l ra
ney,
68
.9%
Ni,
28
.2%
Cu,
2.9
% A
l
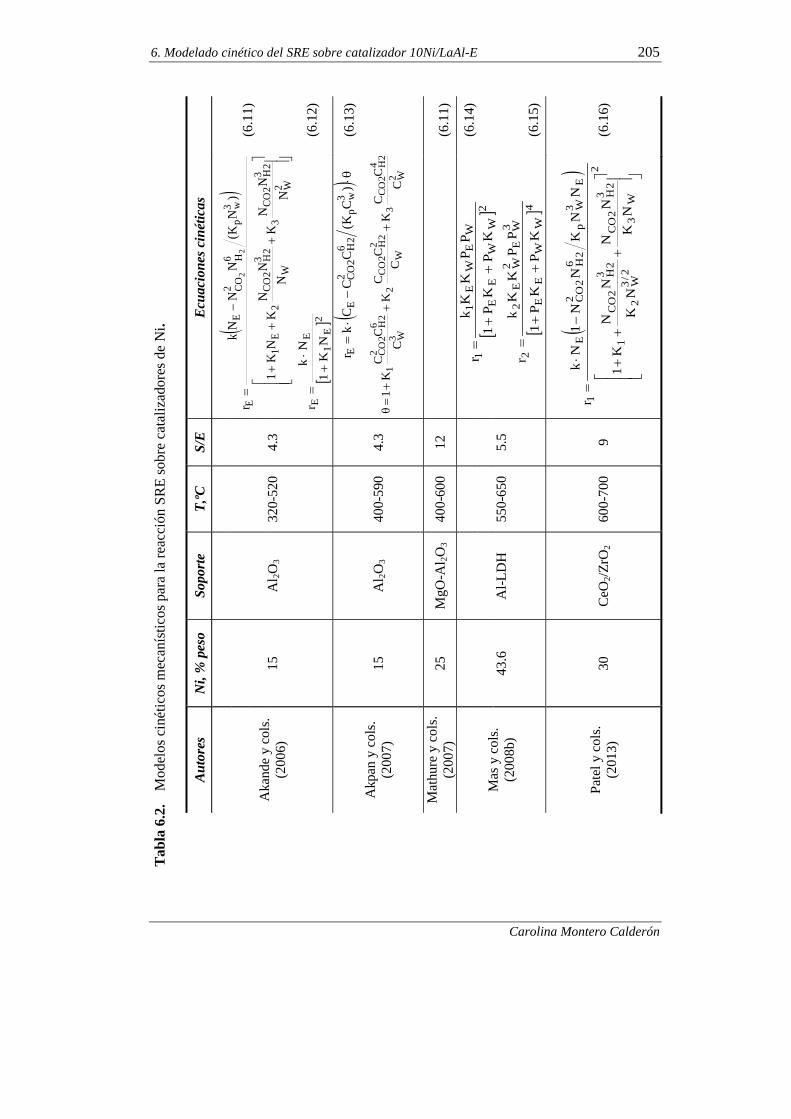
6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 205
Carolina Montero Calderón
Ta
bla
6.2
. M
odel
os
cinét
icos
mec
anís
tico
s par
a la
rea
cció
n S
RE
sobre
cat
aliz
ado
res
de
Ni.
Au
tore
s N
i, %
pes
o
So
po
rte
T,º
C
S/E
E
cua
cio
nes
cin
étic
as
Akan
de
y c
ols
.
(20
06)
15
A
l 2O
3
32
0-5
20
4
.3
2
2 W
32
H2
CO
3W
32
H2
CO
2E
1
3 wp
6 H2 C
OE
E
N
NN
KN
NN
KN
K1
)N
K(N
NN
kr
22
2
E1
EE
NK
1
Nk
r
(6.1
1)
(6.1
2)
Akp
an y
co
ls.
(20
07)
15
A
l 2O
3
40
0-5
90
4
.3
)
CK(
CC
Ck
r3 w
p6
2H
22
CO
EE
CO
24
2 W
4 H2
CO
23
W
2 H2
CO
22
3 W
6 H2
2 CO
21
CK
C
CC
KC
CC
KC
CC
K1
θ
(6.1
3)
Mat
hu
re y
co
ls.
(20
07)
25
M
gO
-Al 2
O3
40
0-6
00
1
2
(6.1
1)
Mas
y c
ols
.
(20
08
b)
43
.6
Al-
LD
H
55
0-6
50
5
.5
2
WW
EE
WE
WE
11
KP
KP
1
PP
KK
kr
4
WW
EE
3 WE
2 WE
22
KP
KP
1
PP
KK
kr
(6.1
4)
(6.1
5)
Pat
el y
co
ls.
(20
13)
30
C
eO2/Z
rO2
60
0-7
00
9
2
W3
32
H2
CO
2/
3 W2
32
H2
CO
1
E3 W
p6
2H
22
CO
E1
NK
NN
NK
NN
K1
NN
KN
N1
Nk
r
(6.1
6)

206
Capítulo 6
Morgenstern y Fornango (2005) plantearon un mecanismo de reacción para un
catalizador de Ni raney que incluye la deshidrogenación de etanol a acetaldehído y
posterior decarbonilación del acetaldehído para obtener metano. Mediante
linealización de la evolución con el tiempo espacial de la concentración de etanol y
acetaldehído, determinaron que la desaparición de etanol sigue una cinética de primer
orden, Ec. (6.2), en el intervalo de 220-280 ºC. Observaron que a partir de 250 ºC
(para la que se consume totalmente el etanol y el acetaldehído) se activa la reacción de
WGS. A pesar de las bajas temperaturas de operación, no tuvieron evidencia de
reacciones de metanación.
Para catalizadores de Ni soportados en Y2O3, Al2O3 y La2O3, a temperaturas
entre 250 y 350 ºC, Sun y cols. (2005) plantearon modelos empíricos con orden de
reacción 1 respecto al etanol y al agua, Ec. (6.3) en Tabla 6.1. Además encontraron
que el catalizador con La2O3 como soporte tenía la mayor actividad.
Akande y cols. (2006), estudiaron el reformado de bio-etanol, C2.12H6.12O1.23
(combinación de los oxigenados derivados de la fermentación: 12% de etanol, acido
láctico, glicerol, maltosa y agua) sobre un catalizador de Ni/Al2O3, en el intervalo 320-
520 ºC, y plantearon dos tipos de modelo: i) modelo empírico, Ec. (6.4) en Tabla 6.1,
en el cual no incluyeron el termino de concentración del agua, debido a que se
encuentra en exceso comparado con los demás compuestos oxigenados y; ii) Modelos
mecanísticos del tipo ER, considerando diferentes ecuaciones de velocidad según cual
fuera la etapa limitante del mecanismo (Figura 6.2).
C2H5OH + (S) ↔ C2H5OH(S)
C2H5OH(S) + (S) ↔ CH4O*(S) + CH2*(S)
CH4O*(S) + H2O ↔ CO2 + 3H2 + (S)
CH2*(S) + 2H2O ↔ CO2+3H2 + (S)
Figura 6.2. Mecanismo propuesto por Akande y cols. (2006) para la reacción de
reformado con vapor de etanol.

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 207
Carolina Montero Calderón
De entre los modelos mecanísticos propuestos por Akande y cols. (2006), el
mejor ajuste (con una desviación absoluta promedio de los datos experimentales del 6
%) se obtuvo con el que considera que la etapa limitante es la disociación del etanol
adsorbido sobre los centros activos (etapa 2 de la Figura 6.2), cuya ecuación cinética
viene dada por la Ec. (6.11) de la Tabla 6.2. Teniendo en cuenta los valores calculados
de las constantes cinéticas y constantes de equilibrio, esta ecuación se simplifica y se
puede expresar mediante la Ec. (6.12). No obstante, con el modelo empírico dado por
la Ec. (6.4) se obtuvo incluso un mejor ajuste, con una desviación absoluta promedio
del 4.5 %.
Un trabajo posterior del mismo grupo (Akpan y cols., 2007), presenta una
comparación entre modelos empíricos y mecanísticos, tanto de LHHW como ER, para
simular un reactor de lecho fijo en el intervalo 400-590 ºC. Concluyen que el más
adecuado es el modelo mecanístico del tipo ER, que asume que la etapa limitante es la
adsorción molecular del etanol, con una expresión de la velocidad de reacción dada
por la Ec. (6.13) de la Tabla 6.2. Con esta ecuación simularon el reactor a 490 ºC,
pudiendo predecir adecuadamente exactitud los perfiles de concentración de etanol
tanto en dirección axial como radial.
Mathure y cols. (2007) aplicaron tanto modelos empíricos como los modelos
del tipo ER planteados por Akande y cols. (2006) (según el mecanismo de la Figura
6.2) para describir la cinética del SRE sobre un catalizador comercial de Ni/MgO-
Al2O3. Concluyeron que un modelo empírico que incluye el efecto de la presión
parcial del agua, Ec. (6.6) en Tabla 6.1, proporciona menor desviación estándar de los
resultados que los modelos mecanísticos, entre los cuales el mejor ajuste se obtuvo
considerando la disociación de etanol adsorbido (etapa 2 de la Figura 6.2) como la
etapa limitante, con la expresión de velocidad dada por la Ec. (6.11) de la Tabla 6.2.
Además, sugirieron que la elevada presencia de coque podría ser atenuada con
presencia de una corriente de hidrógeno adicional en el medio.
Laborde y colaboradores han realizado varios estudios de modelado cinético
con un catalizador obtenido a partir de un precursor Ni(II)-Al(III)-LDH (lamellar
double hydroxide). Trabajando en condiciones de ausencia total de acetaldehído y
etileno (entre 550 y 650 ºC) y alimentando un gas inerte, de modo que solo aparecen

208
Capítulo 6
trazas de CH4 en la corriente de productos (dado que se desplazan completamente las
reacciones de reformado de CH4), el conjunto de ecuaciones que describe el proceso
de reformado puede simplificarse a las siguientes:
C2H5OH + H2O 2CO + 4H2 (6.17)
C2H5OH + 3H2O 2CO2 + 6H2 (6.18)
Para describir la velocidad de estas reacciones, los autores propusieron en
primer lugar ecuaciones cinéticas empíricas de tipo potencial, Ecs. (6.7) y (6.8) de la
Tabla 6.1, que no contemplan un término de concentración de agua, ya que en el
intervalo de relación S/E que utilizaron estos autores (4.5-5.5), la concentración de
agua no afectaba a la conversión de etanol (Mas y cols., 2008a).
Posteriormente, para describir la velocidad de las reacciones de las Ecs. (6.17) y
(6.18), estos mismos autores propusieron un modelo mecanístico del tipo LHHW,
deducido a partir de las reacciones elementales mostradas en la Figura 6.3, en las que
se asume que las adsorciones de CO y CO2 son despreciables (para disminuir el nº de
parámetros cinéticos), que solo hay un tipo de centro activo (Ni0) y que no existe
disociación del agua y etanol adsorbidos. Considerando las reacciones superficiales
sobre 2 centros activos como las etapas limitantes de la velocidad de reacción, las
expresiones cinéticas correspondientes a las reacciones de las Ecs. (6.17) y (6.18)
vienen dadas por las Ecs. (6.14) y (6.15) de la Tabla 6.2, respectivamente.
C2H5OH + (S) ↔ C2H5OH(S) adsorción de etanol
H2O + (S) ↔ H2O(S) adsorción de agua:
C2H5OH(S) + H2O(S) → 2CO + 4H2 + 2(S) reacción (Ec. 6.17)
C2H5OH(S) + 2H2O(S) → 2CO2 + 6H2 + 3(S) reacción (Ec. 6.18)
Figura 6.3. Etapas propuestas por Mas y cols. (2008b) para el reformado con vapor
de etanol.
Para el catalizador de Ni/CeO2/ZrO2, Patel y cols. (2013) observaron poca
selectividad hacia metano y CO, así como una baja velocidad de desactivación, lo que
atribuyeron a la capacidad de almacenamiento de oxígeno en la estructura cristalina

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 209
Carolina Montero Calderón
del soporte de ceria. Para describir la reacción de reformado de etanol plantearon un
modelo empírico potencial, Ec. (6.9) en Tabla 6.1, así como modelos mecanísticos de
tipo ER, de acuerdo al mecanismo de 4 etapas de la Figura 6.4. De entre las
ecuaciones mecanísticas planteadas, el mejor ajuste correspondió a la ecuación que
asume la adsorción disociativa del etanol (etapa 1 de la Figura 6.4) como etapa
controlante de la reacción, con una expresión de velocidad dada por la Ec. (6.16). Sin
embargo, comparando los resultados de ajuste y de energía de activación, este modelo
mecanístico no presentó una mejora significativa respecto al modelo empírico que
propusieron estos mismos autores (Ec. (6.9) en Tabla 6.1).
C2H5OH + 2(S) ↔ C2H5OH(S) + H(S)
C2H5OH(S) + (S) ↔ CH3O(S) + CH2(S)
CH3O(S) + H(S) + H2O(g) ↔ CO2 + 3H2 + 2(S)
CH2(S) + 2H2O ↔ CO2 + 3H2 + (S)
Figura 6.4. Etapas propuestas por Patel y cols. (2013) para el reformado con vapor de
etanol en condiciones de ausencia de CH4.
En un reciente trabajo, Wu y cols. (2014b) plantean dos modelos cinéticos para
la reacción de reformado con vapor de etanol sobre catalizador Ni/Al2O3, en un amplio
intervalo de temperatura (200-600 ºC): un modelo empírico de tipo potencial (Ec.
(6.10) en Tabla 6.1) y un modelo mecanístico de tipo LHHW, que considera las 9
etapas de reacción de la Figura 6.5, asumiendo que la etapa limitante es la
descomposición superficial de metano (etapa 5 de la Figura 6.5). El modelo
mecanístico reduce el error del ajuste respecto del empírico (R2 de 0.918 y de 0.981
para el modelo empírico y mecanístico, respectivamente), si bien con ambos modelos
se obtuvo el mismo valor de la energía de activación.

210
Capítulo 6
C2H5OH + (S) ↔ C2H4O(S) + H2
C2H4O(S) ↔ (S) + C2H4O
C2H4O(S) ↔ CO(S) + CH4
CO(S) ↔ CO + (S)
CH4+2(S) ↔ CH3(S) + H(S)
H2O + 2(S) ↔ H(S) + OH(S)
CO(S) + H(S) + OH(S) ↔ 3(S) + CO2 + H2
2H(S) ↔ 2(S) + H2
CH3(S) + OH(S) ↔ CO(S) + 2H2
Figura 6.5. Etapas propuestas por Wu y cols. (2014b) para el reformado con vapor de
etanol.
Además de los trabajos referidos en las Tablas 6.1 y 6.2, que plantean la
ecuación de velocidad para la reacción global de reformado con vapor de etanol, hay
varios trabajos en la bibliografía que han planteado ecuaciones de velocidad de
reacción para varias de las posibles etapas de reacción secundarias involucradas en el
proceso SRE, con el objetivo de cuantificar la evolución con el tiempo espacial (o
tiempo de contacto en el reactor) de los diversos productos intermedios y
subproductos de la reacción.
Entre estos trabajos, cabe citar los de Laborde y colaboradores, que proponen
modelos cinéticos apropiados para describir la evolución de la composición del medio
de reacción en condiciones de ausencia de acetaldehído y etileno (Mas y cols., 2008b;
Llera y cols., 2012).
Así, según Mas y cols. (2008b), los resultados experimentales de composición
obtenidos en condiciones de ausencia de CH4 (alta relación vapor/etanol, alta
temperatura o dilución de la alimentación), pueden describirse adecuadamente
mediante las reacciones dadas por las Ecs (6.17) y (6.18), y utilizando sus
correspondientes expresiones de velocidad (Ecs. (6.14) y (6.15) de la Tabla 6.2) en los
balances de materia para los componentes del medio de reacción (etanol, CO, CO2 e
H2) asumiendo reactor integral de flujo pistón.

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 211
Carolina Montero Calderón
Sin embargo, cuando la concentración de CH4 no es despreciable, los mismos
autores proponen un mecanismo de reacción del tipo LHHW, basados en el siguiente
esquema de reacción de 4 etapas:
1 C2H5OH CH4 + CO + H2 (6.19)
2 C2H5OH + H2O CH4 + CO2 + 2H2 (6.20)
3 CH4 + H2O ↔ CO + 3H2 (6.21)
4 CH4 + 2H2O ↔2CO2 + 4H2 (6.22)
La Figura 6.6 muestra las reacciones elementales del mecanismo de reacción de
LHHW propuesto por Mas y cols. (2008b), asumiendo que solo hay un tipo de centro
activo (Ni0), que no se produce adsorción de CO y CO2, que no existe disociación del
agua y etanol, que el CH4 puede adsorberse en el mismo tipo de centros activos que el
etanol y agua, y que la reacción limitante involucra la presencia de por lo menos dos
centros activos. Si las etapas limitantes de la velocidad de las reacciones dadas por las
Ecs (6.19)-(6-22) son las de reacción, las correspondientes expresiones cinéticas para
cada una de estas cuatro reacciones vienen dadas por las Ecs. (6.23)-(6.26):
C2H5OH + (S) ↔ C2H5OH(S) adsorción de etanol
H2O + (S) ↔ H2O(S) adsorción de agua
C2H5OH(S) →CO + CH4(S) + 2H2+ (S) reacción, Ec. (6.19)
C2H5OH(S) + H2O(S) → CO2 + 2H2+ CH4(S) + (S) reacción, Ec. (6.20)
CH4(S) ↔ CH4 + (S) desorción de metano
CH4(S) + H2O(S) → CO + 3H2 + 2(S) reacción, Ec. (6.21)
CH4(S) + 2H2O(S) → CO2 + 4H2 + 3(S) reacción, Ec. (6.22)
Figura 6.6. Etapas propuestas por Mas y cols. (2008b) para el reformado con vapor
de etanol en condiciones de presencia de CH4.

212
Capítulo 6
MMWWEE
EE11
KPKPKP1
PKkr
(6.23)
2MMWWEE
WWEE22
KPKPKP1
PKPKkr
(6.24)
2MMWWEE
33
2HCOWMWM33
KPKPKP1
KPPPPKKkr
(6.25)
3MMWWEE
44
2H2CO2WMWWM4
4KPKPKP1
KPPPPKKKkr
(6.26)
En otro trabajo de este mismo grupo, Llera y cols. (2012) proponen un modelo
cinético más complejo, aplicable en condiciones de reacción en las que se obtienen
rendimientos elevados de H2 (650 ºC, > 5 moles de H2 / mol de etanol), y en las que
tanto CO2 como CH4 son considerados como intermedios de reacción, mientras que el
CO es un producto final, por lo que es posible la reacción WGS inversa. Considerando
las mismas 4 etapas de reacción propuestas anteriormente por estos autores, Ecs.
(6.19)-(6.22), y asumiendo que las reacciones superficiales son las etapas limitantes de
la velocidad, obtienen expresiones de velocidad para cada una de las cuatro etapas,
resultando un modelo cinético con un total de 36 parámetros (correspondientes a los
factores preexponenciales, energías de activación y entalpías de las 4 constantes
cinéticas y 14 constantes de adsorción).
Más recientemente, Zhang y cols. (2014b) describen el comportamiento cinético
de un catalizador de Ni esqueletal (Ni-raney, no soportado) para el intervalo 300-400
ºC, mediante modelos mecanísticos del tipo ER (Figura 6.7), asumiendo que el agua
participa en las reacciones en forma molecular, debido a la baja probabilidad de su
disociación en el Ni–raney. Las expresiones cinéticas para las reacciones involucradas
en el proceso están dadas por las Ecs. (6.27)-(6.30). Estos autores determinaron que la
reacción de descomposición de etanol (considerado como la suma de la
deshidrogenación a acetaldehído, seguido de una rápida descomposición de éste), es la
etapa limitante, señalando que este modelo es válido sólo para catalizadores no-
soportados.

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 213
Carolina Montero Calderón
C2H5OH + (S) ↔ C2H5OH(S)
C2H5OH(S) → C2H4O(S) + H2
C2H4O(S) + (S) → CO(S) + CH4(S)
CH4(S) + H2O ↔ CO(S) + 3H2
CO(S) + H2O ↔ CO2(S) + H2
CO(S) ↔ CO + (S)
CO2(S) ↔ CO2 + (S)
CH4(S) ↔ CH4+ (S)
Figura 6.7. Etapas del modelo propuesto por Zhang y cols. (2014b) para describir la
cinética del reformado con vapor de etanol.
2vE12ED CPKkr (6.27)
v
53
32HCO
7
W4CH3SRM C
KK
PP
K
PPkr
(6.28)
v
64
2H2CO
5
WCO4WGS C
KK
PP
K
PPkr
(6.29)
7M62CO5COE1v
K/PK/PK/PPK1
1C
(6.30)
Palma y cols. (2014) han determinado la cinética del proceso para un
catalizador bimetálico de Pt/Ni/CeO2. Basándose en sus observaciones experimentales
y en bibliografía previa, propusieron las reacciones dadas por las Ecs. (6.31)-(6.36)
para desarrollar el modelo cinético, con las ecuaciones cinéticas para cada reacción
dadas por las Ecs. (6.37)-(6.42):
1 C2H5OH C2H4O + H2 (6.31)
2 C2H5OH 3/2CH4 + 1/2CO2 (6.32)
3 C2H4O → CH4 + CO (6.33)
4 C2H4O+H2O→2CO+3H2 (6.34)
5 CO + H2O ↔ H2 + CO2 (6.35)
6 CO2 + 4H2 → CH4 + 2H2O (6.36)
12HAcE11 KPPPkr (6.37)

214
Capítulo 6
25.1
4CH5.0
2COE12 KPPPkr (6.38)
34CHCOAc33 KPPPkr (6.39)
44CHCOWAc44 KPPPPkr (6.40)
2CO2COWWCOCO
42H2COWCOW2CO55
KPKPKP1
KPPPPKKkr
(6.41)
62W4CH
42H2CO66 KPPPPkr (6.42)
Los parámetros a optimizar son las 6 constantes cinéticas, dado que para las
constantes de adsorción se consideraron valores constantes e iguales al valor promedio
en el intervalo 300-500 ºC establecido previamente por Podolski y Young (1974) para
la Ec. (6.41). Es destacable que este modelo sí cuantifica la composición de
acetaldehído en el medio de reacción.
6.1.1.2. Modelos cinéticos para catalizadores de Co y de metales nobles
Entre los trabajos que estudian el modelado cinético para el SRE sobre
catalizadores de metales distintos al Ni pueden citarse los siguientes.
Vaidya y Rodrigues (2006b) estudiaron el proceso con catalizador de Ru/γ-
Al2O3 en el intervalo 600-700 ºC y en condiciones de gran exceso de agua (S/E=10).
Propusieron un mecanismo que conlleva la formación de un complejo activo, por
reacción de etanol adsorbido con agua en fase gas, y la formación de compuestos
intermedios adsorbidos por descomposición de este complejo activo (con constante
cinética k3) la cual se considera la etapa limitante de la velocidad. Los intermedios
adsorbidos reaccionan posteriormente con agua para formar CO2 e H2. Aplicando la
hipótesis de estado pseudoestacionario al etanol adsorbido, deducen la siguiente
expresión para la velocidad de reacción:
3WE21W2E11
WE21
kPPkkPkPkk
PPkkr
(6.43)

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 215
Carolina Montero Calderón
Lu y cols. (2011) propusieron un modelo más complejo para describir los datos
cinéticos de Vaidya y Rodrigues (2006b), considerando que el proceso es la
combinación de varios mecanismos individuales, y deducen las ecuaciones de
velocidad (hasta 8) a partir de un esquema de reacción cíclico. Concluyeron que las
etapas que limitan la velocidad de reacción son: i) la reacción del agua con el etanol
adsorbido; ii) reacción de agua con el acetaldehído adsorbido, y iii) la desorción del
CO. Por debajo de 650 ºC la reacción global también está limitada por la desorción de
etanol y acetaldehído, mientras que por encima de 650 ºC la reacción global también
se vería limitada por la deshidrogenación del etanol adsorbido.
Sahoo y cols. (2007) estudiaron el comportamiento cinético del catalizador
Co/Al2O3 entre 400-700 ºC, y consideraron que las reacciones del proceso son el
reformado con vapor y la descomposición de etanol , así como la WGS. Basándose en
el análisis de productos y en la bibliografía previa, proponen un mecanismo del tipo
LHHW con un total de 17 etapas elementales, y deducen las correspondientes
ecuaciones de velocidad para las 3 reacciones consideradas, asumiendo que las etapas
limitantes de la velocidad son la formación de acetaldehído por deshidrogenación de
las especies etoxi adsorbidas, la descomposición de un intermedio formiato y la
descomposición de acetaldehído, respectivamente. El conjunto de 3 ecuaciones
cinéticas propuestas contempla un total de 3 constantes de velocidad y 8 constantes de
adsorción.
Graschinsky y cols. (2010) propusieron un modelo cinético para un catalizador
de Rh/MgAl2O4/Al2O3, en un intervalo 500-600 ºC. El esquema cinético incluye 4
reacciones: descomposición de etanol, reformado de etanol (según la Ec. (6.20),
utilizada por Mas y cols. (2008b)), reacción WGS y reformado de CH4. Propusieron
un mecanismo de LHHW con 14 etapas elementales de reacción para describir el
esquema de 4 reacciones propuestas, y de la aplicación del método de las velocidades
iniciales concluyeron que en la etapa determinante de la velocidad de reacción están
involucrados 2 centros activos. Con estas premisas, desarrollaron 5 modelos cinéticos
alternativos, siendo el modelo de mejor ajuste el que considera limitante la etapa de
reacción superficial sobre 2 centros activos, cuyas expresiones cinéticas son:

216
Capítulo 6
22CO32/12HW2E1
2/12H4CHE
EDED
yKyyKyK1
yyykr
(6.44)
22CO32/12HW2E1
2H4CHWEERER
yKyyKyK1
yyyykr
(6.45)
22CO3
2/12HW2E1
2/12H2COSRM
2/12HWCOSRM
SRM
yKyyKyK1
yykyyykr
(6.46)
22CO3
2/12HW2E1
22HCOWGS2HW4CHWGS
WGS
yKyyKyK1
yykyyykr
(6.47)
Yun y cols. (2012), estudiaron la cinética del reformado con vapor de etanol
sobre un catalizador de Co-Na/ZnO para utilizarla en la simulación del proceso en
reactores de lecho fijo con membranas de Pd y de Pd/Cu. Establecieron una ecuación
cinética de tipo potencial de orden 0.86 para el etanol e independiente de la
concentración de agua (dado el elevado exceso de agua, S/E =13), con la cual predicen
adecuadamente la conversión de etanol y el flujo molar de H2 obtenido en ambos tipos
de reactores (lecho fijo y de membrana). Sin embargo el modelo no contempla los
flujos molares de los posibles subproductos de reacción.
Ciambelli y cols. (2010) analizaron el mecanismo y la cinética con un
catalizador de Pt/CeO2 en el intervalo 300-450 ºC. A partir de los estudios cinéticos y
mediante ensayos de desorción a temperatura (TPD) establecieron un mecanismo de
reacción que incluye la adsorción disociativa del etanol en la superficie del
catalizador, para formar como intermedio al acetaldehído, la decarbonilación para
formar H2, CH4 y CO y la reacción WGS del CO adsorbido en los centros activos del
Pt. No obstante, el modelado cinético lo realizaron asumiendo un modelo empírico de
tipo potencial, y el ajuste a los datos experimentales condujo a un orden de reacción
0.5 para al etanol y 0 para el agua.

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 217
Carolina Montero Calderón
6.1.2. Metodología del modelado cinético a tiempo cero
6.1.2.1. Cálculo de los parámetros cinéticos de mejor ajuste
El cálculo de los parámetros para cada modelo cinético propuesto se ha
realizado mediante ajuste por regresión no lineal múltiple. La optimización se ha
logrado minimizando una función objetivo, establecida como la suma ponderada de
los cuadrados de los errores (diferencias de los valores de concentración
experimentales y calculados):
c cn
1i
p
1j
2
ji,
*
ji,
n
1i
iii XXwwFO (6.48)
donde: wi es el factor de peso de cada componente i del esquema cinético; i es la
suma del cuadrado de los residuos para cada componente del esquema cinético; *j,iX
es el valor de la concentración de cada componente i (expresada como fracción molar
en base húmeda) para la condición experimental j; Xi,j es el correspondiente valor
calculado integrando el balance de materia para el componente i; nc es el número de
componentes del esquema cinético; y p es el número total de condiciones
experimentales.
Los parámetros de mejor ajuste a optimizar son las constantes cinéticas de cada
reacción j, que siguen la ecuación de Arrhenius. Para disminuir la correlación
existente entre el factor pre-exponencial y la energía de activación, se recurre a la
reparametrización de esta ecuación (Gayubo y cols., 2000, 2007, 2011), expresando la
constante cinética en función de la correspondiente, kj*, a una temperatura de
referencia, T*:
*T
1
T
1
R
Eexpkk
j*jj (6.49)
De igual forma, la constante de equilibrio de adsorción de cada compuesto i que
se adsorbe sobre los centros activos se ha expresado de la forma:

218
Capítulo 6
*T
1
T
1
R
HexpKK i*
ii (6.50)
De esta forma, los parámetros cinéticos a calcular son las constantes cinéticas y
constantes de equilibrio de adsorción a la temperatura de referencia (que se ha fijado
en 600 ºC) y sus correspondientes energías de activación y entalpías de adsorción,
respectivamente.
Dado que no se dispone de repeticiones de datos experimentales y, por tanto, no
se puede hacer una estimación inicial de las varianzas, para estimar los factores de
peso se ha adoptado el criterio de que son inversamente proporcionales a la
composición media de cada componente en el intervalo de condiciones de operación
estudiado (Gayubo y cols., 2011):
p
1ji
i
X
1w (6.51)
En la Tabla 6.3 se relacionan los factores de peso de cada componente
calculados con la Ec. (6.51). Los valores elevados de los factores del acetaldehído y
del etileno se explican porque estos subproductos se encuentran en mucha menor
concentración que el resto de los compuestos en el medio de reacción, dado que son
productos intermedios primarios en el proceso, cuya concentración es solo
significativa para bajo tiempo espacial.
Tabla 6.3. Factores de peso de cada componente del medio de reacción en el
reformado con vapor de etanol sobre catalizador 10Ni/LaAl-E.
Compuesto H2 CO2 CO CH4 C2H4O C2H4 H2O C2H5OH
wi 3.28 13.85 29.33 28.28 74.49 263.35 1.95 41.53

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 219
Carolina Montero Calderón
6.1.2.2. Ecuaciones de velocidad de reacción y balance de materia
Los datos cinéticos se han obtenido en un reactor tubular de lecho catalítico
fluidizado, a través del cual circulan los gases, descrito en el Apartado 2.3. La Figura
6.8 muestra un elemento diferencial del lecho catalítico, de altura dz.
Figura 6.8. Elemento diferencial de volumen del lecho catalítico.
Se ha asumido que el flujo de gas es tipo pistón, sin gradientes radiales de
concentración y en régimen isotermo, debido a que las diferencias de temperatura en
diferentes posiciones radiales y longitudinales son inferiores a 1 °C. El flujo molar
total, F, varía a lo largo del reactor, dado que la reacción transcurre con aumento del
número de moles y en las condiciones en que se ha trabajado en esta Tesis (incluyendo
ensayos con alimentación estequiométrica de reactivos) no puede despreciarse tal
variación. Por consiguiente, la ecuación del balance de materia para cada componente
i del esquema cinético viene dada por la expresión:
volumentiempo
i de molr
Sdz
dFX
Sdz
dXF
Sdz
)Xd(F
dV
dFio
Ti
iT
iTi (6.52)
donde S es la sección del reactor, z es la componente longitudinal y Xi es la fracción
molar del componente i, en base húmeda. Despejando de la Ec. (6.52), se obtiene la
expresión para calcular la evolución de la composición de cada componente a lo largo
del reactor.
X+dXdz
FT
Fi=FT·Xi
Xi
FT+dFT
Fi+dFi
Xi+dXi
Z
W (rb)

220
Capítulo 6
dz
dF
F
X
F
rS
dz
dX T
T
i
T
ioi
(6.53)
De esta manera, el balance de materia queda referido a la fracción molar,
magnitud de fácil comprensión por su significado físico y por su relación con el
rendimiento y balance de componentes.
La velocidad de formación de cada componente i, a tiempo cero (catalizador
fresco), se ha calculado considerando las diferentes etapas del esquema cinético de
reacción propuesto en las que interviene:
j
j
jiio rυr (6.54)
donde (i)j es el coeficiente estequiométrico del componente i en la etapa j del
esquema cinético, y rj es la velocidad de reacción de la etapa j, en la cual las
concentraciones de los componentes se han definido como presión parcial, pi,
considerando todos los componentes del medio de reacción.
6.1.3. Programa de cálculo
Para realizar la integración de las ecuaciones cinéticas y la regresión no lineal
múltiple, se ha desarrollado un programa de cálculo escrito en MATLAB, cuyo
diagrama de flujo se muestra en la Figura 6.9.
El programa principal (denominado “prin_cin0_SRE_M#.m”, donde “M#”
identifica cada uno de los diferentes modelos cinéticos propuestos a discriminar), sirve
de interfaz de entrada y salida de datos y resultados, llama a las subrutinas de
regresión no lineal y éstas a su vez a otras subrutinas necesarias para la integración de
las ecuaciones de conservación. Requiere como datos de entrada:
i) Un archivo de texto que contenga los datos cinéticos correspondientes a todas las
condiciones experimentales utilizadas (datos de temperatura, presión, caudal
molar alimentado de cada componente y de inerte, masa de catalizador, tiempo y

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 221
Carolina Montero Calderón
valores experimentales de las fracciones molares en base húmeda (Xi) de los
componentes del esquema cinético)
ii) La identificación del modelo cinético a ajustar (para definir el sistema de
ecuaciones diferenciales que debe integrarse)
iii) Una estimación inicial de los parámetros cinéticos a optimizar
El programa genera como resultado un fichero de texto con los parámetros
cinéticos de mejor ajuste y sus intervalos de confianza, así como el valor de la función
objetivo (FO), la falta de ajuste del modelo (SSE), así como para cada componente del
medio de reacción (SSEi) y los valores de concentraciones calculados para las
diferentes condiciones experimentales. Proporciona igualmente una representación
gráfica del ajuste final (gráficos de paridad de los componentes del medio de reacción)
para visualizar la bondad del ajuste.
Figura 6.9. Diagrama de bloques del programa de cálculo de los parámetros cinéticos
a tiempo cero.
El programa principal llama a la subrutina de cálculo de regresión no-lineal
multivariable, habiéndose establecido dos posibilidades:
1
Programa principal: prin_cin0_SRE_M#.m
Lectura de datos iniciales: SRE_datos.txt
Asignación de valores
iniciales a los parámetros a
optimar
RESULTADOS
Parámetros
cinéticos,
intervalos de
confianza, Xi
F.O y SSE
Cálculo de la función error-regresión lineal multivariable
Levenberg-
Marquardt:
aju-mul
fminsearch
Levenber-
Marquardt:
aju-mul
Subrutinas
Cálc. del
error
fun_error
F.de
integración
fun_int
Cálculo
derivada
ode15s
Sistema Ec.
diferenciales
der_cin_t0

222
Capítulo 6
i) Llamada a la subrutina “aju_mul” (de desarrollo propio y basada en el método
Levenberg-Marquardt (Vivanco, 2004; Alonso, 2008)), que permite un
acercamiento grosero pero rápido hacia los valores óptimos de los parámetros, y
realiza además el cálculo de los intervalos de confianza de los parámetros a
optimizar.
ii) Llamada a la función propia de MATLAB, “fminsearch”, la cual realiza una
búsqueda más detallada del óptimo (basado en el método SIMPLEX, (Lagarias y
cols., 1999)), aunque requiere de una estimación inicial de los parámetros
relativamente cercana al óptimo.
El procedimiento de cálculo de los parámetros de mejor ajuste para cada
modelo cinético propuesto, ha consistido en una primera búsqueda del óptimo
mediante la subrutina “aju_mul”, seguida de una segunda búsqueda del óptimo con la
subrutina “fminsearch”, partiendo del óptimo calculado previamente. La búsqueda del
óptimo finaliza mediante una nueva llamada a “aju_mul” (partiendo del óptimo
definido por “fminsearch”), para determinar los intervalos de confianza de los
parámetros del ajuste. Estas dos subrutinas evalúan la función objetivo mediante una
llamada a la función “fun_err” (en la que la que la función objetivo a minimizar viene
dada por la Ec. (6.48)).
A su vez, la función “fun_err” llama a la subrutina “fun_int”, para realizar el
cálculo de las composiciones correspondientes a las diferentes condiciones
experimentales estudiadas, mediante integración de la ecuación del balance de
materia, Ec. (6.53), para cada componente i del esquema cinético considerado. El
sistema de ecuaciones diferenciales a integrar viene definido por la función
“der_cin_t0_M#”, donde “M#” identifica a cada uno de los modelos cinéticos
concretos a ajustar.
Para realizar la integración del sistema de ecuaciones se ha utilizado la función
propia de MATLAB ode15s. La función ode15s es un solucionador implícito de
diferenciación numérica de múltiples pasos de orden variable, adecuado para resolver
problemas rígidos (“stiff”) que requieran exactitud (Shampine y cols., 1997; 1999).
Para un problema de tipo “stiff” las soluciones pueden cambiar sobre una escala de

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 223
Carolina Montero Calderón
tiempo que es muy corta comparada con el intervalo de integración, pero la solución
de interés cambia sobre escalas de tiempo mucho más largas. Este sistema de
integración es adecuado para resolver procesos cinéticos en los que pueden aparecer
etapas con una significativa diferencia en la velocidad de reacción (etapas muy rápidas
y/o etapas notablemente más lentas).
El significado estadístico y la fiabilidad de los parámetros estimados mediante
el programa de cálculo descrito, se pueden evaluar mediante la expresión de los
intervalos de confianza. Los intervalos de confianza para los parámetros (q) calculados
vienen dados por la siguiente ecuación:
qq2
2αqq cσ(n)tbL (6.55)
donde cqq es el elemento de la matriz varianza-covarianza de q parámetros; bq la
estimación del parámetro q; t/2 el valor crítico de la estadística de Student´s para un
nivel de confianza ; y 2 es la varianza de las respuestas.
6.1.4. Significación y discriminación de los modelos cinéticos
Habitualmente la discriminación de modelos se lleva a cabo mediante el test F
del error residual de los modelos. De este modo, cuando se comparan dos modelos
(i,j), siendo sus varianzas del residual σEi2
y σEj2 y con σEi
2> σEj
2, la mejora lograda con
el ajuste del modelo j respecto al del modelo i será significativa si:
),(F/SSE
/SSEF jiα12
jE,
2iE,
jj
iiji
(6.56)
donde la suma de cuadrados de los errores residuales (SSE) y los grados de libertad
del residual (ν) se calculan según con la siguiente ecuación:
qnp c (6.57)

224
Capítulo 6
El problema de esta prueba de hipótesis reside en que cuando el número de
grados de libertad de los residuales a comparar es elevado, la prueba pierde potencia.
En nuestro estudio, en todos los casos el valor de ν > 120, garantiza que el valor de F
es en todos los casos es muy cercano a 1 y por consiguiente la potencia de la prueba es
prácticamente nula. Consecuentemente, para llevar a cabo la discriminación de los
modelos se han tomado dos alternativas a la metodología convencional de análisis de
varianza del error residual.
Por un lado, para los modelos con diferente número de parámetros, la
discriminación de modelos se ha llevado a cabo mediante una adaptación del test F
para el error residual entre la diferencia de la suma de los errores al cuadrado de pares
de modelos y la correspondiente al modelo de mejor ajuste, que permite determinar si
alguno de los modelos es significativamente mejor que el resto (Epelde, 2013). De
este modo, para dos modelos dados, (i, j), cuyos grados de libertad del residual son νi
y νj, con valores de νi > νj, y tales que SSEi > SSEj, la mejora lograda con el ajuste del
modelo j respecto al del modelo i es significativa si se verifica que:
)ν,ν(νF)/νν(ν
)/SSESSE(SSEF jjiα1
jji
jjiji
(6.58)
En caso contrario, no es significativa la mejora y se considerará más adecuado el
modelo de menor número de parámetros (modelo i).
Por otro lado, si se comparan dos modelos tales que i = j, es decir con el
mismo número de parámetros en este caso, se ha llevado a cabo la comparación de los
modelos o lo que es lo mismo, de la propia regresión, en vez de los errores residuales.
La suma de cuadrados de la regresión (SSM) se calcula como se indica en la Tabla
6.4. Para pares de modelos con valores de SSMi > SSMj la mejora lograda con el
ajuste del modelo i respecto al del modelo j será significativa si:
)q,(qF/qSSM
/qSSMF jiα12
jM,
2iM,
jj
iiji
(6.59)

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 225
Carolina Montero Calderón
donde σMi2 y σMj
2 son las varianzas estimadas por la regresión de cada modelo. En caso
contrario, no se puede afirmar que la mejora lograda sea significativa, por lo que se
considerará más adecuado el de menor σE2
Tabla 6.4. Análisis de varianza para discriminación entre modelos cinéticos
Suma de cuadrados Grados de libertad Varianza
p
X
)X(SST
2n
1i
p
1j
*j,i
n
1i
2p
1j
*j,i
c
c
- -
2j,i
n
1i
p
1j
*j,i )XX(SSE
c
qnp c
SSE
2E
SSM=SST-SSE q q
SSM2M
El valor de la función crítica de Fisher, F1-(i-j, j) ó F1-(qi,qj), se calcula
mediante las tablas de la función de distribución de Fisher o directamente mediante la
función de MATLAB finv(1-,i-j,j) ó finv(1-,qi,qj). Para un valor de dado
(normalmente comprendido entre 0.05 y 0.01), el porcentaje de confianza con el que
se han estimado los parámetros del modelo es de 100 (1-). En esta Tesis se ha
considerado un valor de = 0.05.
6.1.5. Modelos cinéticos propuestos
Como se ha mencionado en el Apartado 1.5.1, la reacción de reformado con
vapor de etanol cuenta con una estequiometria general definida por la Ec. (1.8). Sin
embargo en el proceso, además de la reacción de reformado tienen lugar reacciones
paralelas que generan, además de H2 y CO2, otros subproductos carbonados
(acetaldehído, acetona, etano y eteno, CO y CH4) que a su vez son promotores de la
formación de coque.

226
Capítulo 6
Para el planteamiento del modelado cinético, en esta Tesis se han considerando
únicamente las reacciones que involucran a los componentes identificados en el medio
de reacción en cantidades significativas con el catalizador 10Ni/LaAl-E, que incluyen
etanol, agua, H2, CO, CO2, CH4, acetaldehído y etileno.
Las diferentes etapas de reacción consideradas se esquematizan en la Figura
6.10, y su estequiometría se muestra en la Figura 6.11. Estas 10 etapas incluyen; 4
reacciones de transformación de etanol (reformado con vapor, descomposición,
deshidrogenación y deshidratación), 2 reacciones de transformación del acetaldehído
(descomposición y reformado con vapor), junto con la reacción WGS, el reformado de
etileno y de metano (esta última es la reacción inversa de la metanación del CO) y la
reacción de metanación de CO2.
Figura 6.10. Esquema cinético propuesto para el proceso SRE sobre catalizador
10Ni/LaAl-E.
HIDROGENO
ETANOL
EtilenoAcetaldehído
MetanoCO
CO2
DESCOMPOSICIÓNDESHIDRATACIÓN
DESHIDROGENACIÓN
SR
SR
METANACIÓNWGS
DESCOMPOSICIÓN
SR SR

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 227
Carolina Montero Calderón
1- Reformado con vapor de etanol (SR-E) C2H5OH + 3H2O → 6H2 + 2CO2
2- Deshidratación de etanol (DHT) C2H5OH → C2H4 + H2O
3- Reformado de etileno (SR-C2=) C2H4 + 2H2O → 4H2 + 2CO
4- Deshidrogenación de etanol (DHG) C2H5OH ↔ C2H4O + H2
5- Descomposición de acetaldehído (D-Ac) C2H4O → CH4 + CO
6- Reacción Water Gas Shift (WGS) CO + H2O ↔ H2 + CO2
7- Reformado con vapor de CH4 (SR-M) CH4 + H2O ↔ 3H2 + CO
8- Reformado con vapor de acetaldehído (SR-Ac) C2H4O + H2O → 2CO + 3H2
9- Metanación de CO2 (M-CO2) CO2 + 4H2 ↔ CH4 + 2H2O
10- Descomposición de etanol (D-E) C2H5OH → H2 + CO + CH4
Figura 6.11. Etapas del esquema cinético propuesto para el proceso SRE sobre
catalizador 10Ni/LaAl-E.
Partiendo del esquema cinético de la Figura 6.10, que involucra el conjunto de
10 reacciones mostradas en la Figura 6.11, se ha planteado en primer lugar un modelo
cinético simplificado, considerando el número mínimo de reacciones necesarias para
poder cuantificar la evolución de la composición observada experimentalmente para
los 8 componentes identificados en el medio de reacción, en función de las variables
de operación (temperatura, relación molar S/E y tiempo espacial). Posteriormente se
han ido considerando etapas adicionales, analizando en cada caso la significación de la
mejora lograda con los modelos más complejos respecto al modelo simplificado.
En la Tabla 6.5 se describen los 8 modelos cinéticos estudiados (M1-M8) junto
con las expresiones correspondientes de la velocidad de reacción de cada etapa. Estas
ecuaciones se han planteado asumiendo reacciones elementales y considerando
reacciones reversibles a aquellas con suficientemente bajo valor de su constante de
equilibrio en el intervalo de temperatura de operación estudiado. Además, se ha
incluido un término “θ” para tener en cuenta la atenuación de las velocidades de
reacción debido a la adsorción de alguno/s de los componentes del medio de reacción.
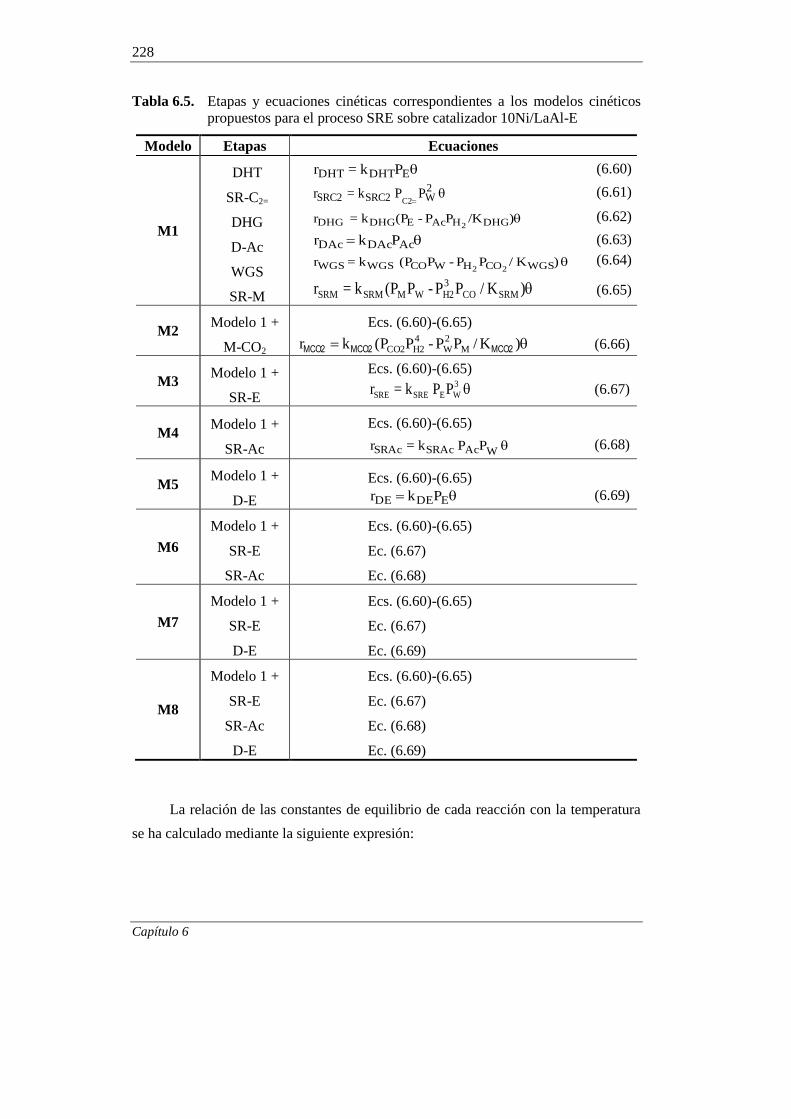
228
Capítulo 6
Tabla 6.5. Etapas y ecuaciones cinéticas correspondientes a los modelos cinéticos
propuestos para el proceso SRE sobre catalizador 10Ni/LaAl-E
Modelo Etapas Ecuaciones
M1
DHT
SR-C2=
DHG
D-Ac
WGS
SR-M
EDHTDHT Pk=r (6.60)
2WSRC2SRC2 PP k=r
C2 (6.61) (6.61)
)/KPP-(Pk= r DHGHAcEDHGDHG 2 (6.62)
Pkr AcDAcDAc (6.63)
)K/ PP-P(P k=r WGSCOHWCOWGSWGS 22 (6.64)
)K/PP-P(Pk=r SRMCO3H2WMSRMSRM (6.65)
M2 Modelo 1 +
M-CO2
Ecs. (6.60)-(6.65)
)K/PP-P(Pkr M2W
4H2CO2 MCO2MCO2MCO2 (6.66)
M3 Modelo 1 +
SR-E
Ecs. (6.60)-(6.65)
3
W ESRESRE PP k=r (6.67)
M4 Modelo 1 +
SR-Ac
Ecs. (6.60)-(6.65)
WAcSRAcSRAc PP k=r (6.68)
M5 Modelo 1 +
D-E
Ecs. (6.60)-(6.65) Pkr EDEDE (6.69)
M6
Modelo 1 +
SR-E
SR-Ac
Ecs. (6.60)-(6.65)
Ec. (6.67)
Ec. (6.68)
M7
Modelo 1 +
SR-E
D-E
Ecs. (6.60)-(6.65)
Ec. (6.67)
Ec. (6.69)
M8
Modelo 1 +
SR-E
SR-Ac
D-E
Ecs. (6.60)-(6.65)
Ec. (6.67)
Ec. (6.68)
Ec. (6.69)
La relación de las constantes de equilibrio de cada reacción con la temperatura
se ha calculado mediante la siguiente expresión:
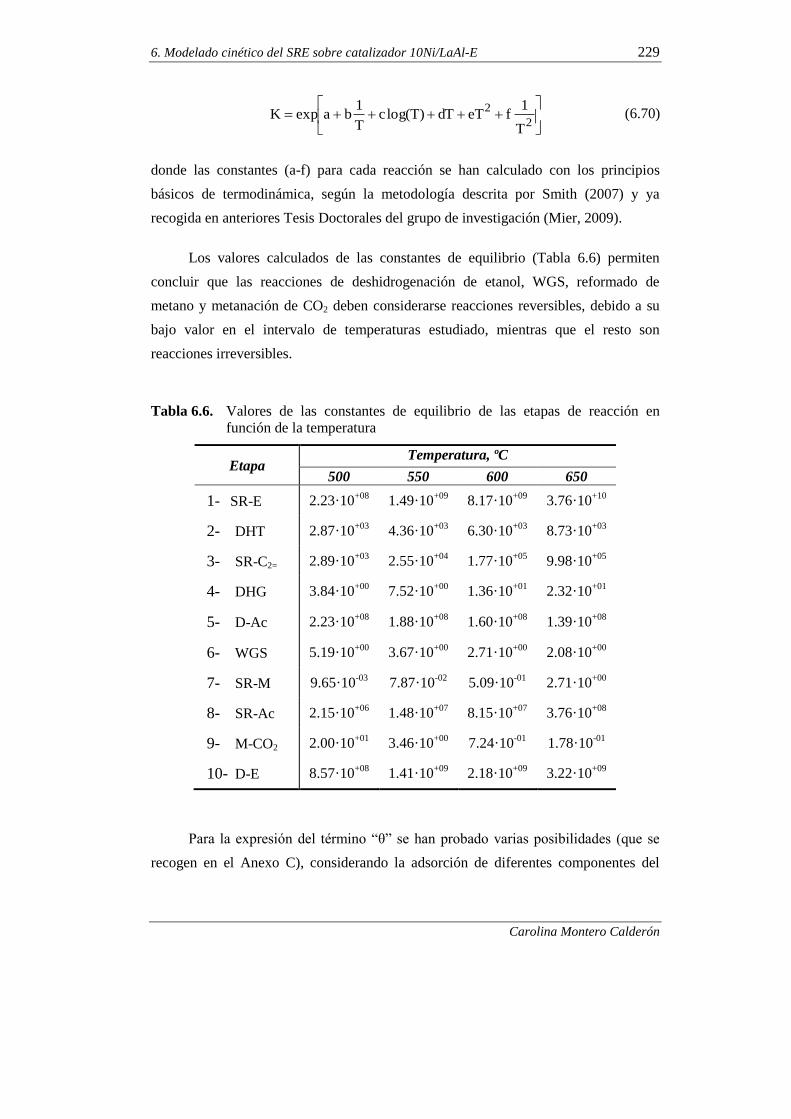
6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 229
Carolina Montero Calderón
2
2
T
1feTdT)Tlog(c
T
1baexpK (6.70)
donde las constantes (a-f) para cada reacción se han calculado con los principios
básicos de termodinámica, según la metodología descrita por Smith (2007) y ya
recogida en anteriores Tesis Doctorales del grupo de investigación (Mier, 2009).
Los valores calculados de las constantes de equilibrio (Tabla 6.6) permiten
concluir que las reacciones de deshidrogenación de etanol, WGS, reformado de
metano y metanación de CO2 deben considerarse reacciones reversibles, debido a su
bajo valor en el intervalo de temperaturas estudiado, mientras que el resto son
reacciones irreversibles.
Tabla 6.6. Valores de las constantes de equilibrio de las etapas de reacción en
función de la temperatura
Etapa Temperatura, ºC
500 550 600 650
1- SR-E 2.23·10+08 1.49·10
+09 8.17·10+09 3.76·10
+10
2- DHT 2.87·10+03 4.36·10
+03 6.30·10+03 8.73·10
+03
3- SR-C2= 2.89·10+03 2.55·10
+04 1.77·10+05 9.98·10
+05
4- DHG 3.84·10+00 7.52·10
+00 1.36·10+01 2.32·10
+01
5- D-Ac 2.23·10+08 1.88·10
+08 1.60·10+08 1.39·10
+08
6- WGS 5.19·10+00 3.67·10
+00 2.71·10+00 2.08·10
+00
7- SR-M 9.65·10-03 7.87·10
-02 5.09·10-01 2.71·10
+00
8- SR-Ac 2.15·10+06 1.48·10
+07 8.15·10+07 3.76·10
+08
9- M-CO2 2.00·10+01 3.46·10
+00 7.24·10-01 1.78·10
-01
10- D-E 8.57·10+08 1.41·10
+09 2.18·10+09 3.22·10
+09
Para la expresión del término “θ” se han probado varias posibilidades (que se
recogen en el Anexo C), considerando la adsorción de diferentes componentes del
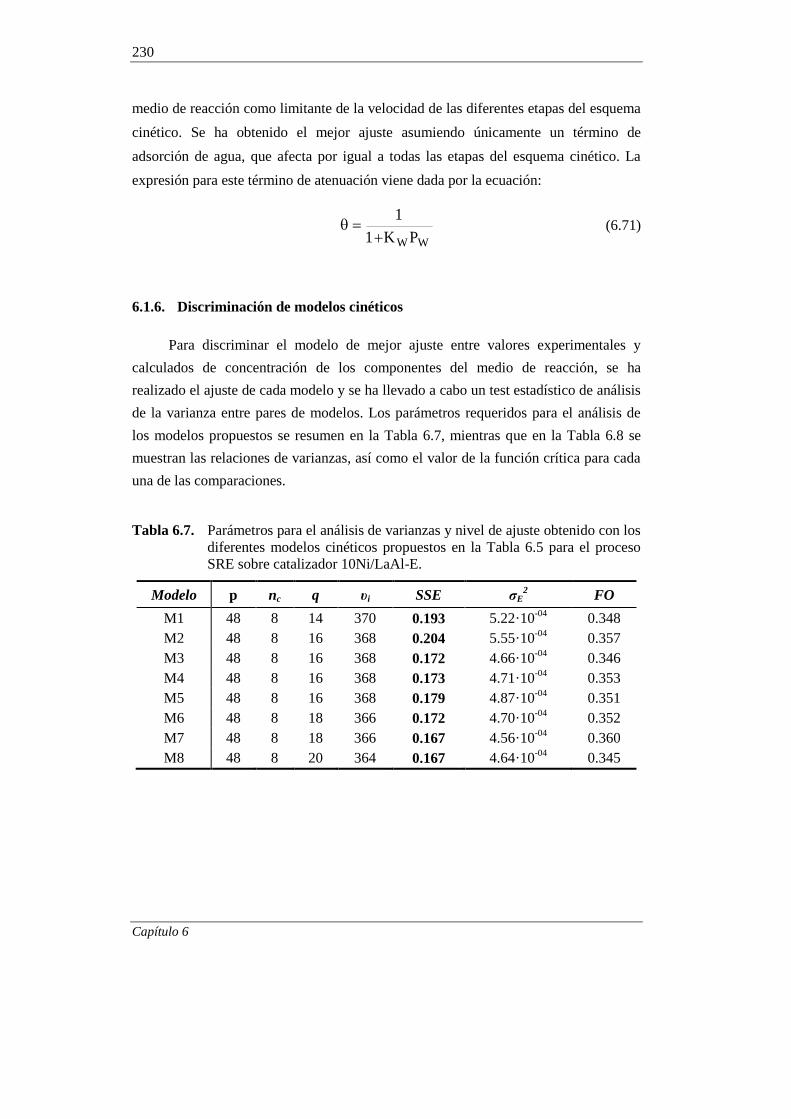
230
Capítulo 6
medio de reacción como limitante de la velocidad de las diferentes etapas del esquema
cinético. Se ha obtenido el mejor ajuste asumiendo únicamente un término de
adsorción de agua, que afecta por igual a todas las etapas del esquema cinético. La
expresión para este término de atenuación viene dada por la ecuación:
WW PK1
1
(6.71)
6.1.6. Discriminación de modelos cinéticos
Para discriminar el modelo de mejor ajuste entre valores experimentales y
calculados de concentración de los componentes del medio de reacción, se ha
realizado el ajuste de cada modelo y se ha llevado a cabo un test estadístico de análisis
de la varianza entre pares de modelos. Los parámetros requeridos para el análisis de
los modelos propuestos se resumen en la Tabla 6.7, mientras que en la Tabla 6.8 se
muestran las relaciones de varianzas, así como el valor de la función crítica para cada
una de las comparaciones.
Tabla 6.7. Parámetros para el análisis de varianzas y nivel de ajuste obtenido con los
diferentes modelos cinéticos propuestos en la Tabla 6.5 para el proceso
SRE sobre catalizador 10Ni/LaAl-E.
Modelo p nc q υi SSE σE2 FO
M1 48 8 14 370 0.193 5.22·10-04 0.348
M2 48 8 16 368 0.204 5.55·10-04 0.357
M3 48 8 16 368 0.172 4.66·10-04 0.346
M4 48 8 16 368 0.173 4.71·10-04 0.353
M5 48 8 16 368 0.179 4.87·10-04 0.351
M6 48 8 18 366 0.172 4.70·10-04 0.352
M7 48 8 18 366 0.167 4.56·10-04 0.360
M8 48 8 20 364 0.167 4.64·10-04 0.345
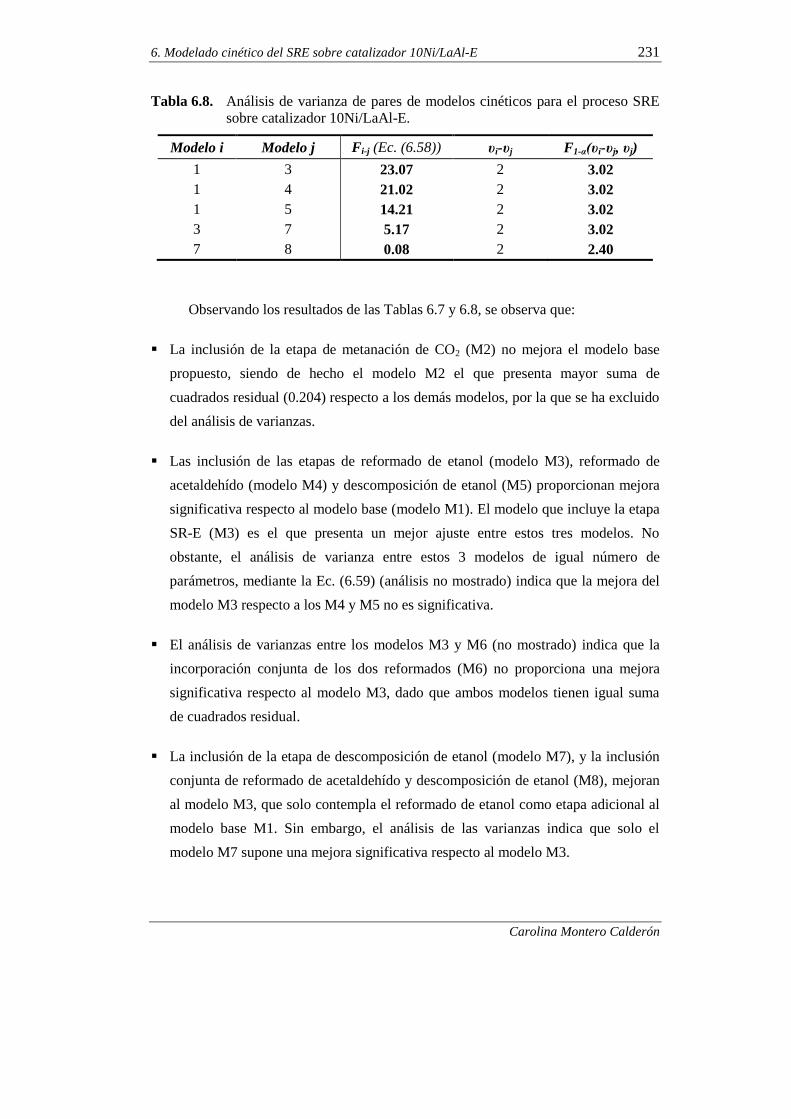
6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 231
Carolina Montero Calderón
Tabla 6.8. Análisis de varianza de pares de modelos cinéticos para el proceso SRE
sobre catalizador 10Ni/LaAl-E.
Modelo i Modelo j Fi-j (Ec. (6.58)) υi-υj F1-α(υi-υj, υj)
1 3 23.07 2 3.02
1 4 21.02 2 3.02
1 5 14.21 2 3.02
3 7 5.17 2 3.02
7 8 0.08 2 2.40
Observando los resultados de las Tablas 6.7 y 6.8, se observa que:
La inclusión de la etapa de metanación de CO2 (M2) no mejora el modelo base
propuesto, siendo de hecho el modelo M2 el que presenta mayor suma de
cuadrados residual (0.204) respecto a los demás modelos, por la que se ha excluido
del análisis de varianzas.
Las inclusión de las etapas de reformado de etanol (modelo M3), reformado de
acetaldehído (modelo M4) y descomposición de etanol (M5) proporcionan mejora
significativa respecto al modelo base (modelo M1). El modelo que incluye la etapa
SR-E (M3) es el que presenta un mejor ajuste entre estos tres modelos. No
obstante, el análisis de varianza entre estos 3 modelos de igual número de
parámetros, mediante la Ec. (6.59) (análisis no mostrado) indica que la mejora del
modelo M3 respecto a los M4 y M5 no es significativa.
El análisis de varianzas entre los modelos M3 y M6 (no mostrado) indica que la
incorporación conjunta de los dos reformados (M6) no proporciona una mejora
significativa respecto al modelo M3, dado que ambos modelos tienen igual suma
de cuadrados residual.
La inclusión de la etapa de descomposición de etanol (modelo M7), y la inclusión
conjunta de reformado de acetaldehído y descomposición de etanol (M8), mejoran
al modelo M3, que solo contempla el reformado de etanol como etapa adicional al
modelo base M1. Sin embargo, el análisis de las varianzas indica que solo el
modelo M7 supone una mejora significativa respecto al modelo M3.
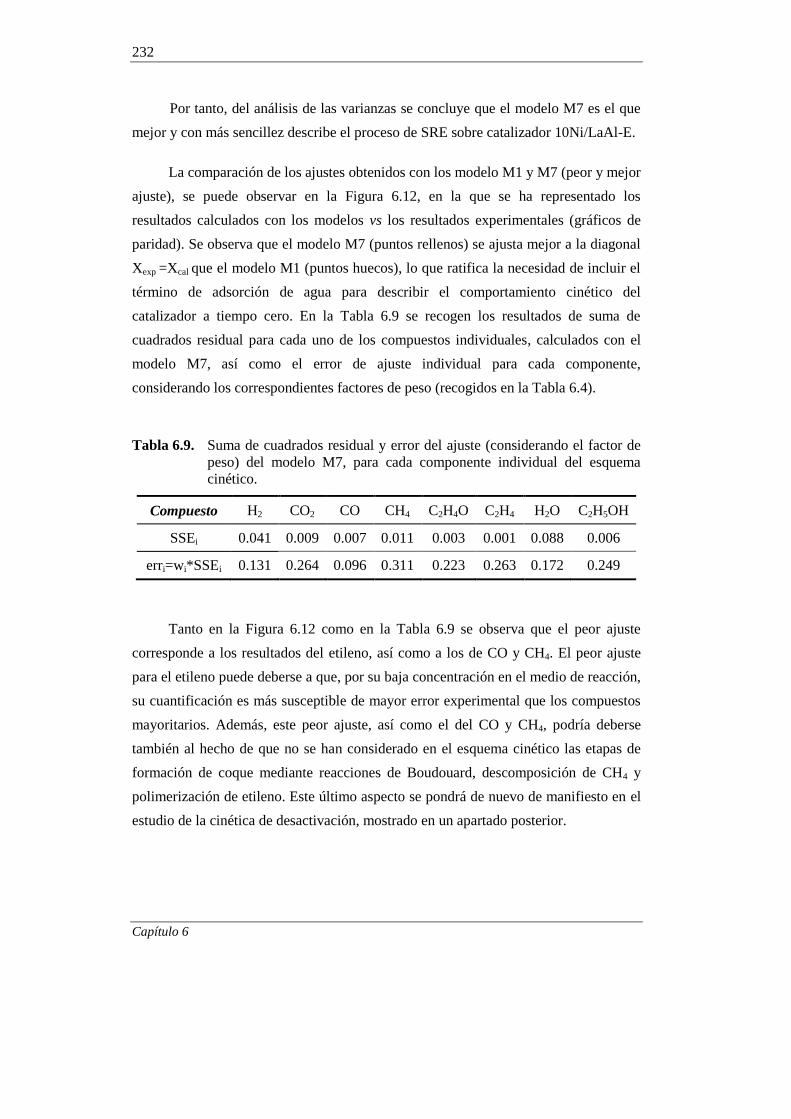
232
Capítulo 6
Por tanto, del análisis de las varianzas se concluye que el modelo M7 es el que
mejor y con más sencillez describe el proceso de SRE sobre catalizador 10Ni/LaAl-E.
La comparación de los ajustes obtenidos con los modelo M1 y M7 (peor y mejor
ajuste), se puede observar en la Figura 6.12, en la que se ha representado los
resultados calculados con los modelos vs los resultados experimentales (gráficos de
paridad). Se observa que el modelo M7 (puntos rellenos) se ajusta mejor a la diagonal
Xexp =Xcal que el modelo M1 (puntos huecos), lo que ratifica la necesidad de incluir el
término de adsorción de agua para describir el comportamiento cinético del
catalizador a tiempo cero. En la Tabla 6.9 se recogen los resultados de suma de
cuadrados residual para cada uno de los compuestos individuales, calculados con el
modelo M7, así como el error de ajuste individual para cada componente,
considerando los correspondientes factores de peso (recogidos en la Tabla 6.4).
Tabla 6.9. Suma de cuadrados residual y error del ajuste (considerando el factor de
peso) del modelo M7, para cada componente individual del esquema
cinético.
Compuesto H2 CO2 CO CH4 C2H4O C2H4 H2O C2H5OH
SSEi 0.041 0.009 0.007 0.011 0.003 0.001 0.088 0.006
erri=wi*SSEi 0.131 0.264 0.096 0.311 0.223 0.263 0.172 0.249
Tanto en la Figura 6.12 como en la Tabla 6.9 se observa que el peor ajuste
corresponde a los resultados del etileno, así como a los de CO y CH4. El peor ajuste
para el etileno puede deberse a que, por su baja concentración en el medio de reacción,
su cuantificación es más susceptible de mayor error experimental que los compuestos
mayoritarios. Además, este peor ajuste, así como el del CO y CH4, podría deberse
también al hecho de que no se han considerado en el esquema cinético las etapas de
formación de coque mediante reacciones de Boudouard, descomposición de CH4 y
polimerización de etileno. Este último aspecto se pondrá de nuevo de manifiesto en el
estudio de la cinética de desactivación, mostrado en un apartado posterior.
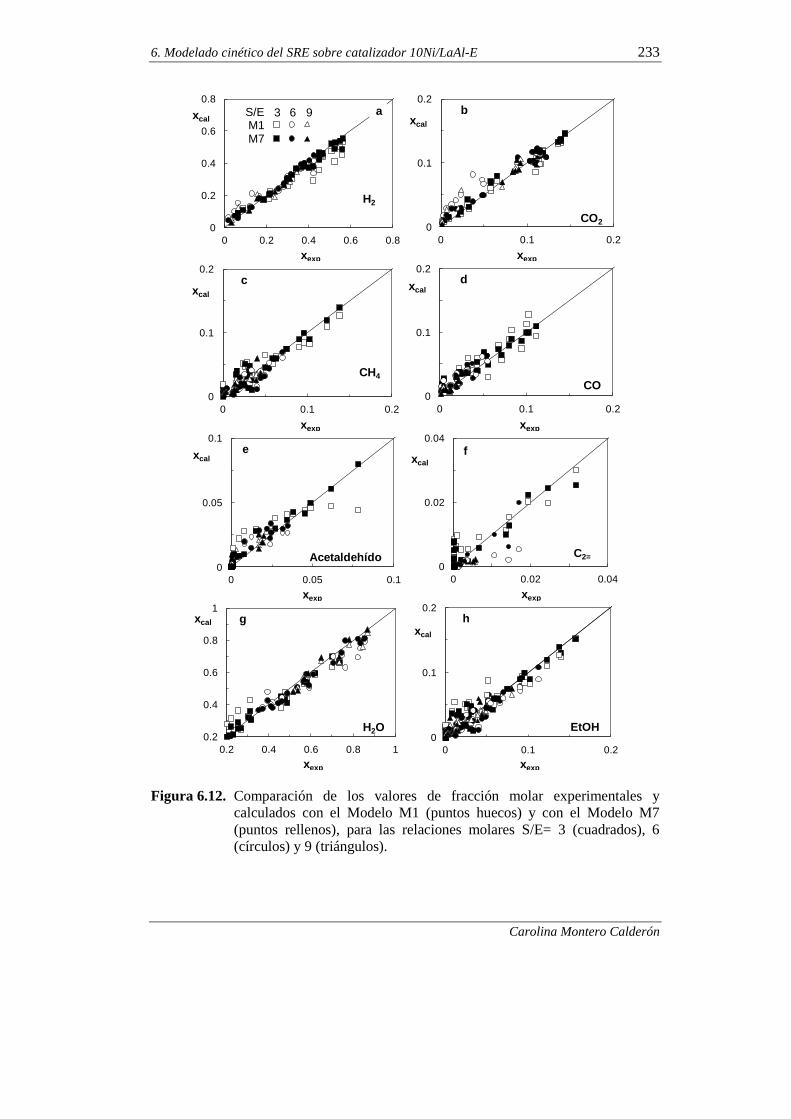
6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 233
Carolina Montero Calderón
Figura 6.12. Comparación de los valores de fracción molar experimentales y
calculados con el Modelo M1 (puntos huecos) y con el Modelo M7
(puntos rellenos), para las relaciones molares S/E= 3 (cuadrados), 6
(círculos) y 9 (triángulos).
0
0.2
0.4
0.6
0.8
0 0.2 0.4 0.6 0.8
xcal
xexp
H2
3 6 9S/EM1M7
a
0
0.1
0.2
0 0.1 0.2
xcal
xexp
CO2
b
0
0.1
0.2
0 0.1 0.2
xcal
xexp
CH4
c
0
0.1
0.2
0 0.1 0.2
xcal
xexp
CO
d
0
0.05
0.1
0 0.05 0.1
xcal
xexp
Acetaldehído
e
0
0.02
0.04
0 0.02 0.04
xcal
xexp
C2=
f
0.2
0.4
0.6
0.8
1
0.2 0.4 0.6 0.8 1
xcal
xexp
H2O
g
0
0.1
0.2
0 0.1 0.2
xcal
xexp
EtOH
h
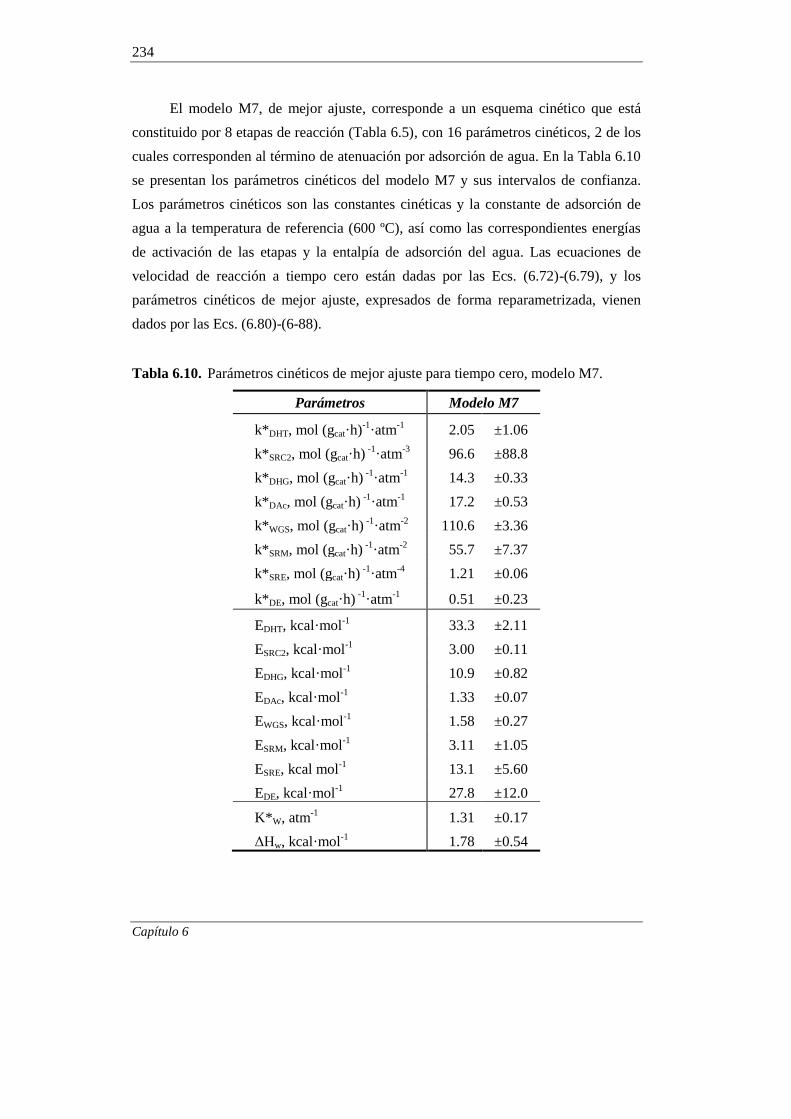
234
Capítulo 6
El modelo M7, de mejor ajuste, corresponde a un esquema cinético que está
constituido por 8 etapas de reacción (Tabla 6.5), con 16 parámetros cinéticos, 2 de los
cuales corresponden al término de atenuación por adsorción de agua. En la Tabla 6.10
se presentan los parámetros cinéticos del modelo M7 y sus intervalos de confianza.
Los parámetros cinéticos son las constantes cinéticas y la constante de adsorción de
agua a la temperatura de referencia (600 ºC), así como las correspondientes energías
de activación de las etapas y la entalpía de adsorción del agua. Las ecuaciones de
velocidad de reacción a tiempo cero están dadas por las Ecs. (6.72)-(6.79), y los
parámetros cinéticos de mejor ajuste, expresados de forma reparametrizada, vienen
dados por las Ecs. (6.80)-(6-88).
Tabla 6.10. Parámetros cinéticos de mejor ajuste para tiempo cero, modelo M7.
Parámetros Modelo M7
k*DHT, mol (gcat·h)-1
·atm-1 2.05 ±1.06
k*SRC2, mol (gcat·h) -1
·atm-3 96.6 ±88.8
k*DHG, mol (gcat·h) -1
·atm-1 14.3 ±0.33
k*DAc, mol (gcat·h) -1
·atm-1 17.2 ±0.53
k*WGS, mol (gcat·h) -1
·atm-2 110.6 ±3.36
k*SRM, mol (gcat·h) -1
·atm-2 55.7 ±7.37
k*SRE, mol (gcat·h) -1
·atm-4 1.21 ±0.06
k*DE, mol (gcat·h) -1
·atm-1 0.51 ±0.23
EDHT, kcal·mol-1 33.3 ±2.11
ESRC2, kcal·mol-1 3.00 ±0.11
EDHG, kcal·mol-1 10.9 ±0.82
EDAc, kcal·mol-1 1.33 ±0.07
EWGS, kcal·mol-1 1.58 ±0.27
ESRM, kcal·mol-1 3.11 ±1.05
ESRE, kcal mol-1 13.1 ±5.60
EDE, kcal·mol-1 27.8 ±12.0
K*W, atm-1 1.31 ±0.17
ΔHw, kcal·mol-1 1.78 ±0.54

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 235
Carolina Montero Calderón
WW
EDHT0DHT
PK1
Pk=r
(6.72)
WW
2WSRC2
0SRC2PK1
PP kr C2
(6.73)
WW
DHGHAcEDHG
0DHGPK1
)/KPP-(Pk= r 2
(6.74)
WW
AcDAc0DAc
PK1
Pkr
(6.75)
WW
WGSCOHWCOWGS
0WGSPK1
)K/ PP-P(P k=r 22
(6.76)
WW
SRMCO3H2WMSRM
0SRMPK1
)K/PP-P(Pk=r
(6.77)
WW
3WESRE
0SREPK1
PP k=r
(6.78)
WW
EDE0DE
PK1
Pkr
(6.79)
873
1
T
1
R
11.2 33.3- 06.1 2.05k DHT (6.80)
873
1
T
1
R
0.11 3.00- 88.8 96.6kSRC2 (6.81)
873
1
T
1
R
0.82 10.9- 0.33 14.3k DHG (6.82)
873
1
T
1
R
0.07 1.33- 0.53 17.2k DAc (6.83)
873
1
T
1
R
27.0 1.58- 36.3 6.101k WGS (6.84)
873
1
T
1
R
05.1 3.11- 37.7 7.55kSRM (6.85)
873
1
T
1
R
60.5 13.1- 06.0 21.1kSRE (6.86)
873
1
T
1
R
0.21 27.8- 23.0 51.0k DE (6.87)
873
1
T
1
R
0.54 1.78 0.17 1.31K W (6.88)

236
Capítulo 6
6.1.7. Incorporación de las rutas térmicas en el modelado cinético
Se ha planteado el balance de materia referido al volumen (o longitud) del
reactor, en vez de a la masa de catalizador, para poder diferenciar y tener en cuenta
tanto la contribución catalítica (cj
r ), como la contribución térmica (tj
r ) a la velocidad
de reacción de cada etapa (rj), dado que, como se mostró en el Apartado 4.1, para
temperatura elevada (600 ºC y especialmente a 650 ºC) las rutas térmicas tienen una
contribución no despreciable a la velocidad de reacción de determinadas etapas del
esquema cinético de reacción. Estas rutas térmicas serán especialmente importantes en
el estudio de la cinética de desactivación, para cuantificar los rendimientos de
productos de reacción obtenidos a elevada temperatura y bajo tiempo espacial (cuando
el catalizador sufre una rápida desactivación).
Así, para unas condiciones dadas de temperatura y relación S/E en la
alimentación, se ha considerado que la contribución de las rutas térmicas a cada etapa
de reacción es constante, y corresponde a la circulación en la longitud efectiva de
reactor (esto es, la zona caliente del reactor, que es de 116 mm), mientras que la
contribución catalítica es función de la masa de catalizador utilizada (es decir, del
tiempo espacial). Por ello, para una masa de catalizador dada, se ha definido un
parámetro de densidad del lecho, rb, como el cociente de la masa de catalizador por
unidad de volumen efectivo de reactor, de modo que la velocidad de reacción de cada
etapa del esquema cinético es la suma de la contribución térmica y catalítica, según la
Ec. (6.89)
tj
cjbj rrr r (6.89)
donde ρb es la densidad del lecho.
El modelado del proceso SRE considerando el efecto térmico se ha desarrollado
en dos fases:
i) Selección de un modelo para cuantificar únicamente la contribución de las rutas
térmicas, mediante el ajuste de los diferentes modelos propuestos a datos
experimentales de composición del medio de reacción obtenidos en ausencia de
catalizador.

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 237
Carolina Montero Calderón
ii) Determinación del modelo cinético global para el proceso SRE, integrando la
contribución catalítica y la térmica, considerando para ello conjuntamente el
modelo cinético de mejor ajuste previamente seleccionado, M7, junto con el
modelo para las rutas térmicas seleccionado en la etapa (i), y recalculando
conjuntamente los parámetros de mejor ajuste para ambos modelos.
El procedimiento seguido y los resultados obtenidos en la etapa (i) se muestran
en el Anexo D. Se concluye que el modelo cinético que mejor cuantifica el efecto
térmico en el proceso SRE está constituido por 7 etapas de reacción con 4 constantes
cinéticas. Las ecuaciones de velocidad de reacción de cada etapa para cuantificar este
efecto, (rj)t, están dadas por las siguientes expresiones:
E2t
DHT Pk=r (6.90)
)/KPP-(Pk= r DHGHAcE1t
DHG 2(6.91)
Ac3t
DAc Pkr (6.92)
)K/ PP-P(P k=r WGSCOHWCO4t
WGS 22(6.93)
)K/PP-P(Pk=r SRMCO3H2WM4
tSRM (6.94)
3WE3
tSRE PP k=r (6.95)
E3t
DE Pkr (6.96)
Teniendo en cuenta que la velocidad de reacción de cada etapa del esquema
cinético debe considerar la contribución catalítica y la térmica, (Ec. (6.89)), la
velocidad de reacción de las etapa del esquema cinético correspondiente al modelo M7
(recogidas en la Tabla 6.5) debe combinar las Ecs. (6.72)-(6.79), correspondientes a la
contribución catalítica, con las Ecs. (6.90)-(6.96), correspondientes a la contribución
térmica:
E2WW
EDHTb0DHT Pk
PK1
Pk=r
r (6.97)
WW
2WSRC2
b0SRC2PK1
PP kr C2
r (6.98)

238
Capítulo 6
rDHG
HAcE1
WW
DHG
HAcEDHG
b0DHGK
PP-Pk
PK1
K
PP-Pk
= r 2
2
(6.99)
Ac3WW
AcDAcb0DAc Pk
PK1
Pkr
r (6.100)
rWGS
COHWCO4
WW
WGS
COHWCOWGS
b0WGSK
PP-PP k
PK1
K
PP-PP k
=r 22
22
(6.101)
rSRM
CO3H2
WM4WW
SRM
CO3H2
WMSRM
b0SRMK
PP-PPk
PK1
K
PP-PPk
=r (6.102)
3WE3
WW
3WESRE
b0SRE PP kPK1
PP k=r
r (6.103)
E3WW
EDEb0DE Pk
PK1
Pkr
r (6.104)
A este modelo cinético global, dado por las Ecs. (6.97)-(6.104), se le ha
denominado modelo M0. Se ha llevado a cabo el ajuste de este modelo y los
resultados se muestran en la Tabla 6.11, que incluye el valor de los parámetros
cinéticos de mejor ajuste junto con sus intervalos de confianza, así como el valor de la
función objetivo error, la suma de cuadrados residual total y la suma de cuadrados
residual de cada componente individual.
En la Figura 6.13, que muestra los gráficos de paridad para cada componente
individual,y se puede visualizar el grado de ajuste global obtenido con este modelo
M0, en comparación con el modelo M7 que no incluye el efecto térmico. Se observa
que en general el ajuste de ambos modelos es muy similar, si bien el modelo global
M0 ajusta ligeramente mejor tanto el etanol como algunos componentes minoritarios,
especialmente el etileno, así como el CH4 y en menor medida el CO.
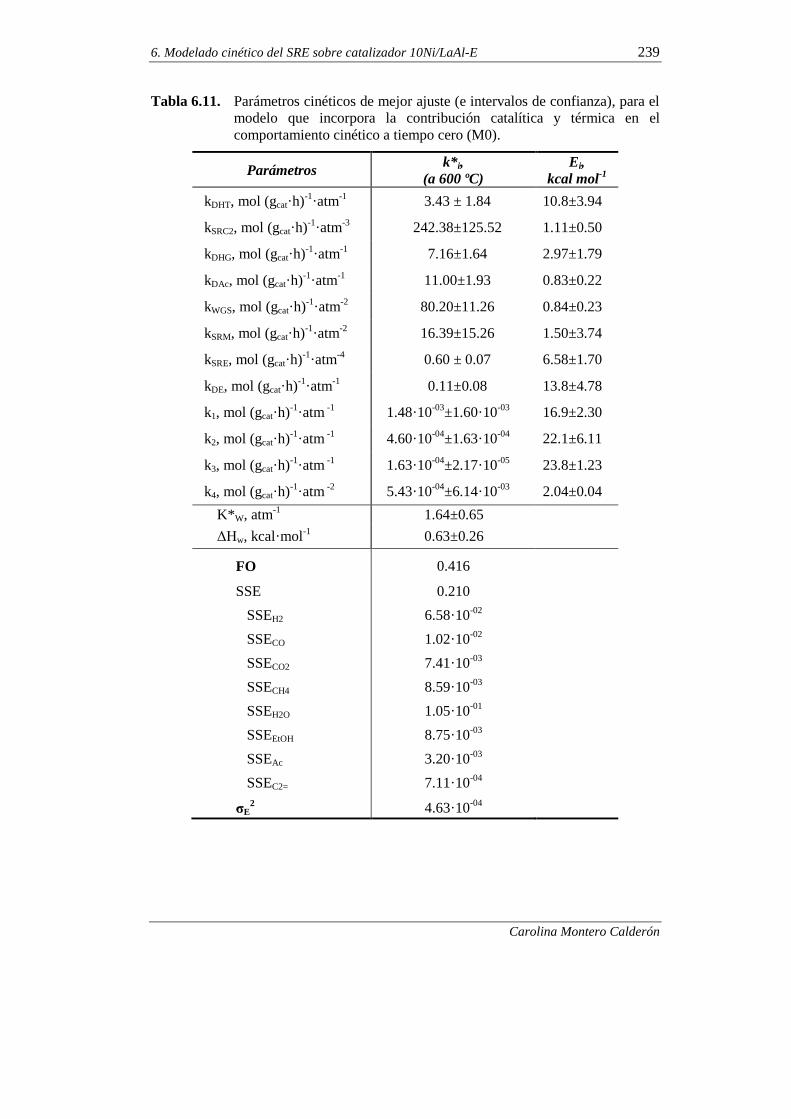
6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 239
Carolina Montero Calderón
Tabla 6.11. Parámetros cinéticos de mejor ajuste (e intervalos de confianza), para el
modelo que incorpora la contribución catalítica y térmica en el
comportamiento cinético a tiempo cero (M0).
Parámetros k*i,
(a 600 ºC) Ei,
kcal mol-1
kDHT, mol (gcat·h)-1
·atm-1 3.43 ± 1.84 10.8±3.94
kSRC2, mol (gcat·h)-1
·atm-3 242.38±125.52 1.11±0.50
kDHG, mol (gcat·h)-1
·atm-1 7.16±1.64 2.97±1.79
kDAc, mol (gcat·h)-1
·atm-1 11.00±1.93 0.83±0.22
kWGS, mol (gcat·h)-1
·atm-2 80.20±11.26 0.84±0.23
kSRM, mol (gcat·h)-1
·atm-2 16.39±15.26 1.50±3.74
kSRE, mol (gcat·h)-1
·atm-4 0.60 ± 0.07 6.58±1.70
kDE, mol (gcat·h)-1
·atm-1 0.11±0.08 13.8±4.78
k1, mol (gcat·h)-1
·atm -1
1.48·10-03
±1.60·10-03 16.9±2.30
k2, mol (gcat·h)-1
·atm -1
4.60·10-04
±1.63·10-04 22.1±6.11
k3, mol (gcat·h)-1
·atm -1
1.63·10-04
±2.17·10-05 23.8±1.23
k4, mol (gcat·h)-1
·atm -2
5.43·10-04
±6.14·10-03 2.04±0.04
K*W, atm-1 1.64±0.65
ΔHw, kcal·mol-1 0.63±0.26
FO 0.416
SSE
SSEH2
SSECO
SSECO2
SSECH4
SSEH2O
SSEEtOH
SSEAc
SSEC2=
0.210
6.58·10-02
1.02·10-02
7.41·10-03
8.59·10-03
1.05·10-01
8.75·10-03
3.20·10-03
7.11·10-04
σE2 4.63·10
-04
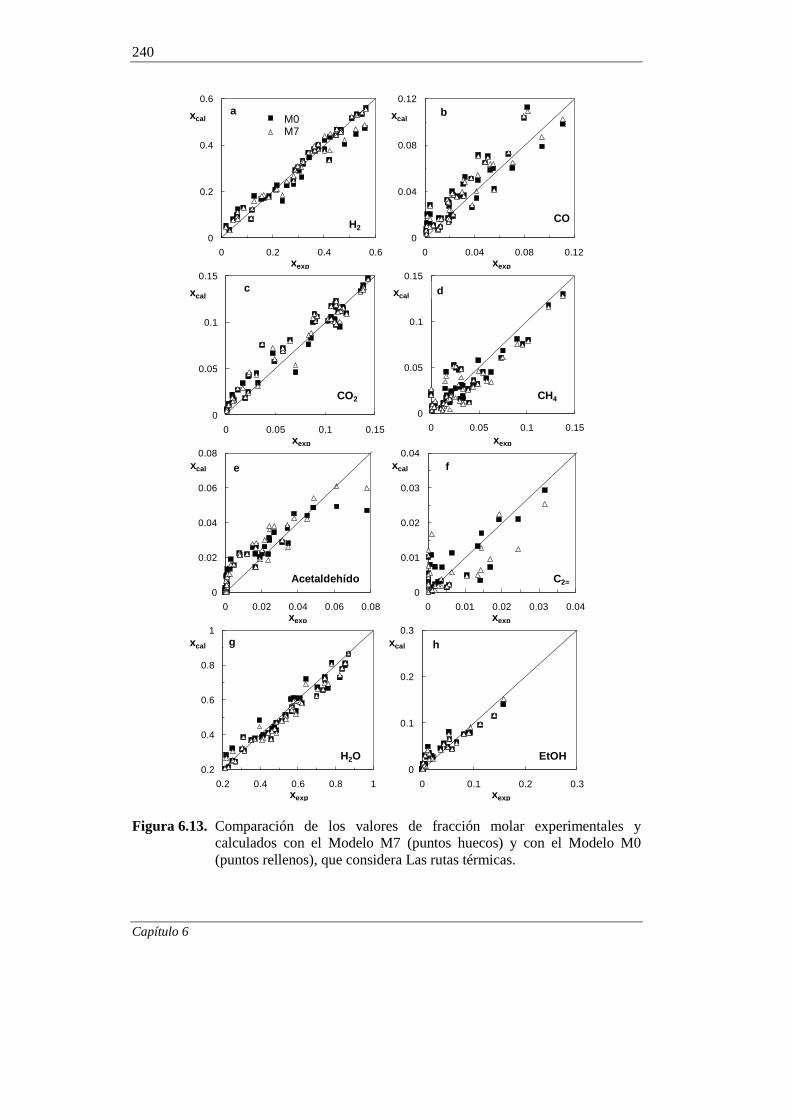
240
Capítulo 6
Figura 6.13. Comparación de los valores de fracción molar experimentales y
calculados con el Modelo M7 (puntos huecos) y con el Modelo M0
(puntos rellenos), que considera Las rutas térmicas.
0
0.2
0.4
0.6
0 0.2 0.4 0.6
xcal
xexp
H2
M0M7
a
0
0.04
0.08
0.12
0 0.04 0.08 0.12
xcal
xexp
CO
b
0
0.05
0.1
0.15
0 0.05 0.1 0.15
xcal
xexp
CO2
c
0
0.05
0.1
0.15
0 0.05 0.1 0.15
xcal
xexp
CH4
d
0
0.02
0.04
0.06
0.08
0 0.02 0.04 0.06 0.08
xcal
xexp
e
Acetaldehído
0
0.01
0.02
0.03
0.04
0 0.01 0.02 0.03 0.04
xcal
xexp
f
C2=
0.2
0.4
0.6
0.8
1
0.2 0.4 0.6 0.8 1
xcal
xexp
g
H2O
0
0.1
0.2
0.3
0 0.1 0.2 0.3
xcal
xexp
h
EtOH
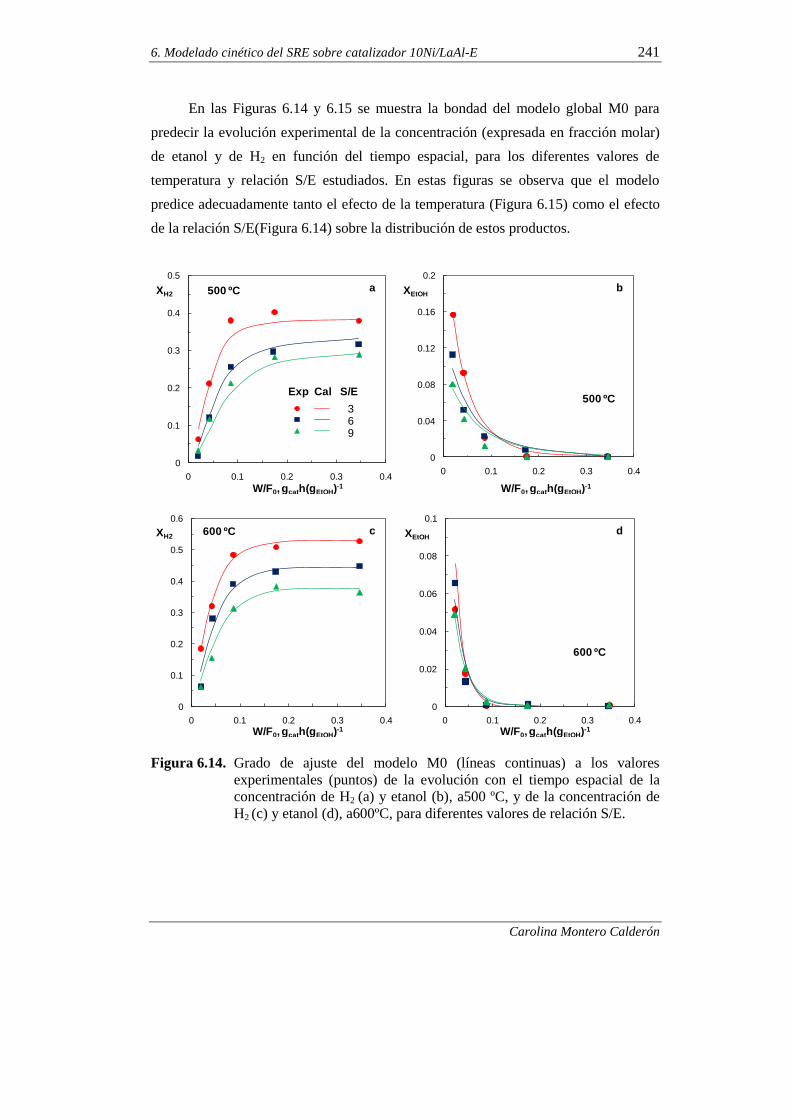
6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 241
Carolina Montero Calderón
En las Figuras 6.14 y 6.15 se muestra la bondad del modelo global M0 para
predecir la evolución experimental de la concentración (expresada en fracción molar)
de etanol y de H2 en función del tiempo espacial, para los diferentes valores de
temperatura y relación S/E estudiados. En estas figuras se observa que el modelo
predice adecuadamente tanto el efecto de la temperatura (Figura 6.15) como el efecto
de la relación S/E(Figura 6.14) sobre la distribución de estos productos.
Figura 6.14. Grado de ajuste del modelo M0 (líneas continuas) a los valores
experimentales (puntos) de la evolución con el tiempo espacial de la
concentración de H2 (a) y etanol (b), a500 ºC, y de la concentración de
H2 (c) y etanol (d), a600ºC, para diferentes valores de relación S/E.
0
0.1
0.2
0.3
0.4
0.5
0 0.1 0.2 0.3 0.4
XH2
W/F0, gcath(gEtOH)-1
Exp Cal S/E
369
a500 ºC
0
0.04
0.08
0.12
0.16
0.2
0 0.1 0.2 0.3 0.4
XEtOH
W/F0, gcath(gEtOH)-1
b
500 ºC
0
0.1
0.2
0.3
0.4
0.5
0.6
0 0.1 0.2 0.3 0.4
XH2
W/F0, gcath(gEtOH)-1
c600 ºC
0
0.02
0.04
0.06
0.08
0.1
0 0.1 0.2 0.3 0.4
XEtOH
W/F0, gcath(gEtOH)-1
d
600 ºC
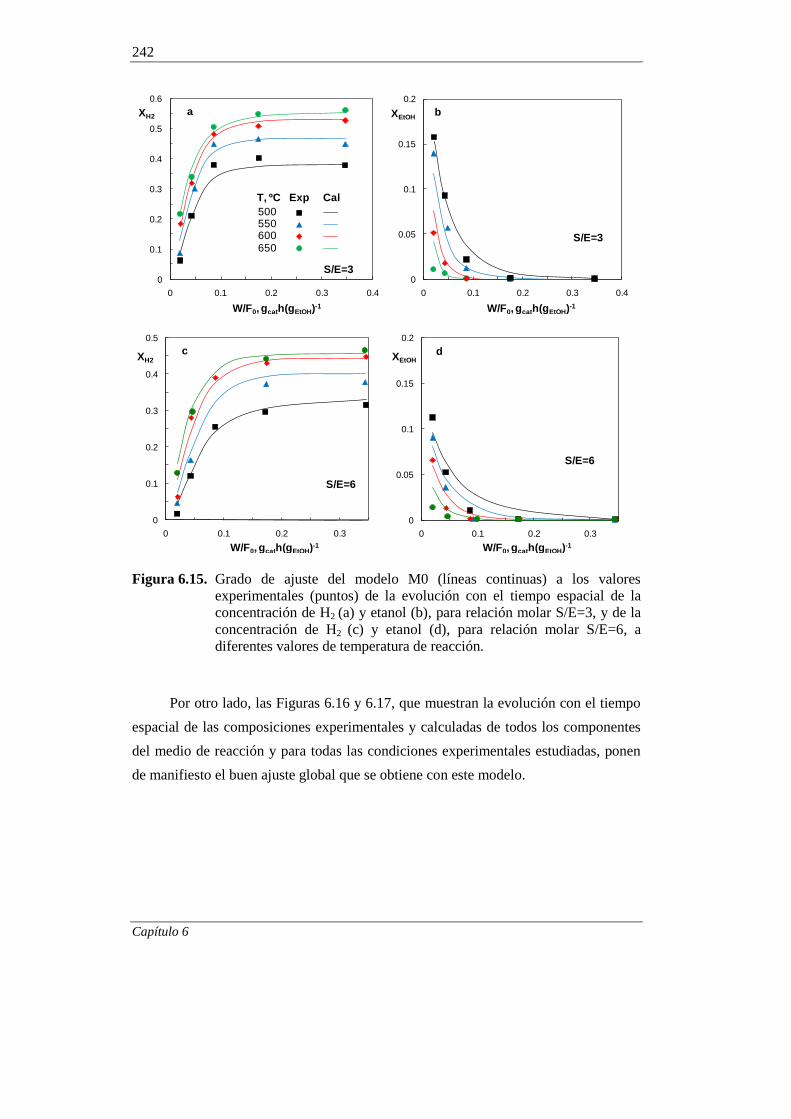
242
Capítulo 6
Figura 6.15. Grado de ajuste del modelo M0 (líneas continuas) a los valores
experimentales (puntos) de la evolución con el tiempo espacial de la
concentración de H2 (a) y etanol (b), para relación molar S/E=3, y de la
concentración de H2 (c) y etanol (d), para relación molar S/E=6, a
diferentes valores de temperatura de reacción.
Por otro lado, las Figuras 6.16 y 6.17, que muestran la evolución con el tiempo
espacial de las composiciones experimentales y calculadas de todos los componentes
del medio de reacción y para todas las condiciones experimentales estudiadas, ponen
de manifiesto el buen ajuste global que se obtiene con este modelo.
0
0.1
0.2
0.3
0.4
0.5
0.6
0 0.1 0.2 0.3 0.4
XH2
W/F0, gcath(gEtOH)-1
T, ºC Exp Cal
500550600650
a
S/E=30
0.05
0.1
0.15
0.2
0 0.1 0.2 0.3 0.4
XEtOH
W/F0, gcath(gEtOH)-1
S/E=3
b
0
0.1
0.2
0.3
0.4
0.5
0 0.1 0.2 0.3
XH2
W/F0, gcath(gEtOH)-1
c
S/E=6
0
0.05
0.1
0.15
0.2
0 0.1 0.2 0.3
XEtOH
W/F0, gcath(gEtOH)-1
d
S/E=6
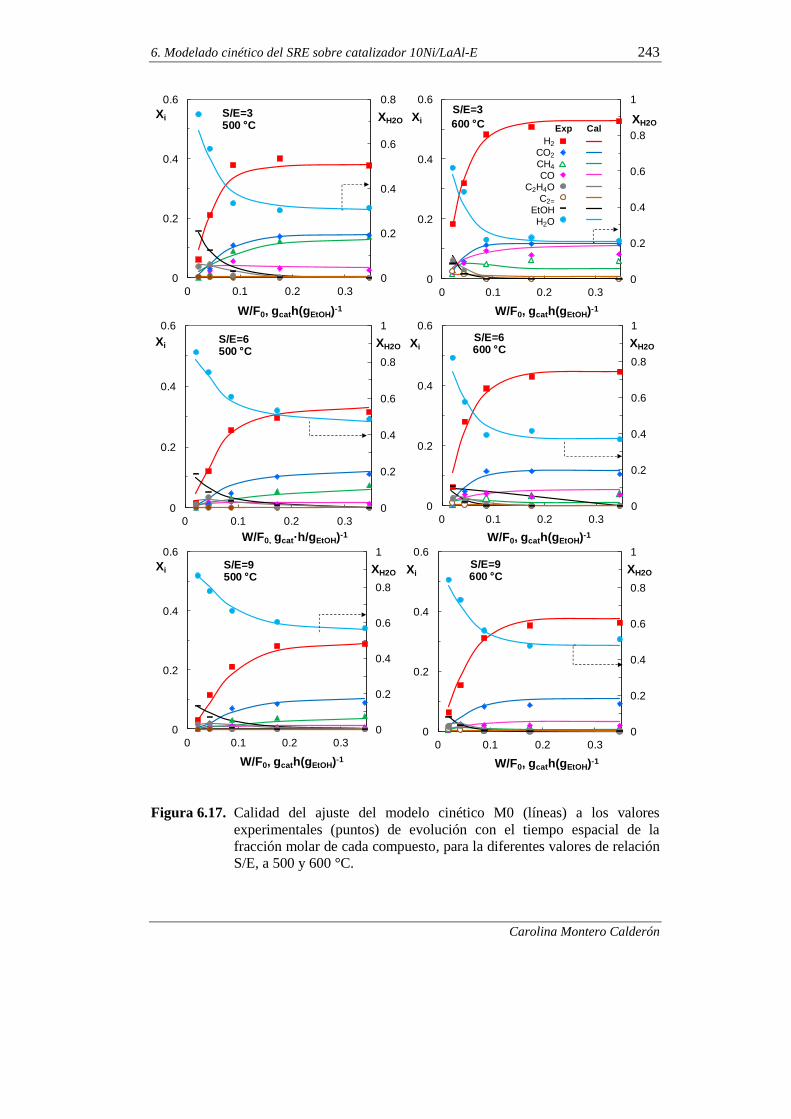
6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 243
Carolina Montero Calderón
Figura 6.17. Calidad del ajuste del modelo cinético M0 (líneas) a los valores
experimentales (puntos) de evolución con el tiempo espacial de la
fracción molar de cada compuesto, para la diferentes valores de relación
S/E, a 500 y 600 °C.
0
0.2
0.4
0.6
0.8
0
0.2
0.4
0.6
0 0.1 0.2 0.3
XH2OXi
W/F0, gcath(gEtOH)-1
S/E=3500 C
0
0.2
0.4
0.6
0.8
1
0
0.2
0.4
0.6
0 0.1 0.2 0.3
XH2OXi
W/F0, gcath(gEtOH)-1
S/E=3
600 C
H2
CO2
CH4
CO
C2H4O
C2=
EtOH
H2O
Exp Cal
0
0.2
0.4
0.6
0.8
1
0
0.2
0.4
0.6
0 0.1 0.2 0.3
XH2OXi
W/F0, gcat·h/gEtOH)-1
S/E=6500 C
0
0.2
0.4
0.6
0.8
1
0
0.2
0.4
0.6
0 0.1 0.2 0.3
XH2OXi
W/F0, gcath(gEtOH)-1
S/E=6600 C
0
0.2
0.4
0.6
0.8
1
0
0.2
0.4
0.6
0 0.1 0.2 0.3
XH2OXi
W/F0, gcath(gEtOH)-1
S/E=9500 C
0
0.2
0.4
0.6
0.8
1
0
0.2
0.4
0.6
0 0.1 0.2 0.3
XH2OXi
W/F0, gcath(gEtOH)-1
S/E=9600 C
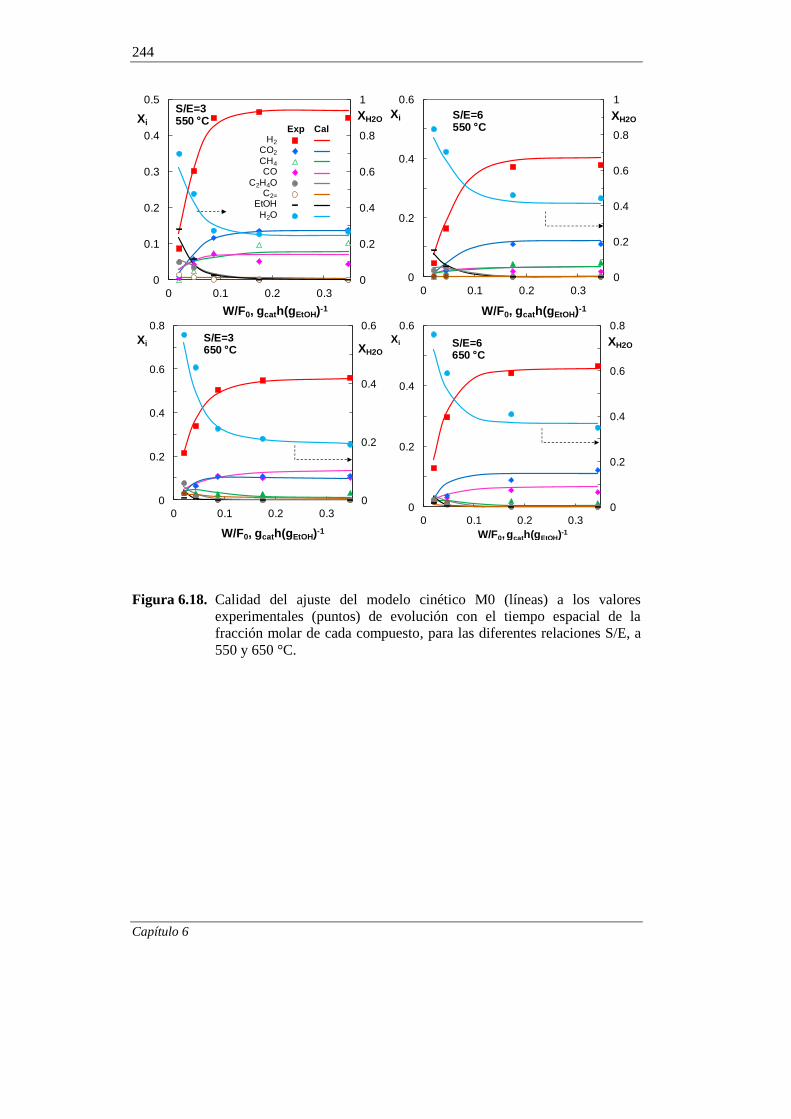
244
Capítulo 6
Figura 6.18. Calidad del ajuste del modelo cinético M0 (líneas) a los valores
experimentales (puntos) de evolución con el tiempo espacial de la
fracción molar de cada compuesto, para las diferentes relaciones S/E, a
550 y 650 °C.
0
0.2
0.4
0.6
0.8
1
0
0.1
0.2
0.3
0.4
0.5
0 0.1 0.2 0.3
XH2OXi
W/F0, gcath(gEtOH)-1
Exp CalH2
CO2
CH4
CO
C2H4OC2=
EtOH
H2O
S/E=3550 C
0
0.2
0.4
0.6
0.8
1
0
0.2
0.4
0.6
0 0.1 0.2 0.3
XH2OXi
W/F0, gcath(gEtOH)-1
S/E=6550 C
0
0.2
0.4
0.6
0
0.2
0.4
0.6
0.8
0 0.1 0.2 0.3
XH2O
Xi
W/F0, gcath(gEtOH)-1
S/E=3650 C
0
0.2
0.4
0.6
0.8
0
0.2
0.4
0.6
0 0.1 0.2 0.3
XH2OXi
W/F0, gcath(gEtOH)-1
S/E=6650 C

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 245
Carolina Montero Calderón
6.2. MODELADO CINÉTICO DE LA DESACTIVACIÓN POR COQUE
Se ha propuesto inicialmente un modelo de desactivación no selectiva, con una
misma actividad para todas las etapas del esquema cinético considerado. Esta
actividad se ha definido como el cociente de las velocidades de reacción a tiempo t y a
tiempo cero:
cjo
cj
r
ra= (6.105)
donde rjc y rjo
c se refieren exclusivamente a la contribución catalítica a la velocidad de
reacción de cada etapa del esquema cinético, puesto que la contribución por rutas
térmicas a la velocidad de reacción no está afectada por la desactivación. Por tanto, la
velocidad de reacción a tiempo t para cada etapa del esquema cinético vendrá dada por
la siguiente expresión:
tj
cjobj rarr +⋅ρ= (6.106)
Para cuantificar la disminución con el tiempo de la actividad se han considerado
ecuaciones de desactivación que son función de la concentración de los precursores de
coque en el medio de reacción, y que tienen la expresión general siguiente:
di a)C,T(
dt
da ⋅ψ=− (6.107)
donde d es el orden de desactivación y ψ(T,Ci) es la función de desactivación, que
incluye el efecto de la temperatura y de la composición del medio de reacción sobre la
velocidad de desactivación. Este tipo de ecuación de desactivación ha sido
previamente utilizada para describir la desactivación para el reformado con vapor de
DME sobre un catalizador bifuncional (metal/ácido) (Oar-Arteta, 2014), así como en
procesos que transcurren sobre catalizadores ácidos, como los procesos MTG y MTO
(Benito y cols., 1996; Aguayo y cols., 1997; 1999; Gayubo y cols., 2002; Mier y cols.,
2011), la transformación de etanol en hidrocarburos (Aguayo y cols., 2002; Gayubo y
cols., 2011, 2012), y en la síntesis de DME en una etapa sobre catalizadores
bifuncionales (Sierra y cols., 2010; Ereña y cols., 2011; Ateka, 2014).

246
Capítulo 6
Sin embargo, dada la diferente naturaleza de las reacciones involucradas en el
esquema cinético en el proceso SRE, y la marcada diferencia en la evolución de los
rendimientos de los productos de reacción con el tiempo observada
experimentalmente, también se puede plantear un modelo de desactivación selectiva,
en el que la pérdida de actividad de los centros activos afecte de diferente forma a las
diferentes etapas del esquema cinético, de modo que se consideren diferentes
actividades (para diferentes etapas del esquema cinético) y diferentes expresiones para
la ecuación cinética de cada una de las actividades definidas.
Con estas premisas, el modelado de la desactivación en esta Tesis se ha
abordado en dos pasos:
i) Asumiendo un modelo de desactivación no selectiva, se ha realizado una primera
discriminación de modelos cinéticos para determinar el/los precursor/es del coque
responsables de la pérdida de actividad observada en el catalizador, así como para
determinar el orden de desactivación de la ecuación cinética
ii) Una vez determinado el/los componentes del medio de reacción responsables de
la disminución de actividad y el orden de desactivación, se ha analizado la
posible mejora que supone considerar un modelo de desactivación selectivo, que
contemple diferente velocidad de desactivación para una o varias de las etapas del
esquema cinético, utilizando para cada una de las actividades consideradas el
mismo tipo de expresiones de velocidad de desactivación (precursores y orden de
desactivación) determinado previamente en la etapa (i)
6.2.1. Metodología de cálculo de los parámetros cinéticos
La discriminación de modelos cinéticos para la desactivación y el cálculo de los
parámetros cinéticos de mejor ajuste para cada modelo propuesto, se ha llevado a cabo
mediante regresión no lineal múltiple. La optimización se ha realizado minimizando
una función objetivo para la desactivación, FOd, definida como la suma ponderada de
los cuadrados de los errores entre los valores de composición experimentales y
calculados:

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 247
Carolina Montero Calderón
∑ ∑ ∑∑= = ==
−−=φ=
c tc n
1i
p
1j
n
1k
2
ij,0ijk*ijki
n
1iiid errXXwwFO
(6.108)
donde nt es el número de tiempos de reacción para los que se ha determinado la
composición del medio de reacción para cada condición experimental j (definida por
un valor dado de temperatura, relación molar S/E y tiempo espacial); err0ij, es la
diferencia entre el valor experimental y calculado de concentración a tiempo cero para
cada compuesto i en la condición experimental j.
En el cálculo del error entre el valor experimental y calculado de concentración
de cada compuesto para el tiempo de reacción k, se resta el error correspondiente a
tiempo cero, err0,ij, con el fin de que el grado de ajuste del modelo cinético a tiempo
cero, previamente calculado, no interfiera en el ajuste correspondiente al modelo
cinético para la desactivación.
El organigrama simplificado para el cálculo de los parámetros cinéticos de cada
modelo en el lenguaje de programación MATLAB es igual al mostrado anteriormente
para el cálculo de la cinética a tiempo cero (Figura 6.9), salvo que en este caso la
función para el cálculo de las ecuaciones diferenciales incluye la ecuación cinética
para la desactivación.
El cálculo de la evolución con el tiempo de las concentraciones de los
componentes del medio de reacción para cada condición experimental, requiere la
resolución de las ecuaciones de conservación de materia de cada compuesto,
conjuntamente con la/s ecuación/es cinética/s para la desactivación, lo que a su vez
requiere integrar conjuntamente las ecuaciones de velocidad de reacción con respecto
al tiempo espacial y la/s ecuación/es cinética/s de desactivación con respecto del
tiempo. De este modo, se tiene en cuenta la historia pasada del catalizador en cada
posición del reactor, condición necesaria cuando la desactivación depende de la
composición en el medio de reacción.
Para llevar a cabo la integración se ha utilizado un procedimiento de cálculo
basado en la cuadratura de Gauss-Legendre, que ha sido desarrollado en anteriores
estudios del grupo de investigación (Epelde, 2013; Gamero 2013), que permite una
resolución 100 veces más rápida que utilizando el método convencional pdepe de

248
Capítulo 6
resolución de ecuaciones diferenciales parciales (Ateka, 2014). El procedimiento de
integración define un mallado bidimensional (en función del tiempo espacial y del
tiempo de reacción) y calcula los valores de las variables dependientes (composición y
actividad) para todo el mallado establecido. Por tanto, calcula la evolución de la
actividad a lo largo del reactor (como correspondería a un lecho fijo) y con el tiempo.
Sin embargo, debe tenerse en cuenta que los datos cinéticos se han obtenido en un
lecho fluidizado, lo que implica que para un valor dado del tiempo espacial, la
actividad de todo el lecho catalítico es la misma, dado que se asume mezcla perfecta
para el sólido.
Por ello, para calcular las velocidades de reacción para un valor dado de tiempo
espacial, correspondiente a una posición z de la malla en que se ha discretizado esta
variable independiente, τz, el valor de la actividad del lecho catalítico se ha calculado
con la siguiente expresión:
z
0z
z
da
aτ
τ=∫τ
τ
τ (6.109)
es decir, es el valor medio de los valores de actividad calculados para todas las
posiciones de la malla correspondientes a tiempos espaciales inferiores al valor de τz.
Al igual que en el cálculo de la cinética a tiempo cero, las constantes cinéticas
de desactivación se han expresado en forma reparametrizada, por lo que los
parámetros cinéticos a optimizar son la constante cinética de desactivación a una
temperatura de referencia (600 ºC) y sus correspondientes energías de activación.
La discriminación entre modelos se ha llevado a cabo de forma similar a la
discriminación del modelo cinético a tiempo cero (Apartado 6.1.4), con la única
diferencia de que en el cálculo de los grados de libertad no se incluye el término de nc
(nº de componentes del esquema cinético). No se ha considerado su inclusión en la Ec.
(6.56), dado que no se considera que contribuya a los grados de libertad en el estudio
de la desactivación, puesto que la distribución de productos ha quedado fijada ya por
el modelo cinético a tiempo cero previamente establecido.

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 249
Carolina Montero Calderón
6.2.2. Modelos cinéticos de desactivación no selectiva
Se han establecido modelos de desactivación de diferente complejidad (Tabla
6.12), considerando que la ecuación cinética de desactivación depende de la
concentración de los componentes del medio que son precursores de la formación del
coque encapsulante y pirolítico (esto es, etanol, acetaldehído y etileno). No se
considera la intervención de CO y CH4, que son precursores de la formación de coque
fibrilar, dado que este tipo de coque no origina una pérdida significativa de la
actividad del catalizador.
Se han estudiado en primer lugar modelos sencillos de desactivación
“separable”, en los que la dependencia de la temperatura se considera en una constante
cinética de desactivación, separadamente de la dependencia de la concentración de los
posibles precursores de coque, y asumiendo orden de desactivación = 1. Entre éstos, se
han ajustado ecuaciones con una constante cinética de desactivación, que incluyen
bien un solo precursor de coque (modelos MD1_1a (PAc), MD1_1b (PC2=) o MD1_1c
(PE)), dos precursores (MD1_2a (PAc y PE), MD1_2b (PAc y PC2=) o MD1_2c (PC2= y
PE)), y tres precursores (MD1_3). Se han considerado también modelos con dos o tres
constantes cinéticas, para considerar la diferente contribución de cada posible
precursor de coque a la desactivación (Modelos MD2a, MD2b, MD2c y MD3).
Tras seleccionar el mejor modelo cinético entre los anteriores (el que
proporciona un mejor ajuste con la mayor sencillez, según se muestra a continuación)
se probó el ajuste obtenido con un mayor orden de desactivación (Modelos MD1d2_1c
y Md1d3_1c), así como la consideración de un modelo “no separable”, que incluye la
adsorción de agua (θ) en el denominador (MD1d2_1cW), dado el efecto atenuante del
agua observado sobre la deposición de coque.
La discriminación del modelo de mejor ajuste se ha realizado mediante un test
estadístico de análisis de la varianza entre pares de modelos, como el mostrado para el
cálculo de la cinética a tiempo cero. Los resultados del ajuste de cada modelo y los
parámetros necesarios para el análisis de varianzas de pares de modelos se muestran
en la Tabla 6.13. Estos resultados ponen de manifiesto que entre los modelos que
consideran orden de desactivación d=1, el de mejor ajuste es el modelo MD1_1c, que

250
Capítulo 6
considera como único precursor de la desactivación al etanol. Este modelo
proporciona incluso un mejor ajuste que los modelos de mayor número de parámetros,
por lo que estos últimos son descartados (no siendo es estos casos necesaria la
aplicación del análisis de varianzas).
Tabla 6.12. Modelos cinéticos de desactivación no selectiva propuestos para el proceso SRE sobre catalizador 10Ni/LaAl-E.
Modelo Ecuación
MD1_1a a)P(kdt
daAc1d=− (6.110)
MD1_1b a)P(kdt
da2C1d ==− (6.111)
MD1_1c a)P(kdt
daE1d=− (6.112)
MD1_2a a)PP(kdt
daEAc1d +=− (6.113)
MD1_2b a)PP(kdt
daAc2C1d +=− = (6.114)
MD1_2c a)PP(kdt
daE2C1d +=− = (6.115)
MD1_3 a)PPP(kdt
daE2CAc1d ++=− = (6.116)
MD2a a)PkPk(dt
daE2dAc1d +=− (6.117)
MD2b a)PkPk(dt
da2C2dAc1d =+=− (6.118)
MD2c a)PkPk(dt
daE2d2C1d +=− = (6.119)
MD3 a)PkPkPk(dt
daE3d2C2dAc1d ++=− = (6.120)
MD1d2_1c 2E1d a)P(k
dt
da=− (6.121)
MD1d3_1c 3E1d a)P(k
dt
da=− (6.122)
MD1d2_1cW WWd
2E1d
PK1
a)P(k
dt
da
+=− (6.123)
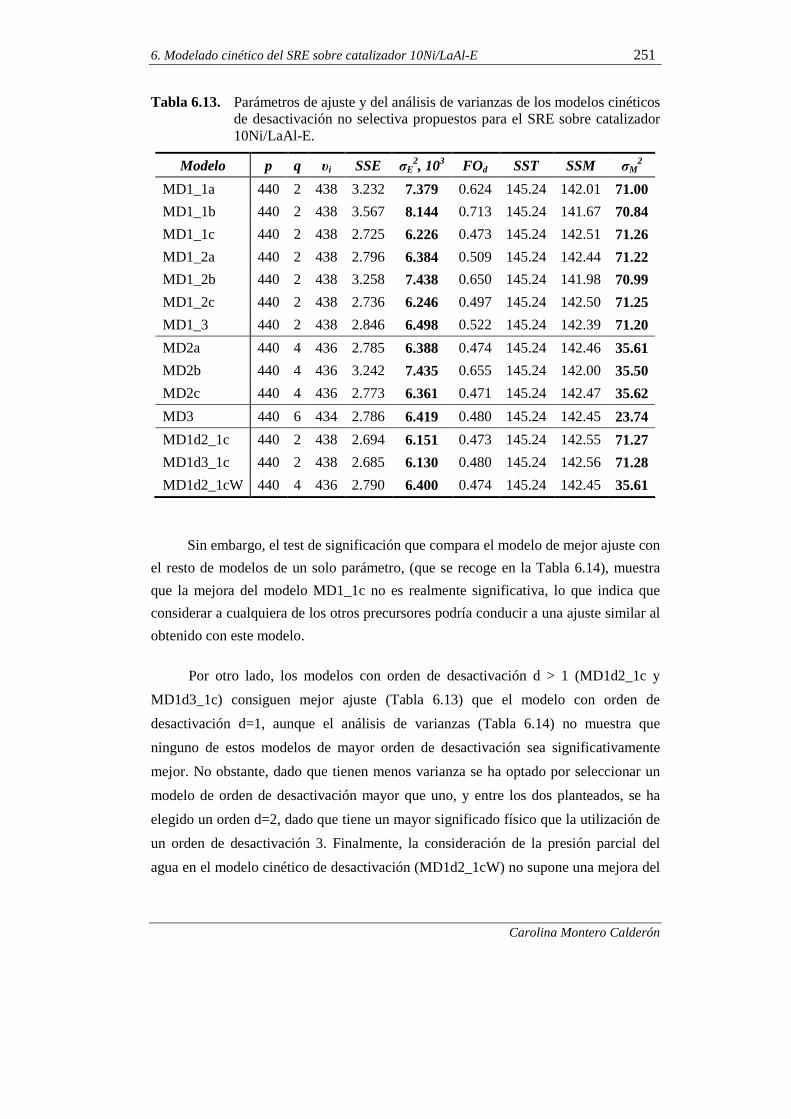
6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 251
Carolina Montero Calderón
Tabla 6.13. Parámetros de ajuste y del análisis de varianzas de los modelos cinéticos de desactivación no selectiva propuestos para el SRE sobre catalizador 10Ni/LaAl-E.
Modelo p q υi SSE σE2, 103 FOd SST SSM σM
2
MD1_1a 440 2 438 3.232 7.379 0.624 145.24 142.01 71.00
MD1_1b 440 2 438 3.567 8.144 0.713 145.24 141.67 70.84
MD1_1c 440 2 438 2.725 6.226 0.473 145.24 142.51 71.26
MD1_2a 440 2 438 2.796 6.384 0.509 145.24 142.44 71.22
MD1_2b 440 2 438 3.258 7.438 0.650 145.24 141.98 70.99
MD1_2c 440 2 438 2.736 6.246 0.497 145.24 142.50 71.25
MD1_3 440 2 438 2.846 6.498 0.522 145.24 142.39 71.20
MD2a 440 4 436 2.785 6.388 0.474 145.24 142.46 35.61
MD2b 440 4 436 3.242 7.435 0.655 145.24 142.00 35.50
MD2c 440 4 436 2.773 6.361 0.471 145.24 142.47 35.62
MD3 440 6 434 2.786 6.419 0.480 145.24 142.45 23.74
MD1d2_1c 440 2 438 2.694 6.151 0.473 145.24 142.55 71.27
MD1d3_1c 440 2 438 2.685 6.130 0.480 145.24 142.56 71.28
MD1d2_1cW 440 4 436 2.790 6.400 0.474 145.24 142.45 35.61
Sin embargo, el test de significación que compara el modelo de mejor ajuste con
el resto de modelos de un solo parámetro, (que se recoge en la Tabla 6.14), muestra
que la mejora del modelo MD1_1c no es realmente significativa, lo que indica que
considerar a cualquiera de los otros precursores podría conducir a una ajuste similar al
obtenido con este modelo.
Por otro lado, los modelos con orden de desactivación d > 1 (MD1d2_1c y
MD1d3_1c) consiguen mejor ajuste (Tabla 6.13) que el modelo con orden de
desactivación d=1, aunque el análisis de varianzas (Tabla 6.14) no muestra que
ninguno de estos modelos de mayor orden de desactivación sea significativamente
mejor. No obstante, dado que tienen menos varianza se ha optado por seleccionar un
modelo de orden de desactivación mayor que uno, y entre los dos planteados, se ha
elegido un orden d=2, dado que tiene un mayor significado físico que la utilización de
un orden de desactivación 3. Finalmente, la consideración de la presión parcial del
agua en el modelo cinético de desactivación (MD1d2_1cW) no supone una mejora del
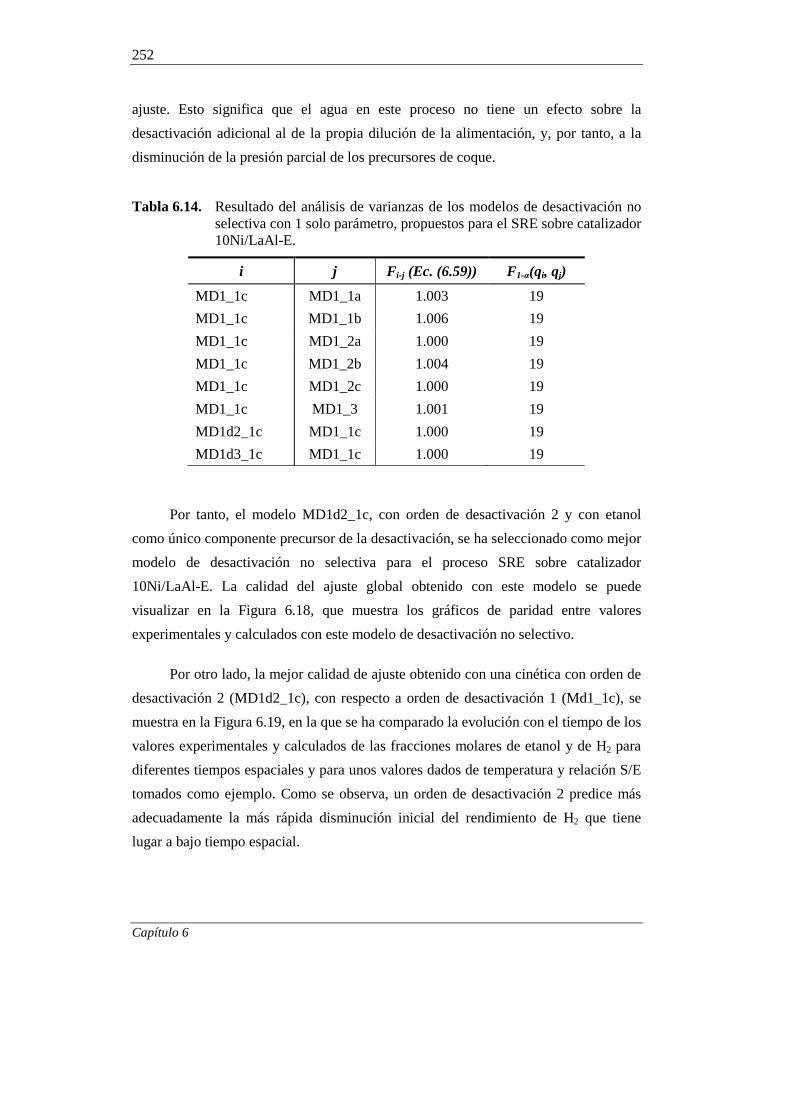
252
Capítulo 6
ajuste. Esto significa que el agua en este proceso no tiene un efecto sobre la
desactivación adicional al de la propia dilución de la alimentación, y, por tanto, a la
disminución de la presión parcial de los precursores de coque.
Tabla 6.14. Resultado del análisis de varianzas de los modelos de desactivación no selectiva con 1 solo parámetro, propuestos para el SRE sobre catalizador 10Ni/LaAl-E.
i j F i-j (Ec. (6.59)) F1-α(qi, qj)
MD1_1c MD1_1a 1.003 19
MD1_1c MD1_1b 1.006 19
MD1_1c MD1_2a 1.000 19
MD1_1c MD1_2b 1.004 19
MD1_1c MD1_2c 1.000 19
MD1_1c MD1_3 1.001 19
MD1d2_1c MD1_1c 1.000 19
MD1d3_1c MD1_1c 1.000 19
Por tanto, el modelo MD1d2_1c, con orden de desactivación 2 y con etanol
como único componente precursor de la desactivación, se ha seleccionado como mejor
modelo de desactivación no selectiva para el proceso SRE sobre catalizador
10Ni/LaAl-E. La calidad del ajuste global obtenido con este modelo se puede
visualizar en la Figura 6.18, que muestra los gráficos de paridad entre valores
experimentales y calculados con este modelo de desactivación no selectivo.
Por otro lado, la mejor calidad de ajuste obtenido con una cinética con orden de
desactivación 2 (MD1d2_1c), con respecto a orden de desactivación 1 (Md1_1c), se
muestra en la Figura 6.19, en la que se ha comparado la evolución con el tiempo de los
valores experimentales y calculados de las fracciones molares de etanol y de H2 para
diferentes tiempos espaciales y para unos valores dados de temperatura y relación S/E
tomados como ejemplo. Como se observa, un orden de desactivación 2 predice más
adecuadamente la más rápida disminución inicial del rendimiento de H2 que tiene
lugar a bajo tiempo espacial.
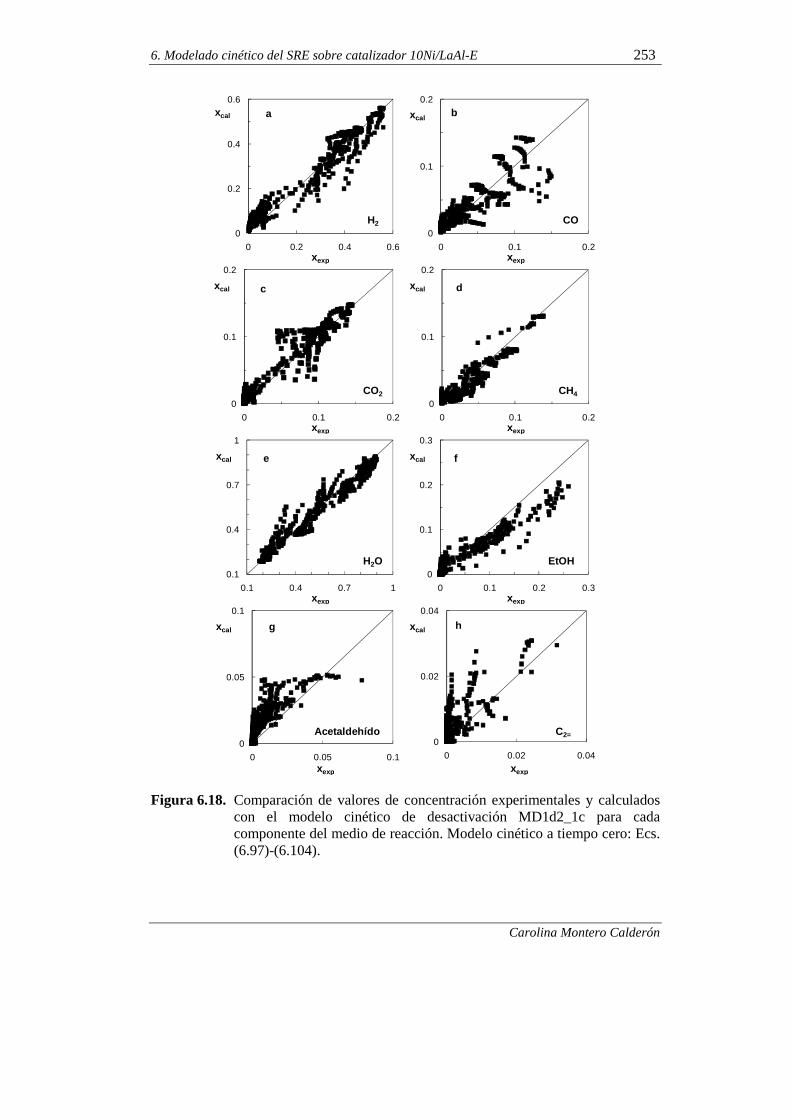
6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 253
Carolina Montero Calderón
0
0.2
0.4
0.6
0 0.2 0.4 0.6
xcal
xexp
H2
a
0
0.1
0.2
0 0.1 0.2
xcal
xexp
CO
b
0
0.1
0.2
0 0.1 0.2
xcal
xexp
CO2
c
0
0.1
0.2
0 0.1 0.2
xcal
xexp
CH4
d
0.1
0.4
0.7
1
0.1 0.4 0.7 1
xcal
xexp
H2O
e
0
0.1
0.2
0.3
0 0.1 0.2 0.3
xcal
xexp
EtOH
f
0
0.05
0.1
0 0.05 0.1
xcal
xexp
Acetaldehído
g
0
0.02
0.04
0 0.02 0.04
xcal
xexp
C2=
h
Figura 6.18. Comparación de valores de concentración experimentales y calculados con el modelo cinético de desactivación MD1d2_1c para cada componente del medio de reacción. Modelo cinético a tiempo cero: Ecs. (6.97)-(6.104).
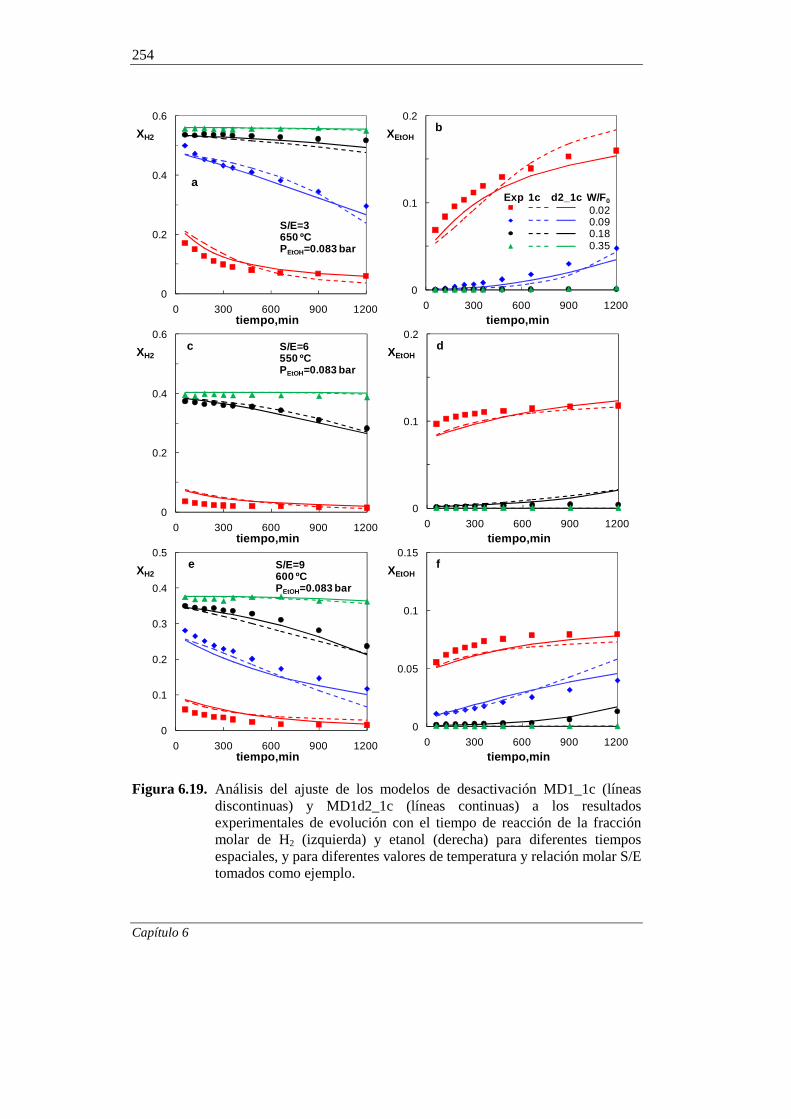
254
Capítulo 6
0
0.2
0.4
0.6
0 300 600 900 1200
XH2
tiempo,min
S/E=3650 ºCPEtOH=0.083 bar
a
0
0.1
0.2
0 300 600 900 1200
XEtOH
tiempo,min
0.020.090.180.35
Exp 1c d2_1c W/F0
b
0
0.2
0.4
0.6
0 300 600 900 1200
XH2
tiempo,min
c S/E=6550 ºCPEtOH=0.083 bar
0
0.1
0.2
0 300 600 900 1200
XEtOH
tiempo,min
d
0
0.1
0.2
0.3
0.4
0.5
0 300 600 900 1200
XH2
tiempo,min
e S/E=9600 ºCPEtOH=0.083 bar
0
0.05
0.1
0.15
0 300 600 900 1200
XEtOH
tiempo,min
f
Figura 6.19. Análisis del ajuste de los modelos de desactivación MD1_1c (líneas discontinuas) y MD1d2_1c (líneas continuas) a los resultados experimentales de evolución con el tiempo de reacción de la fracción molar de H2 (izquierda) y etanol (derecha) para diferentes tiempos espaciales, y para diferentes valores de temperatura y relación molar S/E tomados como ejemplo.

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 255
Carolina Montero Calderón
6.2.3. Modelo cinético de desactivación selectiva
Los resultados de los gráficos de paridad de la Figura 6.18 ponen de manifiesto
que el ajuste no es completamente satisfactorio, especialmente para los componentes
minoritarios del medio de reacción. Para la mejora del grado de ajuste, se han
considerado modelos de desactivación selectiva, que incorporan diferentes actividades
para una o varias de las reacciones del esquema cinético.
cjo
cj
jr
ra = (6.124)
dando lugar a diferentes ecuaciones de desactivación para cada reacción:
djij
ja)C,T(
dt
da⋅ψ=− (6.125)
6.2.3.1. Análisis de la necesidad de un modelo de desactivación selectiva
La incorporación de una actividad más en el modelo cinético conlleva la
dificultad añadida de tener que resolver una ecuación de desactivación adicional, lo
que implica a su vez un aumento considerable del tiempo de cálculo requerido. Por
ello, la consideración de los nuevos modelos se ha realizado de la siguiente forma:
i) Se ha analizado el ajuste que se obtiene definiendo un modelo con 2 actividades:
una misma actividad (a1) para la mayoría de las etapas del esquema cinético y
otra actividad diferente (a2) para una o varias de las etapas restante. Así, se han
definido 5 modelos selectivos, que se muestran en la Tabla 6.15, en función de
que la etapa con diferente actividad sea la deshidrogenación (MD1d2-DHG), la
deshidratación (MD1d2-DHT), la reacción WGS (MD1d2-WGS), las etapas de
descomposición (MD1d2-DSC), o las etapas de reformado con vapor ((MD1d2-
SR).
ii) El cálculo de la actividad a2 se ha realizado de forma simplificada a partir del
valor de la actividad a1, según la expresión:
256
Capítulo 6
n12 aa = (6.126)
donde un valor de n<1 indica una más lenta disminución de la actividad a2
respecto de a1, mientras que para n>1 sucede lo contrario. Esta forma
simplificada de cálculo de un modelo de desactivación selectivo evita tener que
definir una ecuación diferencial adicional, lo que supone un ahorro considerable
de tiempo de cálculo, y ha sido utilizada anteriormente para describir la más lenta
desactivación de la etapa de deshidratación de metanol respecto a las etapas de
formación e interconversión de hidrocarburos en los procesos MTG y MTO
(Gayubo y cols., 2002, 2003).
Tabla 6.15. Modelos de desactivación selectiva propuestos para el proceso SRE sobre catalizador 10Ni/LaAl-E.
Modelo Etapas actividad Ecuación
MD1d2-DHG DHG a2 (6.126) Resto etapas a1 (6.121)
MD1d2-DHT DHT a2 (6.126) Resto etapas a1 (6.121)
MD1d2-WGS WGS a2 (6.126) Resto etapas a1 (6.121)
MD1d2-DSC D-Ac, D-E a2 (6.126) Resto etapas a1 (6.121)
MD1d2-SR SR-E, SR-C2=, SR-M a2 (6.126) Resto etapas a1 (6.121)
El resultado del ajuste obtenido con cada uno de los modelos de desactivación
selectiva se muestra en la Tabla 6.16. Como se observa, todos los modelos mejoran el
ajuste respecto al modelo no selectivo (con un menor valor de la FO y de la suma de
cuadrados residuales). El mejor ajuste (con menor suma de cuadrados residual)
corresponde al modelo que considera diferente desactivación (más lenta, dado que
n<1) para las etapas de reformado, seguido del que considera desactivación más rápida
(n>1) para la etapa de deshidrogenación. Cabe mencionar el valor de n≈0 obtenido
para el modelo MD1d2-DSC, lo que indica que para las etapas de descomposición la
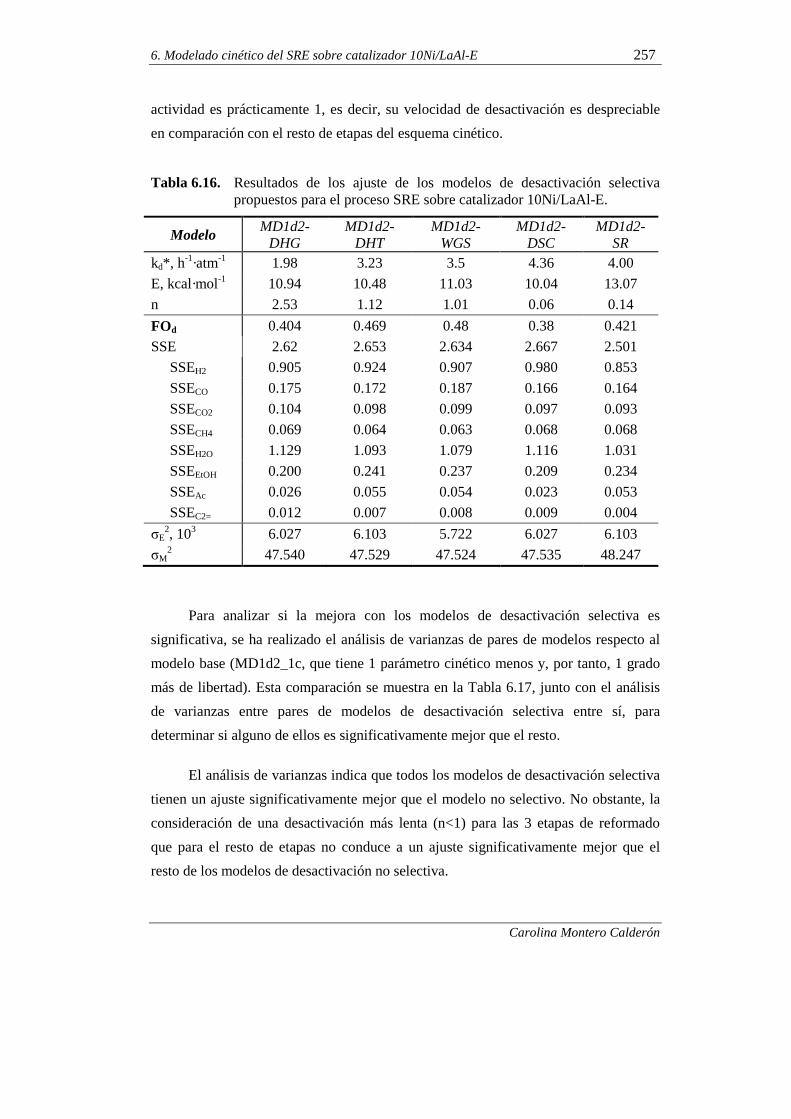
6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 257
Carolina Montero Calderón
actividad es prácticamente 1, es decir, su velocidad de desactivación es despreciable
en comparación con el resto de etapas del esquema cinético.
Tabla 6.16. Resultados de los ajuste de los modelos de desactivación selectiva propuestos para el proceso SRE sobre catalizador 10Ni/LaAl-E.
Modelo MD1d2-
DHG MD1d2-
DHT MD1d2-
WGS MD1d2-
DSC MD1d2-
SR kd*, h
-1·atm-1 1.98 3.23 3.5 4.36 4.00
E, kcal·mol-1 10.94 10.48 11.03 10.04 13.07
n 2.53 1.12 1.01 0.06 0.14
FOd 0.404 0.469 0.48 0.38 0.421
SSE 2.62 2.653 2.634 2.667 2.501
SSEH2 0.905 0.924 0.907 0.980 0.853
SSECO 0.175 0.172 0.187 0.166 0.164
SSECO2 0.104 0.098 0.099 0.097 0.093
SSECH4 0.069 0.064 0.063 0.068 0.068
SSEH2O 1.129 1.093 1.079 1.116 1.031
SSEEtOH 0.200 0.241 0.237 0.209 0.234
SSEAc 0.026 0.055 0.054 0.023 0.053
SSEC2= 0.012 0.007 0.008 0.009 0.004
σE2, 103 6.027 6.103 5.722 6.027 6.103
σM2 47.540 47.529 47.524 47.535 48.247
Para analizar si la mejora con los modelos de desactivación selectiva es
significativa, se ha realizado el análisis de varianzas de pares de modelos respecto al
modelo base (MD1d2_1c, que tiene 1 parámetro cinético menos y, por tanto, 1 grado
más de libertad). Esta comparación se muestra en la Tabla 6.17, junto con el análisis
de varianzas entre pares de modelos de desactivación selectiva entre sí, para
determinar si alguno de ellos es significativamente mejor que el resto.
El análisis de varianzas indica que todos los modelos de desactivación selectiva
tienen un ajuste significativamente mejor que el modelo no selectivo. No obstante, la
consideración de una desactivación más lenta (n<1) para las 3 etapas de reformado
que para el resto de etapas no conduce a un ajuste significativamente mejor que el
resto de los modelos de desactivación no selectiva.

258
Capítulo 6
Tabla 6.17. Resultados del análisis de varianza de pares de modelos de desactivación propuestos para el proceso SRE sobre catalizador 10Ni/LaAl-E.
i j F i-j (Ec. (6.58)) F1-α(υi-υj, υj)
MD1d2_DHG MD1d2_1c 17.565 3.863
MD1d2_DHT MD1d2_1c 11.877 3.863
MD1d2_WGS MD1d2_1c 15.149 3.863
MD1d2_DSC MD1d2_1c 9.487 3.863
MD1d2_SR MD1d2_1c 39.235 3.863
F i-j (Ec. (6.59)) F1-α(qi, qj) MD1d2_SR MD1d2_DHG 1.015 9.277 MD1d2_SR MD1d2_DHT 1.015 9.277 MD1d2_SR MD1d2_WGS 1.015 9.277 MD1d2_SR MD1d2_DSC 1.015 9.277
6.2.3.2. Propuesta de un modelo de desactivación selectiva
A tenor de los resultados mostrados en la Tabla 6.16 y 6.17, se decidió probar el
nivel de ajuste obtenido con un modelo de desactivación selectivo más complejo,
MD1d2-4act, que incorpora 4 actividades diferentes: a1 para las etapas de SR, a2 para
la etapa de deshidrogenación, a3 para la WGS y la deshidratación y a4 para las etapas
de descomposición. La ecuación cinética de desactivación para las actividades a1, a2 y
a3 está dada por la expresión:
→=→=
−→==−
=
y WGS DHT3j
DHG2j
M-SRy C-SR E,SR1j
a)P(kdt
da 22jEdj
j(6.127)
siendo a4=1 (desactivación insignificante para la descomposición de etanol y
acetaldehído).
Los resultados del ajuste de este modelo más complejo de desactivación
selectiva, junto con el análisis de comparación de varianza con respecto al modelo
selectivo más sencillo de mejor ajuste (MD1d2-SR) se muestran en la Tabla 6.18.
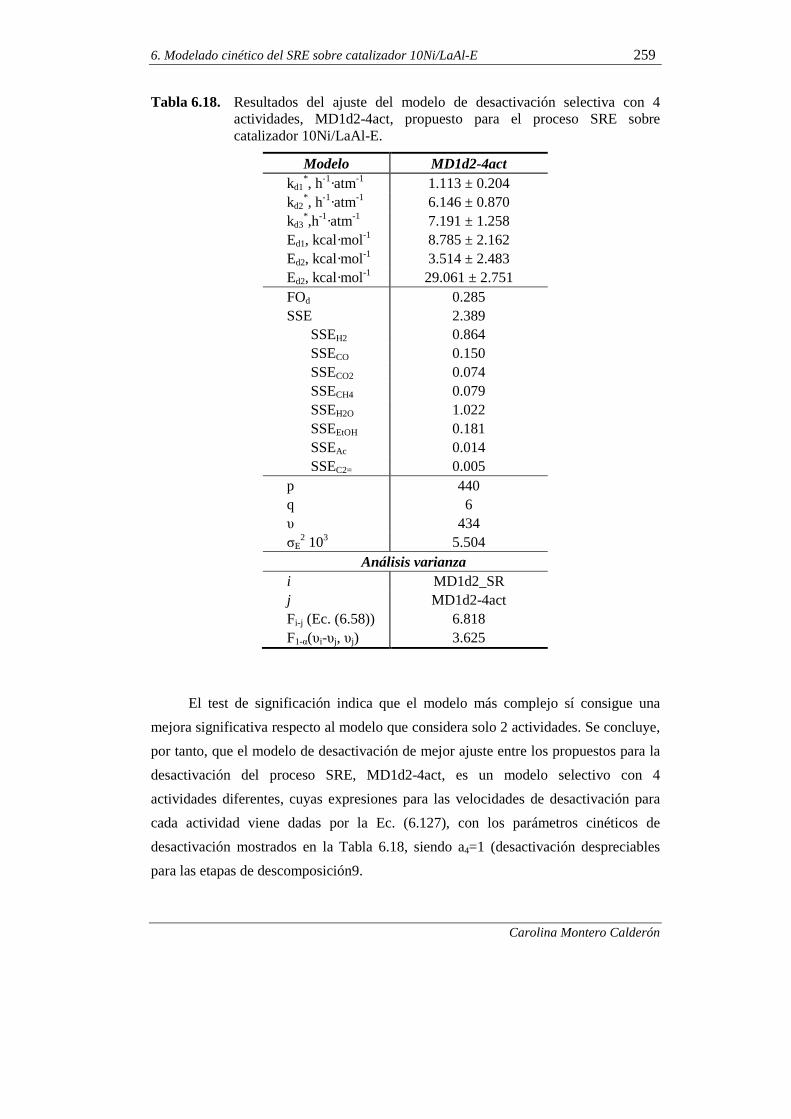
6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 259
Carolina Montero Calderón
Tabla 6.18. Resultados del ajuste del modelo de desactivación selectiva con 4 actividades, MD1d2-4act, propuesto para el proceso SRE sobre catalizador 10Ni/LaAl-E.
Modelo MD1d2-4act kd1
*, h-1·atm-1 1.113 ± 0.204 kd2
*, h-1·atm-1 6.146 ± 0.870 kd3
*,h-1·atm-1 7.191 ± 1.258 Ed1, kcal·mol-1 8.785 ± 2.162 Ed2, kcal·mol-1 3.514 ± 2.483 Ed2, kcal·mol-1 29.061 ± 2.751 FOd 0.285 SSE 2.389
SSEH2 0.864 SSECO 0.150 SSECO2 0.074 SSECH4 0.079 SSEH2O 1.022 SSEEtOH 0.181 SSEAc 0.014 SSEC2= 0.005
p 440 q 6 υ 434 σE
2 103 5.504 Análisis varianza
i MD1d2_SR j MD1d2-4act Fi-j (Ec. (6.58)) 6.818 F1-α(υi-υj, υj) 3.625
El test de significación indica que el modelo más complejo sí consigue una
mejora significativa respecto al modelo que considera solo 2 actividades. Se concluye,
por tanto, que el modelo de desactivación de mejor ajuste entre los propuestos para la
desactivación del proceso SRE, MD1d2-4act, es un modelo selectivo con 4
actividades diferentes, cuyas expresiones para las velocidades de desactivación para
cada actividad viene dadas por la Ec. (6.127), con los parámetros cinéticos de
desactivación mostrados en la Tabla 6.18, siendo a4=1 (desactivación despreciables
para las etapas de descomposición9.

260
Capítulo 6
La calidad del ajuste global obtenido con este modelo a los datos
experimentales en el conjunto de condiciones experimentales estudiado se muestra en
la Figura 6.20, que muestra los gráficos de paridad de valores experimentales y
calculados de la fracción molar de cada componente del medio de reacción. Se incluye
en la figura los datos correspondientes al ajuste obtenido con el modelo no selectivo,
para visualizar el nivel de mejora obtenido.
Se observa en estos gráficos que este modelo predice adecuadamente la
evolución con el tiempo de la conversión de etanol y del rendimiento de H2, así como
de los productos minoritarios acetaldehído y etileno. Sin embargo, el ajuste de los
componentes carbonados CO, CO2 y CH4 requiere aún de mejora, no siendo suficiente
la consideración de la desactivación selectiva para obtener un ajuste aceptable de los
valores calculados a los experimentales para estos componentes. Esta falta de ajuste
puede deberse al hecho de que no se han considerado en el esquema cinético las
reacciones responsable de la formación del elevado contenido de coque de naturaleza
fibrilar observado en la mayoría de las condiciones experimentales estudiadas
(reacción de Boudouard y descomposición de CH4), ni la reacción de gasificación del
coque. Si bien el coque de naturaleza fibrilar no contribuye de manera significativa a
la desactivación del catalizador, la consideración de las reacciones de su formación sí
puede contribuir significativamente a los balances de materia del CO y CH4,
particularmente con el transcurso del tiempo de reacción, dado que el contenido de
coque depositado es muy elevado en gran parte de las condiciones experimentales
utilizadas.
En cualquier caso, la consideración de estas reacciones en el esquema cinético y
el ajuste de sus correspondientes parámetros cinéticos requieren la resolución del
balance de materia para el coque, lo que tiene una gran complejidad adicional para el
programa de ajuste (con un tiempo de cálculo mucho más elevado), al aumentar el
número de ecuaciones diferenciales simultáneas a resolver. Este ajuste ha quedado
fuera del alcance de esta Tesis, y queda pendiente para futuros trabajos de mejora del
modelado cinético.
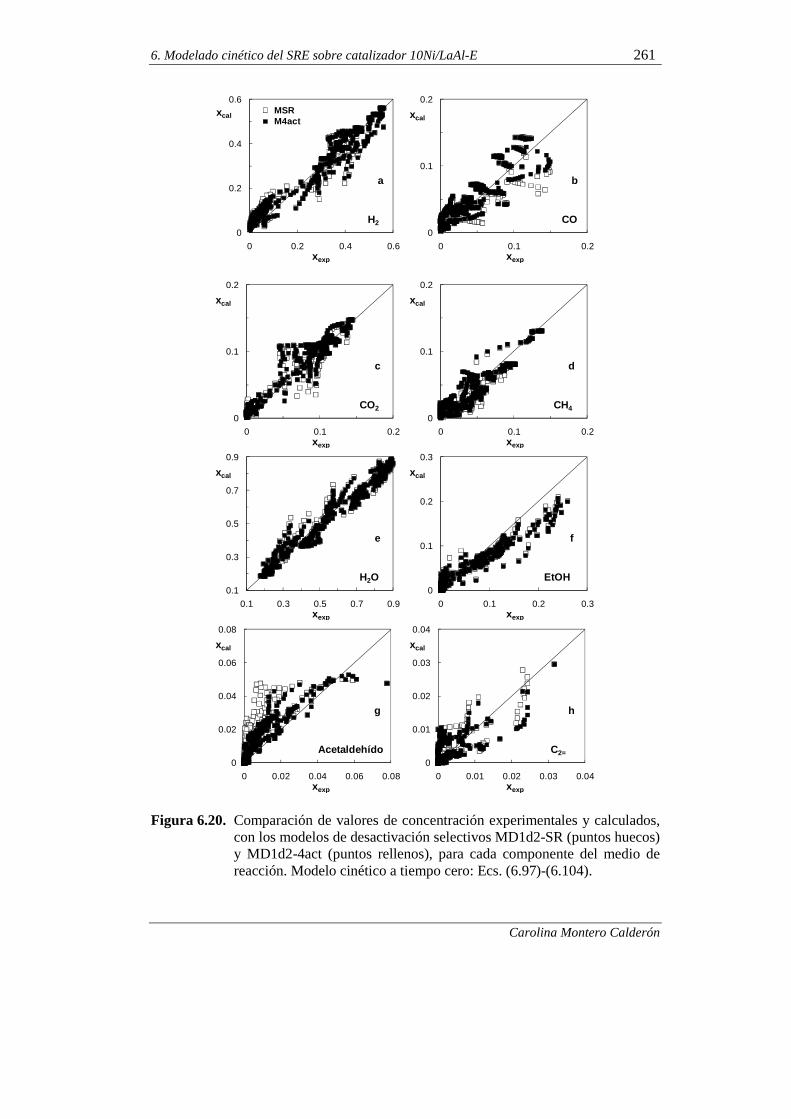
6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 261
Carolina Montero Calderón
0
0.2
0.4
0.6
0 0.2 0.4 0.6
xcal
xexp
H2
a
MSRM4act
0
0.1
0.2
0 0.1 0.2
xcal
xexp
CO
b
0
0.1
0.2
0 0.1 0.2
xcal
xexp
CO2
c
0
0.1
0.2
0 0.1 0.2
xcal
xexp
CH4
d
0.1
0.3
0.5
0.7
0.9
0.1 0.3 0.5 0.7 0.9
xcal
xexp
H2O
e
0
0.1
0.2
0.3
0 0.1 0.2 0.3
xcal
xexp
EtOH
f
0
0.02
0.04
0.06
0.08
0 0.02 0.04 0.06 0.08
xcal
xexp
Acetaldehído
g
0
0.01
0.02
0.03
0.04
0 0.01 0.02 0.03 0.04
xcal
xexp
C2=
h
Figura 6.20. Comparación de valores de concentración experimentales y calculados, con los modelos de desactivación selectivos MD1d2-SR (puntos huecos) y MD1d2-4act (puntos rellenos), para cada componente del medio de reacción. Modelo cinético a tiempo cero: Ecs. (6.97)-(6.104).

262
Capítulo 6
6.3. MODELO CINÉTICO PROPUESTO
Atendiendo a los resultados de los apartados anteriores se propone para el
proceso SRE sobre catalizador 10Ni/LaAl-E un modelo cinético basado en el esquema
cinético de la Figura 6.21, con las siguientes ecuaciones cinéticas para cada reacción
individual, contemplando la desactivación selectiva.
( ) E23WW
EDHTbDHT Pka
PK1
Pkr +
+ρ= (6.128)
( ) 1WW
2WSRC2
bSRC2 aPK1
PP kr C2
+ρ= =
= (6.129)
( )
+
+
ρDHG
HAcE12
WW
DHG
HAcEDHG
bDHG K
PP-Pka
PK1
K
PP-Pk
= r 2
2
(6.130)
( ) Ac3WW
AcDAcbDAc Pk
PK1
Pkr +
+ρ= (6.131)
( )
+
+
ρWGS
COHWCO43
WW
WGS
COHWCOWGS
bWGS K
PP-PP ka
PK1
K
PP-PPk
=r 22
22
(6.132)
( )
+
+
SRM
CO3H2
WM41WW
SRM
CO3H2
WMSRM
bSRM K
PP-PPka
PK1
K
PP-PPk
ρ=r (6.133)
( ) 3WE31
WW
3WESRE
bSRE PP kaPK1
PP k=r +
+ρ (6.134)
( ) E3WW
EDEbDE Pk
PK1
Pkr +
+ρ= (6.135)
donde las actividades se calculan con las siguientes ecuaciones cinéticas para la
desactivación:

6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 263
Carolina Montero Calderón
21E1d
1 a)P(kdt
da=− (6.136)
22E2d
2 a)P(kdt
da=− (6.137)
23E3d
3 a)P(kdt
da=− (6.138)
Los parámetros cinéticos para las ecuaciones de velocidad de reacción se
muestran en la Tabla 6.11 (para la reacción a tiempo cero) y Tabla 6.18 (para la
desactivación).
1- Reformado con vapor de etanol (SR-E) C2H5OH + 3H2O → 6H2 + 2CO2
2- Deshidratación de etanol (DHT) C2H5OH → C2H4 + H2O
3- Reformado de etileno (SR-C2=) C2H4 + 2H2O → 4H2 + 2CO
4- Deshidrogenación de etanol (DHG) C2H5OH ↔ C2H4O + H2
5- Descomposición de acetaldehído (D-Ac) C2H4O → CH4 + CO
6- Reacción Water Gas Shift (WGS) CO + H2O ↔ H2 + CO2
7- Reformado con vapor de CH4 (SR-M) CH4 + H2O ↔ 3H2 + CO
8- Descomposición de etanol (D-E) C2H5OH → H2 + CO + CH4
Figura 6.21. Etapas del esquema cinético para el proceso SRE sobre catalizador 10Ni/LaAl-E.
En las Figuras 6.22-6.25 se muestran los valores experimentales y calculados de
evolución con el tiempo de la composición de todos los compuestos del medio de
reacción, para diferentes conjuntos de condiciones de operación tomados como
ejemplo.
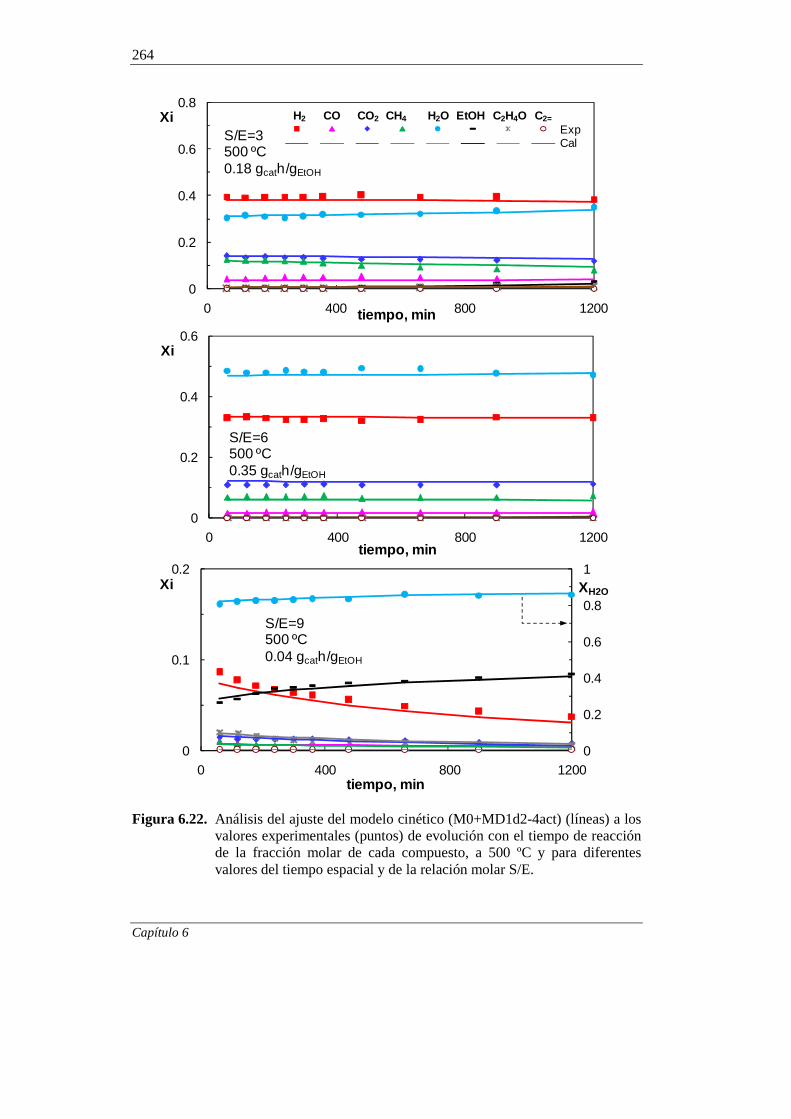
264
Capítulo 6
0
0.2
0.4
0.6
0.8
0 400 800 1200
Xi
tiempo, min
S/E=3500 ºC0.18 gcath/gEtOH
ExpCal
H2 CO CO2 CH4 H2O EtOH C2H4O C2=
0
0.2
0.4
0.6
0 400 800 1200
Xi
tiempo, min
S/E=6500 ºC0.35 gcath/gEtOH
0
0.2
0.4
0.6
0.8
1
0
0.1
0.2
0 400 800 1200
XH2OXi
tiempo, min
S/E=9500 ºC0.04 gcath/gEtOH
Figura 6.22. Análisis del ajuste del modelo cinético (M0+MD1d2-4act) (líneas) a los valores experimentales (puntos) de evolución con el tiempo de reacción de la fracción molar de cada compuesto, a 500 ºC y para diferentes valores del tiempo espacial y de la relación molar S/E.
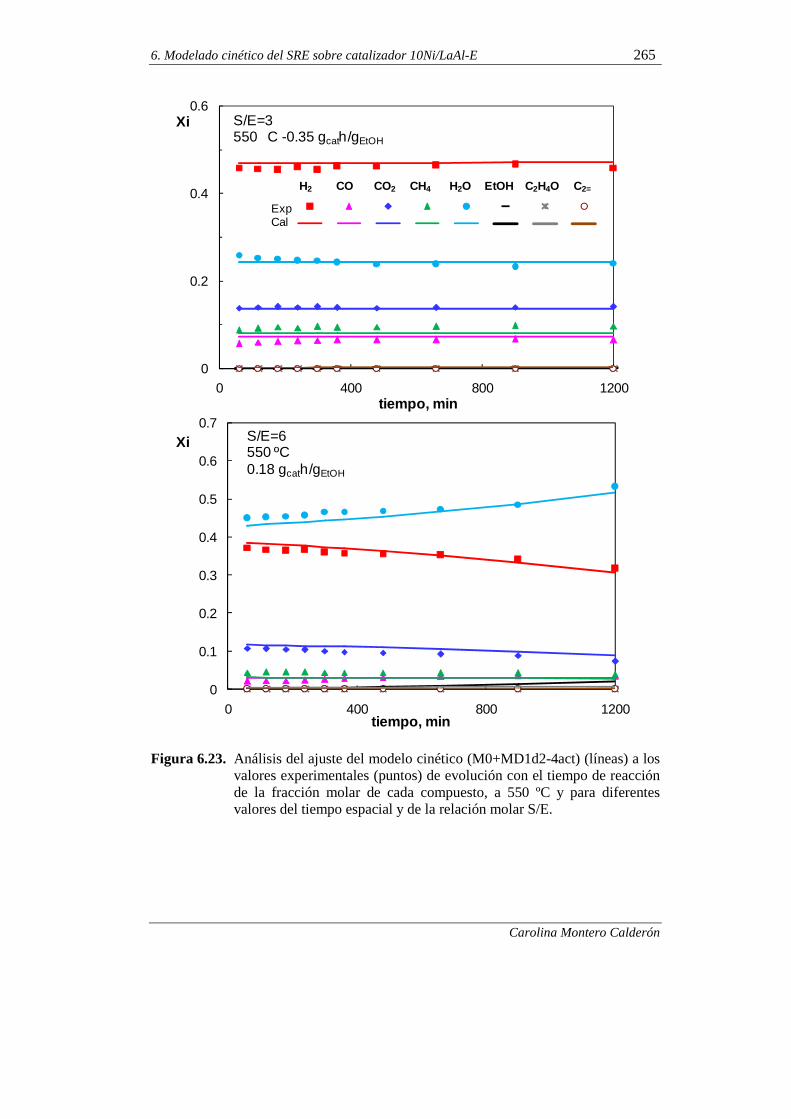
6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 265
Carolina Montero Calderón
0
0.2
0.4
0.6
0 400 800 1200
Xi
tiempo, min
S/E=3550 C -0.35 gcath/gEtOH
H2 CO CO2 CH4 H2O EtOH C2H4O C2=
ExpCal
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0 400 800 1200
Xi
tiempo, min
S/E=6550 ºC0.18 gcath/gEtOH
Figura 6.23. Análisis del ajuste del modelo cinético (M0+MD1d2-4act) (líneas) a los valores experimentales (puntos) de evolución con el tiempo de reacción de la fracción molar de cada compuesto, a 550 ºC y para diferentes valores del tiempo espacial y de la relación molar S/E.
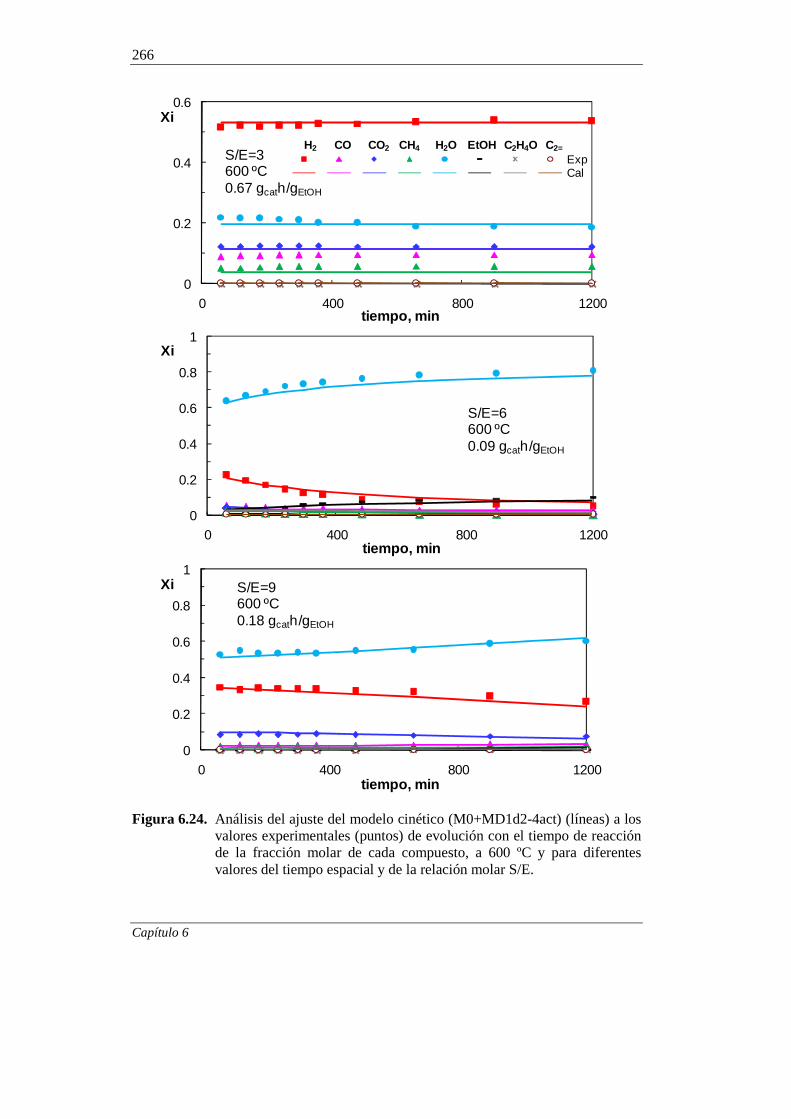
266
Capítulo 6
0
0.2
0.4
0.6
0 400 800 1200
Xi
tiempo, min
S/E=3600 ºC0.67 gcath/gEtOH
H2 CO CO2 CH4 H2O EtOH C2H4O C2=
ExpCal
0
0.2
0.4
0.6
0.8
1
0 400 800 1200
Xi
tiempo, min
S/E=6600 ºC0.09 gcath/gEtOH
0
0.2
0.4
0.6
0.8
1
0 400 800 1200
Xi
tiempo, min
S/E=9600 ºC0.18 gcath/gEtOH
Figura 6.24. Análisis del ajuste del modelo cinético (M0+MD1d2-4act) (líneas) a los valores experimentales (puntos) de evolución con el tiempo de reacción de la fracción molar de cada compuesto, a 600 ºC y para diferentes valores del tiempo espacial y de la relación molar S/E.
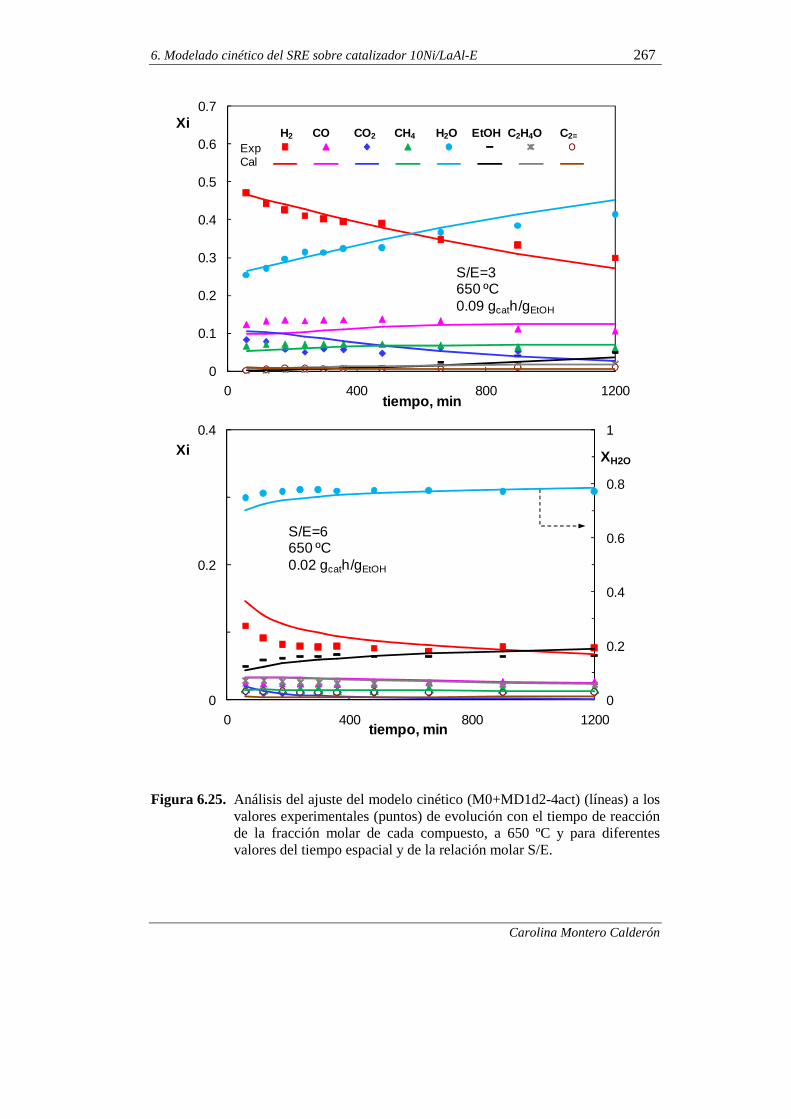
6. Modelado cinético del SRE sobre catalizador 10Ni/LaAl-E 267
Carolina Montero Calderón
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0 400 800 1200
Xi
tiempo, min
S/E=3650 ºC0.09 gcath/gEtOH
ExpCal
H2 CO CO2 CH4 H2O EtOH C2H4O C2=
0
0.2
0.4
0.6
0.8
1
0
0.2
0.4
0 400 800 1200
XH2OXi
tiempo, min
S/E=6650 ºC0.02 gcath/gEtOH
Figura 6.25. Análisis del ajuste del modelo cinético (M0+MD1d2-4act) (líneas) a los valores experimentales (puntos) de evolución con el tiempo de reacción de la fracción molar de cada compuesto, a 650 ºC y para diferentes valores del tiempo espacial y de la relación molar S/E.


7. RESUMEN


271
Carolina Montero Calderón
7. RESUMEN
En esta Tesis se ha estudiado la producción de H2 mediante reformado catalítico
con vapor de etanol (proceso SRE) utilizando un catalizador con 10 % en peso de Ni
soportado en La2O3-αAl2O3, (denominado 10Ni/LaAl), habiéndose abordado 3
aspectos clave para progresar hacia el desarrollo tecnológico del proceso, como son: i)
optimización de la preparación del catalizador, buscando no solo máxima actividad,
selectividad de H2 y estabilidad, sino también un comportamiento reproducible para la
operación en ciclos de reacción-regeneración; ii) análisis detallado del efecto de las
condiciones de operación sobre los índices de reacción, prestando especial atención al
estudio del catalizador con diferentes niveles de desactivación, para delimitar las
causas de la misma y; iii) desarrollo de un modelo cinético del proceso, contemplando
la desactivación del catalizador.
La caracterización de los catalizadores frescos, desactivados y regenerados se
llevado a cabo mediante las siguientes técnicas de análisis:
a. Adsorción-desorción de N2, para determinar la estructura micro-mesoporosa
(superficie específica BET y distribución de volumen de poros).
b. Microscopía electrónica de barrido (SEM), para determinar la morfología de los
catalizadores, y microscopía de electrones retrodispersados (BSE) para observar
las partículas de Ni en los catalizadores desactivados.
c. Difracción de rayos X (XRD), para identificar las fases o compuestos cristalinos
presentes en el catalizador sintetizado y el tamaño medio de cristal de los centros
metálicos (mediante la fórmula de Debye-Scherrer).
d. Espectrometría de emisión atómica por plasma de acoplamiento inductivo (ICP-
AES), para determinar la composición del catalizador.
e. Reducción a temperatura programada (TPR), para determinar el tipo de especies
metálicas reducibles presentes en el catalizador y la temperatura a la que ocurre
dicha reducción.
f. Quimisorción de H2, para determinar la dispersión y la superficie metálica del Ni.
g. Espectroscopía fotoeléctrica de rayos X (XPS), para determinar la composición
atómica de la superficie de los catalizadores.

272
Capítulo 7
h. Oxidación a temperatura programada (TPO), para determinar el contenido de
coque depositado sobre los catalizadores desactivados.
i. Espectroscopía infrarroja (FTIR), para determinar cualitativamente los enlaces
presentes en el coque depositado en el catalizador desactivado y su abundancia
relativa.
j. Espectroscopía Raman, para determinación del grado de cristalinidad y desarrollo
del coque.
El comportamiento cinético del catalizador se ha analizado utilizando un equipo
automatizado de reacción ubicado en los laboratorios de investigación del grupo
ProCatVaRes, que dispone de un reactor de lecho fluidizado conectado en línea
(mediante una línea termostatizada) con un micro-cromatógrafo de gases (Micro GC
3000 de Agilent), para el análisis en continuo de los productos de reacción. Estos
estudios se han llevado a cabo en condiciones de operación (caudales de reactivos y
dilución del lecho catalítico con un sólido inerte (CSi) con adecuadas propiedades
fluidodinámicas) determinadas en trabajos previos, para asegurar una adecuada
fluidodinámica del lecho.
Se ha optimizado la temperatura de calcinación del catalizador para obtener la
mayor cantidad de especies de Ni reducibles, y, por tanto, mayor actividad, para lo
cual se han ensayado tres temperaturas de calcinación 550, 650 y 850º. Se ha
analizado el efecto de un proceso de equilibrado o estabilización del catalizador
(mediante un ciclo previo de reacción (reformado a alta temperatura, de hasta 700 ºC)-
regeneración (por combustión con aire a 550 ºC)) sobre la reproducibilidad de su
comportamiento cinético en ciclos de reacción-regeneración.
Se han llevado a cabo ensayos dinámicos “en blanco”, sin catalizador,
(utilizando secuencias de escalones crecientes de temperatura, de corta duración, entre
500-700 ºC), para determinar la influencia de las rutas térmicas en el proceso SRE.
Con el catalizador estabilizado se ha realizado un estudio detallado del efecto de
las condiciones de reformado (temperatura, tiempo espacial, relación molar
vapor/etanol y tiempo de reacción) sobre los índices de reacción (conversión,
rendimiento y selectividad de H2), con objeto de delimitar las condiciones óptimas

7. Resumen 273
Carolina Montero Calderón
para maximizar la producción de H2 con el mejor compromiso de actividad-
selectividad-desactivación, así como para obtener datos cinéticos suficientes para
abordar con rigor el modelado cinético del proceso SRE sobre dicho catalizador. El
intervalo de temperatura estudiado ha sido entre 500-650 ºC (para que la posible
contribución térmica no enmascare la contribución del catalizador a los índices de
reacción); el tiempo espacial hasta 0.69 gcath/gEtOH, que asegura alcanzar el equilibrio
termodinámico; la relación molar vapor/etanol entre 3 (relación estequiométrica del
reformado) y 9 (gran dilución con agua); y tiempos de reacción de hasta 200 h, para
observar la posible sinterización del catalizador en las condiciones más adversas
(elevada temperatura y contenido de agua).
Se ha realizado un estudio detallado de caracterización (conjuntando técnicas
analíticas muy diversas) de los catalizadores desactivados en diferentes condiciones de
operación, para: i) determinar la influencia de estas condiciones sobre la deposición de
coque; ii) correlacionar el contenido y naturaleza del coque con el nivel de
desactivación observado en el catalizador y delimitar los posibles precursores según
las condiciones de operación y iii) establecer el mecanismo de formación y evolución
del coque con el tiempo.
El modelado cinético del proceso se ha realizado en dos etapas. En la primera se
ha determinado la cinética a tiempo cero, para a continuación completar el modelo con
la cinética de desactivación, considerando así el efecto del tiempo sobre la distribución
de productos. Para el modelado cinético se ha desarrollado un programa de cálculo en
Matlab que determina los parámetros cinéticos de mejor ajuste de cada modelo
propuesto, mediante regresión no lineal múltivariable, optimizando una función
objetivo error definida como la suma de cuadrados de los errores entre los valores de
composición experimental y calculada con el modelo cinético correspondiente,
considerando los factores de peso de cada compuesto. Para la propuesta de modelos
cinéticos para el proceso a tiempo cero (catalizador fresco), se ha partido de un
esquema cinético que incluye hasta 10 posibles reacciones individuales y para cada
reacción se han contemplado expresiones de velocidad en función de los componentes
presentes en el medio de reacción, bien considerando a las reacciones individuales
como reacciones elementales, o bien utilizando expresiones de velocidad de tipo
mecanístico, que incluyen un término de atenuación de la velocidad por adsorción de

274
Capítulo 7
alguno/os componente/s del medio de reacción. Para el modelado cinético del proceso,
se ha tenido también en cuenta la contribución a la velocidad de reacción del efecto
térmico, utilizando expresiones de velocidad para cada reacción del esquema cinético
que incluyen 2 términos (catalítico y térmico).
Se ha llevado a cabo una optimación de los parámetros cinéticos de cada
modelo propuesto. La discriminación entre modelos se ha realizado mediante análisis
de varianzas entre pares de modelos, viendo si uno de ellos es significativamente
mejor que el resto, obteniendo así el modelo final de mejor ajuste entre los valores
experimentales y calculados de composición, con la menor complejidad.
A partir del conocimiento del origen de la desactivación (deposición de coque)
y del efecto sobre la misma de las condiciones de reacción y concentración de
precursores del coque, se ha abordado el modelado cinético para la desactivación por
coque, realizando en primer lugar una discriminación de modelos cinéticos de
desactivación no selectiva (con una misma actividad para todas las etapas del esquema
cinético), para determinar el/los precursor/es del coque responsables de la pérdida de
actividad observada en el catalizador, así como para determinar el orden de
desactivación. Posteriormente, se ha analizado la posible mejora que supone
considerar un modelo de desactivación selectiva, que contempla una velocidad de
desactivación diferente para una o varias de las reacciones del esquema cinético,
utilizando para cada una de las actividades consideradas el mismo tipo de expresiones
de velocidad de desactivación (precursores y orden de desactivación) determinado
previamente con el modelo no selectivo. El procedimiento de cálculo de los
parámetros de cada modelo y la discriminación de modelos cinéticos se ha llevado a
cabo de forma análoga al modelado de la cinética a tiempo cero, utilizando un
programa de cálculo propio escrito en Matlab.
Como resultado de la Tesis Doctoral se dispone de las herramientas
(catalizador, conocimiento del efecto de las condiciones de operación y modelo
cinético) que permiten abordar estudios de escalado y optimización del proceso de
reformado con vapor de etanol.

8. CONCLUSIONES


277
Carolina Montero Calderón
8. CONCLUSIONES
1. Sobre la preparación del catalizador:
1.1. La temperatura de calcinación tiene un importante efecto en la selectividad y
estabilidad del catalizador 10Ni/LaAl utilizado en el proceso SRE.
� Al aumentar la temperatura de calcinación en el intervalo 550-850 ºC, como
consecuencia del cambio en las propiedades del catalizador: i) disminuyen la
conversión de etanol y el rendimiento de H2, dado que cobran importancia
las reacciones secundarias de deshidratación y deshidrogenación del etanol y
están menos favorecidas las reacciones de reformado (de etanol, de etano y
de metano) y descomposición del acetaldehído; ii) aumenta el contenido de
coque depositado en el catalizador y, en consecuencia, es mayor la velocidad
de desactivación.
� Las diferencias en el comportamiento cinético del catalizador se deben a la
diferente naturaleza de las especies metálicas de Ni resultantes de la
calcinación. Una alta temperatura de calcinación favorece la formación de
NiAl 2O4, que tiene más fuerte interacción con el soporte que los óxidos
NiOx, por lo que disminuye la reducibilidad y se favorece la formación de
cristalitas de Ni0 mayores tras la reducción, que son menos activas para el
reformado y promueven la formación y crecimiento de depósitos
carbonosos.
� 550 ºC es la temperatura más adecuada de calcinación, dado que produce
mayor cantidad de NiOx dispersos, que se reducen para formar
nanopartículas de Ni0 altamente dispersas, dando lugar a un catalizador más
activo y selectivo (con el mayor rendimiento de H2) y más estable (con la
menor deposición de coque).
1.2. La operación en ciclos de reacción-regeneración exige el equilibrado del
catalizador:
� El catalizador equilibrado mediante un ciclo previo de reacción-regeneración
(reacción con escalones decrecientes de temperatura a 700, 600 y 500 ºC, de

278
Capítulo 8
2 h de duración cada uno, y regeneración por combustión con aire a 550 ºC
durante 2 h), denominado 10Ni/LaAl-E, tiene un comportamiento
reproducible en ciclos sucesivos de reacción-regeneración, si bien su
actividad es notablemente menor que la del catalizador sin equilibrar
(10Ni/LaAl-F). Este último, por contra, sufre una pérdida irreversible de
actividad en los sucesivos ciclos de reacción, que se va atenuando, tendiendo
a un comportamiento estable.
� El diferente comportamiento del catalizador equilibrado se debe a la menor
dispersión y superficie metálica (evidenciado por quimisorción de H2) y
mayor tamaño de cristal de Ni0 (~ 1.7 veces, calculado mediante XRD), que
el catalizador sin equilibrar, lo que indica que el tratamiento de equilibrado
induce un cierto nivel de sinterización del Ni, responsable de la menor
actividad del catalizador respecto al no equilibrado, a pesar de la mayor
reducibilidad de las especies de Ni restantes en el catalizador equilibrado
(evidenciada mediante TPR).
� La falta de reproducibilidad en la operación cíclica del catalizador sin
equilibrar, debe atribuirse a que en las sucesivas etapas de reacción-
regeneración está teniendo lugar una progresiva sinterización del Ni, que se
va acumulando en los sucesivos ciclos hasta alcanzar un comportamiento
estable.
2. Sobre el efecto de las variables de operación
2.1. El efecto de las variables de operación tiene una difícil interpretación, debido a
la complejidad del esquema de reacción, con numerosas reacciones individuales
(ver Conclusión 4.1) y por la contribución de rutas térmicas (no catalíticas) a
estas reacciones:
� La contribución de las rutas térmicas a los valores de conversión y
rendimientos de productos en el proceso SRE es prácticamente despreciable
por debajo de 600 ºC, pero debe ser tenida en cuenta especialmente a partir
de 650 ºC, alcanzándose una fracción molar (en base seca) de 36 % de H2 a
700 ºC y relación molar S/E de 9.

8. Conclusiones 279
Carolina Montero Calderón
� El aumento de temperatura promueve, en primer lugar, la reacción de
deshidrogenación de etanol para formar acetaldehído (cuya presencia ya es
significativa a 500 ºC) (etapa 4 de la conclusión 4.1), y a continuación la
deshidratación para generar etileno (cuya formación es apreciable a partir de
550 ºC) (etapa 2), mientras que las reacciones de descomposición (de etanol
y de acetaldehído) (etapas 8 y 6, respectivamente) son importantes solo a
partir de 650 ºC y su velocidad aumenta exponencialmente al aumentar la
temperatura.
� La reacción WGS (etapa 6), el reformado con vapor de metano (etapa 7) y el
reformado directo con vapor del etanol (etapa 1) no están favorecidas por el
aumento de temperatura en el intervalo 500-700 ºC, como se deduce de la
similar concentración de CO y CH4 obtenida a altas temperaturas, y la casi
insignificante formación de CO2.
2.2. La temperatura tiene gran influencia en el comportamiento cinético del
catalizador 10Ni/LaAl-E, de modo que aumentan notablemente la conversión y
el rendimiento de H2 al aumentar la temperatura, debido al aumento de la
velocidad de reacción y porque el proceso es favorecido al ser muy
endotérmico:
� A 500 ºC la reacción más rápida del esquema cinético es la deshidrogenación
a acetaldehído (etapa 4 de la conclusión 4.1), que es mucho más rápida que
la de reformado de etanol (etapa 1), y esta a su vez es más rápida que las
reacciones de descomposición de etanol (etapa 8) y de acetaldehído (etapa
5), siendo lentas las reacciones WGS (etapa 6), reformado de metano (etapa
7) y especialmente la deshidratación a etileno (etapa 2).
� El aumento de temperatura promueve principalmente la reacción de
deshidratación del etanol, y las reacciones de descomposición de etanol y de
acetaldehído.
� El reformado con vapor de metano tiene una importante contribución al
esquema global de reacción, y el notable aumento de su conversión de
equilibrio al aumentar la temperatura (por ser una reacción muy

280
Capítulo 8
endotérmica) contribuye al notable aumento observado en el rendimiento de
H2.
� El rendimiento de CO2 es elevado y casi constante (~0.46) en el intervalo
500-600 ºC, y disminuye ligeramente por encima de 600 ºC, debido al
desplazamiento del equilibrio termodinámico de la reacción WGS, lo que
atenúa el aumento del rendimiento de H2 por encima de 600 ºC.
2.3. El tiempo espacial tiene un notable efecto en la conversión y distribución de
productos.
� La evolución de la conversión y rendimiento de productos con el tiempo
espacial permite identificar 2 zonas diferenciadas: por debajo de 0.09
gcat/hgEtOH hay una gran influencia del tiempo espacial, con muy rápido
aumento de la conversión y de los rendimientos de H2, CO, CO2 y CH4, y
disminución de los rendimientos de etileno y acetaldehído (productos
mayoritarios a bajo tiempo espacial), mientras que para tiempo espacial
mayor de 0.09 gcat/hgEtOH, la conversión de etanol es prácticamente completa
y hay pocas variaciones de rendimiento de productos.
� El efecto del tiempo espacial es menos importante a 650 ºC, por la apreciable
contribución de las rutas térmicas a las reacciones del esquema cinético.
� A tiempo espacial de 0.09 gcat/hgEtOH se alcanza el máximo rendimiento de
H2 y CO, mientras que los rendimientos de CO2 y de CH4 continúan
aumentando, si bien más atenuadamente, sobre todo a alta temperatura.
� A T ≥ 600 ºC se alcanza el equilibrio termodinámico para tiempo espacial de
0.09 gcat/hgEtOH, mientras que a T≤ 550 ºC se requiere un tiempo espacial de
0.35 gcat/hgEtOH para alcanzar las condiciones de equilibrio termodinámico.
2.4. La relación molar vapor/etanol en la alimentación (S/E) tiene un papel
importante en la conversión y en la selectividad de productos de reacción.
� El aumento de la relación S/E favorece la selectividad de H2, al desplazar
gran parte de las reacciones del esquema cinético que lo generan, como el
reformado de etanol y de metano, así como la reacción WGS. La
selectividad de CO2 también aumenta con la relación S/E, aunque en menor
medida que la selectividad H2, por el desplazamiento de la reacción WGS.

8. Conclusiones 281
Carolina Montero Calderón
La selectividad de CO, CH4, así como de etileno y acetaldehído, disminuye
al aumentar la relación S/E.
� La conversión de etanol disminuye al aumentar la relación S/E, siendo las
diferencias más apreciables cuando más lejos se opera del equilibrio (bajo
tiempo espacial y baja temperatura), lo que indica que el agua contribuye a
atenuar la velocidad de las reacciones del esquema cinético de conversión de
etanol. En consecuencia, al operar lejos del equilibrio termodinámico el
aumento del rendimiento de H2 con la relación S/E es poco apreciable,
porque en estas condiciones se solapan el efecto atenuante del agua sobre la
conversión de etanol con el efecto beneficioso sobre la selectividad de H2.
3. Sobre la desactivación del catalizador
3.1. El catalizador sufre un deterioro de sus propiedades físicas y metálicas con el
transcurso de la reacción, con una magnitud que depende de las condiciones de
reacción.
� La deposición de coque es la única causa de la desactivación para el
catalizador 10Ni/LaAl-E en las condiciones de operación utilizadas, puesto
que el tamaño del cristal de Ni0 medido tras 200 h de reacción en las
condiciones más severas (650 ºC y relación S/E=9) evidencia ausencia de
sinterización.
� Se han identificado dos tipos de coque, formados a partir de diferentes
precursores y que tienen muy diferente incidencia en la desactivación:
i) Coque encapsulante, que quema a baja temperatura (máximo en el perfil
TPO entre 350-430 ºC, para temperatura de reacción entre 550-650 ºC,
respectivamente), cuyos precursores son acetaldehído, etileno y etanol, y
que es el principal responsable de la desactivación. El desplazamiento del
pico de combustión a mayor temperatura al aumentar la temperatura de
reformado puede deberse a que se trata de un coque más evolucionado, o
bien que el mayor contenido de CO presente en el medio de reacción a
alta temperatura contribuya a la formación de cierta cantidad de coque de
naturaleza fibrilar, que quema a mayor temperatura que el encapsulante.

282
Capítulo 8
ii) Coque fibrilar, que quema a alta temperatura, dado que su combustión no
está catalizada por los centros metálicos (máximo de combustión entre
550-650 ºC, para temperatura de reacción entre 550-650 ºC,
respectivamente), cuyos precursores son CO y CH4 y que tiene escasa
incidencia en la desactivación, dado que contenidos muy elevados de este
tipo de coque no originan una gran pérdida de actividad. El
desplazamiento de su pico de combustión hacia mayor temperatura al
aumentar la temperatura de reformado es coherente con la mayor
cristalinidad determinada mediante espectroscopía Raman,
� El coque fibrilar obtenido por reacción de Boudouard a partir de CO
(favorecido a alta temperatura de reformado) es de más difícil combustión
que el formado por descomposición del metano (favorecido a baja
temperatura de reformado). Además, el CO tiene mayor contribución a la
formación del coque fibrilar, dado que el máximo contenido de coque se
obtiene en general para el tiempo espacial intermedio (alrededor de 0.09
gcath/gEtOH), para el cual es máximo el rendimiento de CO.
3.2. El efecto de las variables de operación sobre la evolución con el tiempo de la
conversión y rendimientos de productos, puede explicarse en base al contenido
y, sobre todo, a la naturaleza del coque depositado:
� La rápida desactivación observada para bajo tiempo espacial es consecuencia
de la deposición de coque encapsulante, formado principalmente a partir de
acetaldehído y etanol, y también de etileno a alta temperatura, que son los
componentes mayoritarios en estas condiciones. La deposición de este coque
de naturaleza encapsulante aumenta al aumentar la temperatura, lo que se
traduce en una más rápida desactivación.
� A elevado tiempo espacial, cuando la conversión es elevada y hay ausencia
casi completa de los intermedios acetaldehído y etileno, el coque
depositado es de naturaleza fibrilar, por lo que la desactivación observada
es baja y, además, disminuye al aumentar la temperatura, dado que se
atenúa notablemente la deposición de este tipo de coque al favorecerse su
gasificación.

8. Conclusiones 283
Carolina Montero Calderón
� El aumento de la relación molar S/E tiene como consecuencia una gran
atenuación de la desactivación, especialmente en el intervalo S/E=3-6, lo que
se explica porque un alto contenido de agua disminuye apreciablemente la
concentración de los precursores de coque y, además, promueve la
gasificación del coque, siendo este efecto mucho más importante a elevada
temperatura.
3.3. La interpretación de la disminución de los índices de reacción con el tiempo
exige considerar el mecanismo de formación de coque y la evolución de su
estructura con el tiempo, la cual guarda relación con los cambios de los centros
activos de Ni.
� En condiciones de formación de coque fibrilar se observan 3 etapas
diferenciadas con el tiempo de reacción (1- conversión y rendimientos casi
constantes; 2- rápida desactivación y 3- estado pseudo-estacionario, con muy
lenta desactivación hasta la total desactivación del catalizador), que se
pueden correlacionar con los cambios morfológicos del Ni y del coque. Los
tiempos de transición entre etapas son diferentes, dependiendo de las
condiciones de operación (temperatura, tiempo espacial, relación S/E).
� Para muy bajo tiempo de reacción hay formación incipiente de coque
fibrilar, que tiene elevada concentración de oxigenados y conlleva aumento
del área superficial de micro y mesoporos. En esta primera etapa de reacción
se alcanza el máximo del rendimiento de coque fibrilar, con una elevada
aromaticidad y condensación. Especies de carburo de Ni, formadas a partir
de los precursores CO y CH4, son intermedios en la formación de las fibras
de carbono: el carbono disuelto (proveniente de estos carburos de Ni o de
especies poco reactivas de carbono) difunde a través del Ni para nuclear y
precipitar en la base del cristalito de Ni, y este proceso en continuo conduce
a la formación de un filamento de carbono, que separa las cristalitas de Ni de
la superficie del catalizador, las cuales se ubican en el extremo de las fibras
de carbono, lo que origina la reducción del tamaño de las partículas de Ni0.
� En la segunda etapa de la reacción, con rápida desactivación, continúa
disminuyendo el tamaño de partícula de Ni0 y tiene lugar un importante

284
Capítulo 8
cambio en la formación y naturaleza del coque al cambiar la composición
del medio de reacción, de modo que disminuye el rendimiento de coque
fibrilar, éste se hace parcialmente grafítico y aparece el coque pirolítico, que
es el agente principal de la desactivación en estas condiciones.
� En la tercera etapa (correspondiente a elevado tiempo de reacción)
predomina la formación de coque pirolítico a partir de etanol, con saturación
en la formación de coque (cuyo rendimiento continúa disminuyendo),
bloqueo de centros de Ni0 y alta grafitización del coque remanente.
� Con la regeneración del catalizador por combustión de coque con aire se
produce una re-aglomeración de las partículas de Ni, que son readsorbidas
en el soporte, recuperando las propiedades iniciales.
3.4. El tiempo de reacción tiene un efecto complejo sobre la distribución de
productos, como corresponde a la desactivación de las reacciones individuales
de un esquema de reacción complejo.
� Los rendimientos de H2 y CO2 evolucionan con el tiempo de forma acorde a
la conversión, pero no exactamente en paralelo a ella, mientras que la
evolución de los rendimientos de CO, CH4 y acetaldehído es muy diferente,
y varía con la temperatura, dependiendo de que prevalezca su carácter de
producto intermedio (con un máximo con el tiempo de reacción, caso del
rendimiento de CO a 500 ºC) o final (con continua disminución con el
tiempo, caso del rendimiento de CH4 a 500 ºC) en el esquema cinético de
reacción.
� La diferente evolución con el tiempo de los rendimientos de productos de
reacción evidencia diferente velocidad de desactivación para las etapas del
esquema cinético del proceso. Así, el aumento con el tiempo de los
rendimientos de CO y CH4 observado a 650 ºC indica que las reacciones de
su formación (descomposición de etanol y acetaldehído) se desactivan más
lentamente que las reacciones de su desaparición (WGS y reformado de
metano).

8. Conclusiones 285
Carolina Montero Calderón
3.5. Si bien la combustión del coque permite recuperar el comportamiento cinético
del catalizador, su gasificación no es una estrategia eficaz para la regeneración
del catalizador 10Ni/LaAl en el proceso SRE. Así, una gasificación a 650 ºC no
permite la recuperación total de la actividad del catalizador, dado que
permanece un pequeño contenido de coque residual que requiere mayor
temperatura de gasificación, a la cual se originaría probablemente sinterización
del Ni.
4. Sobre el modelado cinético del proceso
4.1. En el intervalo 550-650 ºC, con relación S/E entre 3 y 9 y tiempos espaciales
hasta 0.69 gcath/gEtOH, la distribución de productos de reacción a tiempo cero se
puede describir con un esquema de reacción con las 8 etapas siguientes:
1- Reformado con vapor de etanol (SR-E) C2H5OH + 3H2O → 6H2 + 2CO2
2- Deshidratación de etanol (DHT) C2H5OH → C2H4 + H2O
3- Reformado con vapor de etileno (SR-C2=) C2H4 + 2H2O → 4H2 + 2CO
4- Deshidrogenación de etanol (DHG) C2H5OH ↔ C2H4O + H2
5- Descomposición de acetaldehído (D-Ac) C2H4O → CH4 + CO
6- Reacción Water Gas Shift (WGS) CO + H2O ↔ H2 + CO2
7- Reformado con vapor de CH4 (SR-M) CH4 + H2O ↔ 3H2 + CO
8- Descomposición de etanol (D-E) C2H5OH → H2 + CO + CH4
Cuyas velocidades de reacción vienen dadas por las ecuaciones siguientes:
( )WW
3WESRE
0SRE PK1
PP k=r
+(6.78)
( )WW
EDHT0DHT PK1
Pk=r
+(6.72)
( )WW
2WSRC2
0SRC2 PK1
PP kr C2
+= = (6.73)
( )WW
DHGHAcEDHG0DHG PK1
)/KPP-(Pk= r 2
+(6.74)
( )WW
AcDAc0DAc PK1
Pkr
+= (6.75)

286
Capítulo 8
( )WW
WGSCOHWCOWGS0WGS PK1
)K/ PP-P(P k=r 22
+(6.76)
( )WW
SRMCO3H2WMSRM
0SRM PK1
)K/PP-P(Pk=r
+(6.77)
( )WW
EDE0DE PK1
Pkr
+= (6.79)
o bien por las ecuaciones de velocidad de reacción siguientes, que contemplan la
contribución a la velocidad de reacción tanto del catalizador como de las rutas
térmicas, que son importantes por encima de 600 ºC.
( ) 3WE3
WW
3WESRE
b0SRE PP kPK1
PP k=r +
+ρ (6.103)
( ) E2WW
EDHTb0DHT Pk
PK1
Pk=r +
+ρ (6.97)
( )WW
2WSRC2
b0SRC2 PK1
PP kr C2
+ρ= = (6.98)
( )
+
+
ρDHG
HAcE1
WW
DHG
HAcEDHG
b0DHG K
PP-Pk
PK1
K
PP-Pk
= r 2
2
(6.99)
( ) Ac3WW
AcDAcb0DAc Pk
PK1
Pkr +
+ρ= (6.100)
( )
+
+
ρWGS
COHWCO4
WW
WGS
COHWCOWGS
b0WGS K
PP-PP k
PK1
K
PP-PP k
=r 22
22
(6.101)
( )
+
+
ρSRM
CO3H2
WM4WW
SRM
CO3H2
WMSRM
b0SRM K
PP-PPk
PK1
K
PP-PPk
=r (6.102)
( ) E3WW
EDEb0DE Pk
PK1
Pkr +
+ρ= (6.104)
Las constantes cinéticas de estas ecuaciones se muestran en la siguiente tabla:
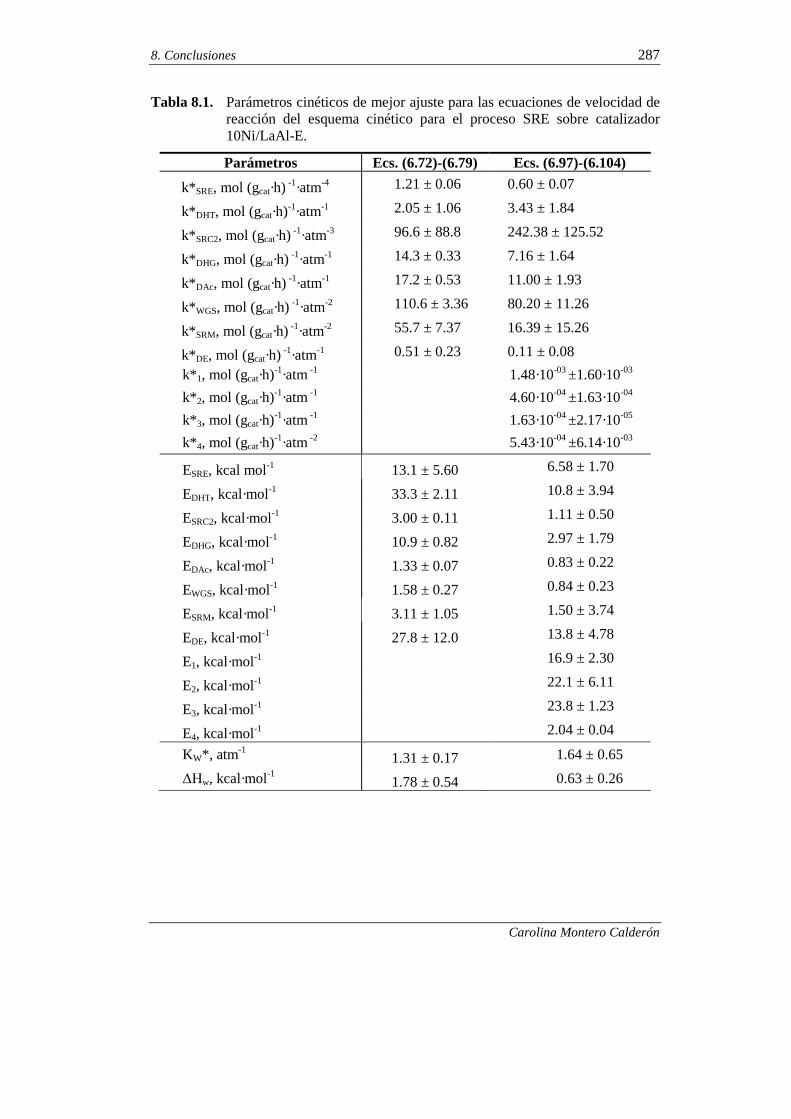
8. Conclusiones 287
Carolina Montero Calderón
Tabla 8.1. Parámetros cinéticos de mejor ajuste para las ecuaciones de velocidad de reacción del esquema cinético para el proceso SRE sobre catalizador 10Ni/LaAl-E.
Parámetros Ecs. (6.72)-(6.79) Ecs. (6.97)-(6.104)
k*SRE, mol (gcat·h) -1·atm-4 1.21 ± 0.06 0.60 ± 0.07
k*DHT, mol (gcat·h)-1·atm-1 2.05 ± 1.06 3.43 ± 1.84
k*SRC2, mol (gcat·h) -1·atm-3 96.6 ± 88.8 242.38 ± 125.52
k*DHG, mol (gcat·h) -1·atm-1 14.3 ± 0.33 7.16 ± 1.64
k*DAc, mol (gcat·h) -1·atm-1 17.2 ± 0.53 11.00 ± 1.93
k*WGS, mol (gcat·h) -1·atm-2 110.6 ± 3.36 80.20 ± 11.26
k*SRM, mol (gcat·h) -1·atm-2 55.7 ± 7.37 16.39 ± 15.26
k*DE, mol (gcat·h) -1·atm-1 0.51 ± 0.23 0.11 ± 0.08
k* 1, mol (gcat·h)-1·atm -1 1.48·10-03 ±1.60·10-03
k* 2, mol (gcat·h)-1·atm -1 4.60·10-04 ±1.63·10-04
k* 3, mol (gcat·h)-1·atm -1 1.63·10-04 ±2.17·10-05
k* 4, mol (gcat·h)-1·atm -2 5.43·10-04 ±6.14·10-03
ESRE, kcal mol-1 13.1 ± 5.60 6.58 ± 1.70
EDHT, kcal·mol-1 33.3 ± 2.11 10.8 ± 3.94
ESRC2, kcal·mol-1 3.00 ± 0.11 1.11 ± 0.50
EDHG, kcal·mol-1 10.9 ± 0.82 2.97 ± 1.79
EDAc, kcal·mol-1 1.33 ± 0.07 0.83 ± 0.22
EWGS, kcal·mol-1 1.58 ± 0.27 0.84 ± 0.23
ESRM, kcal·mol-1 3.11 ± 1.05 1.50 ± 3.74
EDE, kcal·mol-1 27.8 ± 12.0 13.8 ± 4.78
E1, kcal·mol-1 16.9 ± 2.30
E2, kcal·mol-1 22.1 ± 6.11
E3, kcal·mol-1 23.8 ± 1.23
E4, kcal·mol-1 2.04 ± 0.04
KW*, atm-1 1.31 ± 0.17 1.64 ± 0.65
∆Hw, kcal·mol-1 1.78 ± 0.54 0.63 ± 0.26

288
Capítulo 8
4.2. La cuantificación de la evolución de la composición del medio de reacción con
el tiempo requiere un modelo de desactivación selectiva. Se ha establecido un
modelo de desactivación con 4 actividades diferentes para las diferentes etapas
del esquema cinético:
- a1 para las reacciones de reformado de etanol, etileno y metano
- a2 para la deshidrogenación
- a3 para las reacción WGS y la deshidratación
- a4 para la descomposición de etanol y de acetaldehído.
� Por tanto, considerando el modelo cinético a tiempo cero que incluye las
rutas térmicas, las velocidades de reacción a tiempo t para cada reacción
individual del esquema cinético vienen dadas por las siguientes ecuaciones:
( ) 3WE31
WW
3WESRE
bSRE PP kaPK1
PP k=r +
+ρ
(6.134)
( ) E23WW
EDHTbDHT Pka
PK1
Pkr +
+ρ=
(6.128)
( ) 1WW
2WSRC2
bSRC2 aPK1
PP kr C2
+ρ= =
=
(6.129)
( )
+
+
ρDHG
HAcE12
WW
DHG
HAcEDHG
bDHG K
PP-Pka
PK1
K
PP-Pk
= r 2
2
(6.130)
( ) Ac3WW
AcDAcbDAc Pk
PK1
Pkr +
+ρ=
(6.131)
( )
+
+
ρWGS
COHWCO43
WW
WGS
COHWCOWGS
bWGS K
PP-PP ka
PK1
K
PP-PP k
=r 22
22
(6.132)
( )
+
+
ρSRM
CO3H2
WM41WW
SRM
CO3H2
WMSRM
bSRM K
PP-PPka
PK1
K
PP-PPk
=r
(6.133)
( ) E3WW
EDEbDE Pk
PK1
Pkr +
+ρ=
(6.135)

8. Conclusiones 289
Carolina Montero Calderón
� La velocidad de desactivación para la deshidrogenación es notablemente más
rápida que la de las etapas de deshidratación y reacción WGS, y éstas a su
vez tienen desactivación más rápida que las reacciones de reformado, siendo
despreciable la velocidad de desactivación de las etapas de descomposición
(a4≈1).
� Las expresiones para la cinética de desactivación para las actividades a1, a2 y
a3 puede describirse mediante una expresión del tipo:
→=→=
−→==−
=
y WGS DHT3j
DHG2j
M-SRy C-SR E,SR1j
a)P(kdt
da 22jEdj
j (6.127)
Con las siguientes ecuaciones, una vez calculados los parámetros cinéticos:
( ) ( ) 21E
1 a)P(873
1
T
1
R
162.2 8.785- 204.0 113.1
dt
da
−±±=− (8.1)
( ) ( ) 22E
2 a)P(873
1
T
1
R
483.2 3.514- 870.0 146.6
dt
da
−±±=− (8.2)
( ) ( ) 23E
3 a)P(873
1
T
1
R
751.2 29.061- 258.1 191.7
dt
da
−±±=− (8.3)
Con el modelo cinético que integra el modelo cinético a tiempo cero y estas
ecuaciones para la desactivación selectiva se predice adecuadamente la evolución
con el tiempo de la concentración de los reactivos y del producto mayoritario, H2,
y de los minoritarios (acetaldehído y etileno). Un mejor ajuste de los productos
CO y CH4 requiere una mejora de este modelo cinético, mediante la
consideración expresa en el esquema cinético de las reacciones de formación de
coque (por la reacción de Boudouard y descomposición de CH4, así como la
consideración de la descomposición de acetaldehído y de etileno) y de la
gasificación del coque.


9. NOMENCLATURA


293
Carolina Montero Calderón
9. NOMENCLATURA
AM Superficie metálica activa, m2/gcat, Ec. (2.4)
ANi0,
ANi2+
Concentración atómica superficial de especies Ni0 y Ni
2+,
respectivamente
a, aj
Actividad del catalizador para el modelo de desactivación no selectiva y
para la reacción j en el modelo de desactivación selectiva,
respectivamente
ikaNúmero de átomos del k elemento presente en cada molécula de la
especie i, Ec. (B.3)
bq Estimación del parámetro q
CC Contenido de coque, % ó mgc/gcat, Ec. (2.5)
Ci, Ni Concentración y flujo molar del componente i , en las expresiones
cinéticas
cqq Elemento de la matriz varianza-covarianza de q parámetros
D Dispersión metálica, Ec. (2.3)
Dinterior Diámetro interior del lecho fluidizado
D, G Bandas de los espectros Raman correspondientes
d Orden de desactivación, Ec. (6.107)
dMO Tamaño medio del cristalito, Ec. (2.1)
mMd Tamaño de partícula metálica, Ec. (2.4)
EB, Ek
Energía de enlace de los electrones de la muestra (binding energy), y
energía cinética de los electrones emitidos en un proceso de fotoemisión,
Ec. (2.2)
Ej, Edj Energía de activación en la etapa j, reacción y de la constante de
desactivación, kcal/mol
err0ij Diferencia entre el valor experimental y calculado de composición a
tiempo cero para cada compuesto i en la condición experimental j
erri Error del ajuste considerando el factor de peso
FT, Fi Caudal molar total y del componente i, mol/h
F(1-α) Parámetro crítico del test de Fischer
F(i-j) Parámetro crítico de mejora del modelo j respecto al i
FEtOH,e,
FEtOH,s Flujos molares de etanol a la entrada y a la salida del reactor, mol/h
FH2O Caudal molar de agua a la salida del reactor, Ec. (2.8)

294
Capítulo 9
FO, FOd Función objetivo para el cálculo de la cinética a tiempo cero y de la
cinética de desactivación, respectivamente.
Foxigenado,s Caudal de los oxigenados a la salida de reactor, Ec. (2.8)
FT Caudal molar total a la salida del reactor, Ec. (2.8) y Ec. (6.52)
if , 0
ifFugacidad de la especie i en el sistema y en el estado estacionario, Ec.
B1
fi Factor cromatográfico del compuesto i
G Energía libre de Gibbs total del sistema, Ec. (B.1)
0
iG , iG Energía libre de Gibbs estándar y parcial molar de la especie i, Ec. (B.1)
GHSV Velocidad especial horaria del gas, h-1
hv Energía del fotón incidente, Ec. (2.2)
ID, IG Intensidad de las bandas D y G de los espectros Raman
K Constante de equilibrio de la reacción, Ec. (C.1)
Kij, K*ij
Constante de equilibrio de adsorción del componente i en la etapa j, a
temperatura T y a la temperatura de referencia (600 ºC), respectivamente,
atm-1
KW,
K*W
Constante de adsorción del agua a tempertaura T y a la temperatura de
referencia ( 600 ºC), atm-1
k Constante de Scherrer, Ec. (2.1)
kdj, k*dj Constante cinética de desactivación para la reacción j a la temperatura T
y a la temperatura de referencia (600 ºC), h-1
kj, k*j Constante cinética de cada etapa j a la temperatura T y a la temperatura
de referencia (600 ºC), mol (gcat·h)-1
·atm -n
L contenido en masa de metal en el catalizador, Ec. (2.3)
La Tamaño de dominio de coque
Lq Intervalo de confianza de los parámetros calculados
M Peso molecular del metal, Ec. (2.3)
Mcat Masa de catalizador en el ensayo TPO, mg, Ec. (2.5)
MCO2 Masa molar del CO2, g/mol, Ec. (2.5)
Nm Número de átomos superficiales, mol/g, Ec. (2.3)
n Parámetro para cálculo de la actividad en la Ec. (6.126)
nc Número de componentes del medio de reacción.
ni Número de moles de la especie i, Ec. (B.1)
nt Número de tiempos de reacción para los que se determinado la

9. Nomenclatura 295
Carolina Montero Calderón
composición del medio de reacción para cada condición experimental j
np, nr Número de moles productos y reactivos, Ec. (B.9)
O2/E Relación molar oxígeno/etanol
P Presión, atm
P0 Presión en condiciones estándar, Ec. (B.2)
P0 Presión de vapor del adsorbato, atm
PEtOH, Presión parcial inicial de EtOH, atm
PE, PW,
PM Presión parcial de etanol, agua y metano, respectivamente, atm
Pi Presión parcial del compuesto i, atm
PM Peso molecular medio, g/mol
p, q Número de condiciones experimentales y número de parámetros
cinéticos a estimar
Q Caudal total de alimentación, cm3/min
QH2O,
QHe Caudal de agua y de helio, ml/min
Qtotal
Energía total (para el calentamiento, el reformado y el enfriamiento, en el
estudio termodinámico), kJ/mol
R Constante universal de los gases, kcal/molK
Rcoque Rendimiento de coque, %, Ec. (5.3)
Ri Rendimiento del compuesto i en la reacción SRE, referido a los moles
estequiométricos producidos por mol de reactante, Ec.(2.7)
ri, ri0 Velocidad de formación del componente i a tiempo t y a tiempo cero
rjc, rjo
c Contribución catalítica a la velocidad de reacción de cada etapa del
esquema cinético, Ec. (6.105)
rjt Contribución térmica a la velocidad de reacción de cada etapa del
esquema cinético, Ec. (6.106)
(rj)0, rj Velocidad de reacción de la etapa j del esquema cinético a tiempo cero y
a tiempo t, respectivamente
rporo radio de poro, Å
S sección de lecho catalítico, m2
(S) Centro activo
SBET Área superficial BET, m2/g
Si Selectividad del compuesto i, Ec. (2.8)

296
Capítulo 9
Sq Estequiometría de la adsorción, Ec.(2.3) y sección de lecho catalítico, m2
SSE,
SSEi Suma de los cuadrados de los errores total y de cada componente i
SSEi,
SSEj
Suma de los cuadrados de errores del modelo i y modelo j,
respectivamente
SSM Suma de cuadrados de la regresión
S/Ac Relación molar agua/acetaldehído
S/E Relación molar agua/etanol
t /2 Valor crítico de la estadística de Student´s para un nivel de confianza
T, T* Temperatura y temperatura de referencia, K
TC Temperatura de calcinación, ºC
u, umf Velocidad lineal y velocidad mínima de fluidización, m/s
Vads Volumen de N2 adsorbido en la fisisorción, cm3/g
Vm Volumen de la monocapa de N2, cm3/g
Vporos Volumen de poros, cm3/g.
W/F0 Tiempo especial, gcath/gEtOH
wi Factor de peso para cada componente i
w ;asa del catalizador, g
Xcal, Xexp Fracción molar calculada y experimental, respectivamente.
XEtOH,
XAc Conversión de etanol y acetaldehído
Xi Fracción molar en base húmeda del componente i
Xij, Xij* Fracción molar en base húmeda calculada y experimental,
respectivamente, del compuesto i para la condición experimental j
xi* Fracción molar en base seca del componente i en el medio de reacción,
en el ensayo sin catalizador
iy Fracción molar en fase gas del componente i, Ec. (B.2)
z Coordenada longitudinal del reactor, m
Símbolos griegos
α Nivel de confianza.
Ancho a la altura media del pico de difracción de la muestra en el
espectro XRD, Ec. (2.1)

9. Nomenclatura 297
Carolina Montero Calderón
0
fiG Energía de Gibbs estándar de formación de la especie i, kcal/mol
Hc0,EtOH Calor de combustión del etanol, kcal/mol
Hc,r0,
Hc,p0
Entalpía de combustión de reactivos y productos, en condiciones
estándar, kcal·mol-1
Hrº Entalpía de reacción en condiciones estándar, kcal·mol-1
ΔHw Entalpía de adsorción del agua, kcal·mol-1
i Suma del cuadrado de los residuos para cada compuesto i
i Coeficiente de fugacidad de la especie i, Ec. (B.2)
λ Longitud de onda de la radiación utilizada en el análisis XRD, Å, Ec.
(2.1)
k Multiplicador de Lagrange, Ec. (B.3)
i Potencial químico, Ec. (B.1)
Grados de libertad
i
Coeficiente estequiométrico de formación del componente i a partir de
etanol, Ec. (2.7)
( i)j Coeficiente estequiométrico del componente i en la reacción j del
esquema cinético, Ec. (6.54)
Posición del pico de difracción correspondiente al metal a analizar, Ec.
(2.1) y término de atenuación de la velocidad de reacción por adsorción
de los componentes del medio de reacción
b Densidad del lecho, Ec.(6.89)
ρM Densidad del metal, Ec. (2.4)
(T,Ci),
j(T,Ci)
Función de desactivación para el modelo de desactivación no selectiva
y para la reacción j en el modelo de desactivación selectiva,
respectivamente
2 Varianza
σEi2, σEj
2Varianzas del residual de los modelos i y j
σMi2 , σMj
2 Varianzas de la regresión de los modelos i y j
τz Variable independiente correspondiente a una posición z de la malla a
un valor dado de tiempo espacial
Acrónimos
Ac Acetaldehído

298
Capítulo 9
AFEX Pretratamiento de tipo explosión fibrilar
ATR Reformado autotérmico
BJH Método de Barrett, Joyner, y Halenda
BSE Microscopia electrónica de barrido de electrones retrodispersados
CBP Proceso consolidado de transformación de la biomasa
C2= Etileno
CNT Nanotubos de carbono
CZA Catalizador basado en CuO-ZnO-Al2O3
D-Ac Descomposición de acetaldehído
D-E Descomposición de etanol
DHG Deshidrogenación de etanol
DHT Deshidratación de etanol
DME Dimetil éter
DR Reformado con CO2 o reformado seco
DRIFTS Espectroscopía infrarroja de reflectancia difusa
EDS Espectroscopia por dispersión de energía
EDX Espectroscopia por dispersión de rayos X
ER Modelos mecanísticos del tipo Eley Rideal
EtOH, E Etanol
FTIR Espectroscopía infrarroja
GC-MS Cromatógrafo de gases acoplado a masas
GHG Gases de efecto invernadero
GLP Gas licuado de petróleo
ICP-AES Espectrometría de emisión atómica por plasma de acoplamiento
inductivo
LHHW Modelos mecanísticos del tipo de Langmuir-Hinshelwood-Hougen-
Watson
M-CO2 Metanación de CO2
MDEA Metil-dietanol-amina
M Metano
MeOH Metanol
OSR Reformado oxidativo con vapor

9. Nomenclatura 299
Carolina Montero Calderón
OSRE Reformado oxidativo con vapor de etanol
PEMFC Pilas de combustible de membrana polimérica de intercambio
protónico
PO Oxidación parcial
PSA Recuperación por adsorción a presión
r-WGS Reacción reversa del gas de agua
SEM Microscopía electrónica de barrido
SHF Fermentación tras la hidrólisis
SR Reformado con vapor
SRA,
SR-Ac Reformado con vapor de acetaldehído
SRE,
SR-E Reformado con vapor de etanol
SR-C2= Reformado con vapor de etileno
SRD Reformado con vapor de dimetil éter
SR-M Reformado con vapor de metano
SR-MeOH Reformado con vapor de metanol
SSCF Sacarificación simultánea y co-fermentación
SSF Sacarificación simultánea y fermentación
TCD Detector de conductividad térmica
TEM Microscopía electrónica de transmisión
TPD Desorción a temperatura programada
TPO Oxidación a temperatura programada
TPR Reducción a temperatura programada
TPSR Reacción superficial a temperatura programada
WGS Reacción del gas de agua (water gas shift)
W Agua,
XPS Espectroscopía fotoelectrónica de rayos X
XRD Difracción de rayos X


10. BIBLIOGRAFÍA


303
Carolina Montero Calderón
10. BIBLIOGRAFIA
Aguayo AT, Gayubo AG, Ortega JM, Olazar M, Bilbao J. Catalyst deactivation by
coking in the MTG process in fixed and fluidized bed reactors. Catal Today
1997; 37:239-48.
Aguayo AT, Sánchez del Campo AE, Gayubo AG, Tarrío A, Bilbao J. Deactivation by
coke of a catalyst sased on a SAPO-34 in the transformation of methanol into
olefins. J Chem Technol Biotechnol 1999; 74:315-21.
Aguayo AT, Gayubo AG, Atutxa A, Olazar M, Bilbao J. Catalyst deactivation by coke
in the transformation of aqueous ethanol into hydrocarbons. Kinetic modeling
and acidity deterioration of the catalyst. Ind Eng Chem. Res 2002; 41:4216-24.
Akande A, Aboudheir A, Idem R, Dalai A. Kinetic modeling of hydrogen production
by the catalytic reforming of crude ethanol over a co-precipitated catalyst in a
packed bed tubular reactor. Int J Hydrogen Energ 2006; 31:1707-15.
Akpan E, Akande A, Aboudheir A, Ibrahim H, Idem R. Experimental, kinetic and 2-D
reactor modeling for simulation of the production of hydrogen by the catalytic
reforming of concentrated crude ethanol (CRCCE) over a Ni-based commercial
catalyst in a packed-bed tubular reactor. Chem Eng Sci 2007; 62:3112-26.
Alberton AL, Souza M, Schmal M. Carbon formation and its influence on ethanol
steam reforming over Ni/Al2O3 catalysts. Catal Today 2007; 123:257-64.
Alenazey F, Cooper C, Dave C, Elnashaie S, Susu A, Adesina A. Coke removal from
deactivated Co–Ni steam reforming catalyst using different gasifying agents:
An analysis of the gas–solid reaction kinetics. Catal Comm 2009; 10:406-11.
Al-Hamamre Z, Hararah M. Hydrogen production by thermal partial oxidation of
ethanol: Thermodynamics and kinetics study. Int J Hydrogen Energ 2010;
35:5367-77.
Alonso, A. Catalizadores alternativos y modelado cinético del proceso BTO
(Bioetanol a olefinas). Tesis Doctoral 2008. Universidad del País Vasco,
Bilbao.
Andrews J, Shabani B. Re-envisioning the role of hydrogen in a sustainable energy
economy. Int J Hydrogen Energ 2012; 37:1184-203.
Arcoumanis C, Bae C, Crookes R, Kinoshita E. The potential of di-methyl ether
(DME) as an alternative fuel for compression-ignition engines: A review. Fuel
2008; 87:1014-30.
Ateka, A. Innovaciones en el proceso de síntesis de dimetil éter en una etapa con
secuestro de CO2. Tesis Doctoral 2014. Universidad del País Vasco, Bilbao.
Aramburu, B. Desarrollo del proceso de reformado catalítico del líquido de pirólisis de
biomasa vegetal (bio-oil) Tesis Doctoral 2015. Universidad del País Vasco,
Bilbao.

304
Capítulo 10
Banach B, Machocki A, Rybak P, Denis A, Grzegorczyk W, Gac W. Selective
production of hydrogen by steam reforming of bio-ethanol. Catal Today 2011;
176:28-35.
Barattini L, Ramis G, Resini C, Busca G, Sisani M, Costantino U. Reaction path of
ethanol and acetic acid steam reforming over Ni-Zn-Al catalysts. Flow reactor
studies. Chem Eng J 2009; 153:43-49.
Barrera A, Fuentes S, Viniegra M, Avalos-Borja M, Bogdanchikova N, Campa-
Molina J. Structural properties of Al2O3-La2O3 binary oxides prepared by sol-
gel. Mater Res Bull 2007; 42:640-48.
Barroso M, Galetti A, Gomez M, Arrúa L, Abello M. Ni-catalysts supported on
ZnxMg1−xAl2O4 for ethanol steam reforming: Influence of the substitution for
Mg on catalytic activity and stability. Chem Eng J 2013; 222:142-49.
Bartholomew C. Mechanisms of catalyst deactivation. Appl Catal A- Gen 2001;
212:17-60.
Bengaard H, Nørskov J, Sehested J, Clausen B, Nielsen L, Molenbroek A y
cols.Steam Reforming and Graphite Formation on Ni Catalysts. J Catal 2002;
209:365-84.
Benito PL, Gayubo AG, Aguayo AT, Castilla M, Bilbao J. Concentration-dependent
kinetic model for catalyst deactivation in the MTG process. Ind Eng Chem Res
1996; 35:81-89.
Bilal M, Jackson SD. Steam reforming of ethanol at medium pressure over Ru/Al2O3:
Effect of temperature and catalyst deactivation. Cat Sci Technol 2012; 2:2043-
51.
Bilal M, Jackson S. Ethanol steam reforming of ethanol over Rh and Pt catalysts:
effect of temperature and catalyst deactivation. Catal Sci Technol 2013; 3:754-
66.
Biotechnology Industrial Organization. The renewable fuel standard: timeline of a
successful policy, 2014.
Bshish A, Yaakob A, Narayanan B, Ramakrishnan R, Ebshish A. Steam-reforming of
ethanol for hydrogen production. Chem Pap 2011; 65:251-66.
Bussi J, Musso M, Veiga S, Bespalko N, Faccio R, Roger A. Ethanol steam reforming
over NiLaZr and NiCuLaZr mixed metal oxide catalysts. Catal Today 2013;
213:42-49.
Carrera Cerritos R, Fuentes Ramírez R, Aguilera Alvarado A, Martínez Rosales J,
Viveros García T, Galindo Esquivel I. Steam reforming of ethanol over
Ni/Al2O3-La2O3 catalysts synthesized by sol-gel. Ind Eng Chem Res 2011;
50:2576-84.
Carvajal E, Guamán C, Portero P, Salas E, Tufiño C, Bastidas B. Second generation
ethanol from residual biomass: research and perspectives in Ecuador, Biomass
Now - Sustainable Growth and Use. Dr. Miodrag Darko Matovic (Ed.) InTech
2013; ISBN: 978-953-51-1105-4.

10. Bibliografía 305
Carolina Montero Calderón
Cavallaro S, Freni S. Ethanol steam reforming in a molten carbonate fuel cell. A
preliminary kinetic investigation. Int J Hydrogen Energ 1996; 21:465-69.
Chen D, Christensen K., Ochoa E, Yu Z, Tøtdal B, Latorre N, y cols. Synthesis of
carbon nanofibers: effects of Ni crystal size during methane decomposition. J
Catal 2005; 229:82-96.
Chen K, Xue Z, Liu H, Guo A, Wang Z. A temperature-programmed oxidation
method for quantitative characterization of the thermal cokes morphology. Fuel
2013; 113:274-79.
Chen X, Yik E, Butler J, Schwank JW. Gasification characteristics of carbon species
derived from model reforming compound over Ni/Ce–Zr–O catalysts. Catalysis
Today 2014; 233:14-20.
Chiou JYZ, Siang J, Yang S, Ho K, Lee C, Yeh C y cols.Pathways of ethanol steam
reforming over ceria-supported catalysts. Int J Hydrogen Energ 2012;
37:13667-73.
Chiou JYZ, Lai CL, Yu S, Huang H, Chuang C, Wang C. Effect of Co, Fe and Rh
addition on coke deposition over Ni/Ce0.5Zr0.5O2 catalysts for steam reforming
of ethanol. Int J Hydrogen Energ 2014; 39:20689-99.
Choong C, Zhong Z, Huang L, Wang Z, Ang T, Borgna A y cols.Effect of calcium
addition on catalytic ethanol steam reforming of Ni/Al2O3: I. Catalytic stability,
electronic properties and coking mechanism. Appl Catal A-Gen 2011; 407:145-
54.
Ciambelli P, Palma V, Ruggiero A, Iaquaniello G. Platinum catalysts for the low
temperature catalytic steam reforming of ethanol. AIDIC conference series
2009; 83-92.
Ciambelli P, Palma V, Ruggiero A. Low temperature catalytic steam reforming of
ethanol. 2. Preliminary kinetic investigation of Pt/CeO2 catalysts. Appl Catal B-
Environ 2010; 96:190-97.
Comas J, Laborde M, Amadeo N. Thermodynamic analysis of hydrogen production
from ethanol using CaO as a CO2 sorbent. J Power Sources 2004; 138:61-67.
Constantinides A, Mostoufi N. Numerical method for chemical engineers with
MATLAB applications, Prentice Hall: Bew York 1999.
Contreras JL, Salmones J, Colín-Luna JA, Nuño L, Quintana B, Córdova I y
cols.Catalysts for H2 production using the ethanol steam reforming (a review).
Int J Hydrogen Energ 2014; 39:18835-53.
Cormos C. Renewable hydrogen production concepts from bioethanol reforming with
carbon capture. Int J Hydrogen Energ 2014; 39:5597-606.
Coulombe M, Lebeuf M, Fairley N, Walton J, Soucy G. Cryolitic vein XPS imaging
from an industrial grade carbon. J Electron Spectrosc Relat Phenom 2012;
185:588-97.

306
Capítulo 10
Da Silva A, de Souza KR, Jacobs G, Graham U, Davis B, Mattos L y cols.Steam and
CO2 reforming of ethanol over Rh/CeO2 catalyst. Appl Catal B-Environ 2011;
102:94-109.
Dan M, Senila L, Roman M, Mihet M, Lazar MD. From wood wastes to hydrogen -
Preparation and catalytic steam reforming of crude bio-ethanol obtained from
fir wood. Renew Energ 2015; 74:27-36.
De Falco M. Ethanol membrane reformer and PEMFC system for automotive
application. Fuel 2011; 90:739-47.
Djinović P, Osojnik Črnivec IG, Erjavec B, Pintar A. Influence of active metal loading
and oxygen mobility on coke-free dry reforming of Ni-Co bimetallic catalysts.
Appl Catal B-Environ 2012; 125:259-70.
El Doukkali M, Iriondo A, Arias PL, Cambra JF, Gandarias I, Barrio VL.
Bioethanol/glycerol mixture steam reforming over Pt and PtNi supported on
lanthana or ceria doped alumina catalysts. Int J Hydrogen Energ 2012; 37:8298-
309.
Elias K, Lucredio A, Assaf E. Effect of CaO addition on acid properties of Ni-
Ca/Al2O3 catalysts applied to ethanol steam reforming. Int J Hydrogen Energ
2013; 38:4407-17.
Ellabban O, Abu-Rub H, Blaabjerg F. Renewable energy resources: Current status,
future prospects and their enabling technology. Renew Sust Ener Rev 2014;
39:748-64.
Epelde, E. Proceso catalítico de interconversión de olefinas con intensificación de
propeno. Tesis Doctoral 2013. Universidad del País Vasco, Bilbao.
Epelde E, Gayubo A.G, Olazar M, Bilbao J, Aguayo A.T. Modified HZSM-5 zeolites
for intensifying propylene production in the transformation of 1-butene. Chem.
Eng J 2014a; 251:80-91.
Epelde E, Aguayo AT, Olazar M, Bilbao J, Gayubo AG. Modifications in the HZSM-
5 zeolite for the selective transformation of ethylene into propylene. App. Catal
A-Gen 2014b; 479:17-25.
Epelde E, Ibañez M, Aguayo AT, Gayubo AG, Bilbao J, Castaño P. Differences
among the deactivation pathway of HZSM-5 zeolite and SAPO-34 in the
transformation of ethylene or 1-butene to propylene. Micropor Mesopor Mat
2014c; 195:284-93.
Ereña J, Sierra I, Aguayo AT, Ateka A, Olazar M, Bilbao J. Kinetic modelling of
dimethyl ether synthesis from (H2+CO2) by considering catalyst deactivation.
Chem Eng J 2011; 174:660-67.
Ereña J, Vicente J, Aguayo AT, Olazar M, Bilbao J, Gayubo AG. Kinetic behaviour of
catalysts with different CuO-ZnO-Al2O3 metallic function compositions in
DME steam reforming in a fluidized bed. Appl Catal B-Environ 2013; 142-
143:315-22.

10. Bibliografía 307
Carolina Montero Calderón
Erkiaga A. Gasificación con Vapor de Biomasa y Plásticos en Spouted Bed Cónico.
Tesis Doctoral 2014.Universidad del País Vasco, Bilbao.
Estrade-Szwarckopf H. XPS photoemission in carbonaceous materials: A “defect”
peak beside the graphitic asymmetric peak. Carbon 2004; 42:1713-21.
Fatsikostas AN, Kondarides DI, Verykios XE. Production of hydrogen for fuel cells
by reformation of biomass-derived ethanol. Catal Today 2002; 75:145-55.
Fatsikostas AN, Verykios XE. Reaction network of steam reforming of ethanol over
Ni-based catalysts. J Catal 2004; 225:439-52.
Faungnawakij K, Tanaka Y, Shimoda N, Fukunaga T, Kikuchi R, Eguchi K.
Hydrogen production from dimethyl ether steam reforming over composite
catalysts of copper ferrite spinel and alumina. Appl Catal B-Environ 2007;
74:144-51.
Fishtik I, Alexander A, Datta R, Geana D. A thermodynamic analysis of hydrogen
production by steam reforming of ethanol via response reactions. Int J
Hydrogen Energ 2000; 25:31-45.
Gamero, M. Transformación catalítica de metano en olefinas ligeras (vía
clorometano). Tesis Doctoral 2013. Universidad del País Vasco, Bilbao.
García EY, Laborde MA. Hydrogen production by the steam reforming of ethanol:
Thermodynamic analysis. Int J Hydrogen Energ 1991; 16:307-12.
García VM, López E, Serra M, Llorca J. Dynamic modeling of a three-stage low-
temperature ethanol reformer for fuel cell application. J Power Sources 2009;
192:208-15.
Garza M, Magtoto NP, Kelber JA. Characterization of oxidized Ni3Al (1 1 0) and
interaction of the oxide film with water vapor. Surf Sci 2002; 19:259-68.
Gayubo AG, Aguayo AT, Sánchez Del Campo AE, Tarrío AM, Bilbao J. Kinetic
modeling of methanol transformation into olefins on a SAPO-34 catalyst. Ind
Eng Chem Res 2000; 39:292-00.
Gayubo AG, Aguayo AT, Morán AL, Olazar M, Bilbao J. Role of water in the kinetic
modelling of catalyst deactivation in the MTG process. AIChE J 2002; 48:1561-
71.
Gayubo AG, Aguayo AT, Olazar M, Vivanco R, Bilbao J. Kinetics of the Irreversible
Deactivation of the HZSM-5 Catalyst in the MTO Process. Chem Eng Sci 2003;
58:5239-49.
Gayubo AG, Aguayo AT, Atutxa A, Aguado R, Bilbao J. Transformation of
oxygenate components of biomass pyrolysis-oil on a HZSM-5 Zeolite. I.
Alcohols and Phenols. Ind Eng Chem Res 2004a; 43:2610-2618.
Gayubo AG, Aguayo AT, Atutxa A, Aguado R, Olazar M, Bilbao J. Transformation
of oxygenate components of biomass pyrolysis-oil on a HZSM-5 Zeolite. II.
Aldehydes, Ketones and Acids. Ind Eng Chem Res 2004b; 43:2619-26.

308
Capítulo 10
Gayubo AG, Aguayo AT, Alonso A, Bilbao, J. Kinetic modeling of the methanol-to-
olefins process on a silicoaluminophosphate (SAPO-18) catalyst by considering
deactivation and the formation of individual olefins Ind Eng Chem Res 2007;
46:1981-89.
Gayubo AG, Valle B, Aguayo AT, Olazar M, Bilbao J. Olefin production by catalytic
transformation of crude bio-oil in a two-step process. Ind Eng Chem Res 2010a;
49:123-31.
Gayubo AG, Alonso A, Valle B, Aguayo AT, Bilbao J. Kinetic model for the
transformation of bioethanol into olefins over a HZSM-5 zeolite treated with
alkali. Ind Eng Chem Res 2010b; 49:10836-44.
Gayubo AG, Alonso A, Valle B, Aguayo AT, Olazar M, Bilbao J. Kinetic modelling
for the transformation of bioethanol into olefins on a hydrothermally stable Ni-
HZSM-5 catalyst considering the deactivation by coke. Chem Eng J 2011;
167:262-77.
Gayubo AG, Alonso A, Valle B, Aguayo AT, Bilbao J. Deactivation kinetics of a
HZSM-5 zeolite catalyst treated with alkali for the transformation of bio-
ethanol into hydrocarbons. AIChE J 2012; 58:526-37.
Gayubo AG, Vicente J, Ereña J, Montero C, Olazar M, Bilbao J. Comparison of Ni
and Co catalysts for ethanol steam reforming in a fluidized bed reactor. Catal
Lett 2014; 144:1134-43.
Gohier A, Ewels C, Minea T, Djouadi M. Carbon nanotube growth mechanism
switches from tip- to base-growth with decreasing catalyst particle size. Carbon
2008; 46:1331-38.
Gómez-Gualdrón D, Balbuena, P, Characterization of carbon atomistic pathways
during single-walled carbon nanotube growth on supported metal nanoparticles.
Carbon 2013; 57:298-309.
Graschinsky C, Laborde M, Amadeo N, Valant A.L, Bion N, Epron F, Duprez D.
Ethanol Steam Reforming over Rh(1 %)MgAl2O4/Al2O3: A Kinetic Study. Ind
Eng Chem Res 2010; 24:12383-89.
Graschinsky C, Giunta P, Amadeo N, Laborde M. Thermodynamic analysis of
hydrogen production by autothermal reforming of ethanol. Int J Hydrogen
Energ 2012; 37:10118-24.
Gregorini VA, Pasquevich D, Laborde M. Price determination for hydrogen produced
from bio-ethanol in Argentina. Int J Hydrogen Energ 2010; 35:5844-48.
Gupta A, Verma JP. Sustainable bio-ethanol production from agro-residues: A review.
Renew Sust Energ Rev 2015; 41:550-67.
Han S, Bang Y, Yoo J, Seo J, Song I. Hydrogen production by steam reforming of
ethanol over mesoporous Ni-Al2O3-ZrO2 xerogel catalysts: Effect of nickel
content. Int J Hydrogen Energ 2013; 38:8285-92.

10. Bibliografía 309
Carolina Montero Calderón
Hardiman KM, Cooper CG, Adesina AA, Lange R. Post-mortem characterization of
coke-induced deactivated alumina-supported Co-Ni catalysts. Chem Eng Sci
2006; 61:2565-73.
Haryanto A, Fernando S, Murali N, Adhikari S. Current Status of Hydrogen
Production Techniques by Steam Reforming of Ethanol: A Review. Energy
Fuels 2005; 19:2098-106.
He Z, Yang M, Wang X, Zhao Z, Duan A. Effect of the transition metal oxide
supports on hydrogen production from bio-ethanol reforming. Catal Today
2012; 194:2-8.
Helveg S, Sehested J, Rostrup-Nielsen J. Whisker carbon in perspective. Catal Today
2011; 178:42-46.
Hernández L, Kafarov V. Thermodynamic evaluation of hydrogen production for fuel
cells by using bio-ethanol steam reforming: Effect of carrier gas addition. J
Power Sources 2009; 192:195-99.
Hung C, Chen S, Liao Y, Chen C, Wang J. Oxidative steam reforming of ethanol for
hydrogen production on M/Al2O3. Int J Hydrogen Energ 2012; 37:4955-66.
Ibáñez M, Artetxe M, Lopez G, Elordi G, Bilbao J, Olazar M y cols.Identification of
the coke deposited on an HZSM-5 zeolite catalyst during the sequenced
pyrolysis-cracking of HDPE. Appl Catal B-Environ 2014; 148-149:436-45.
IFP Energies Nouvelles, Etapes de production d'éthanol-carburant faisant intervenir
des biocatalyseurs. 2010.
International Energy Agency. World Energy Outlook 2013. 2013.
Ito S, Tomishige K. Steam reforming of ethanol over metal-oxide-promoted Pt/SiO2
catalysts: Effects of strong metal-oxide interaction (SMOI). Catal Commun
2010; 12:157-60.
Jin M, Lau MW, Balan V, Dale BE. Two-step SSCF to convert AFEX-treated
switchgrass to ethanol using commercial enzymes and Saccharomyces
cerevisiae 424A (LNH-ST). Bioresour Technol 2010; 101:8171-78.
Karge HG, Niessen W, Bludau H. In-situ FTIR measurements of diffusion in coking
zeolite catalysts. Appl Cata. A-Gen 1996; 146:339-49.
Karim A, Su Y, Sun J, Yang C, Strohm J, King D, Wang Y. A comparative study
between Co and Rh for steam reforming of ethanol. Appl Catal B-Environ 2010;
96:441-48.
Khila Z, Hajjaji N, Pons M, Renaudin V, Houas A. A comparative study on energetic
and exergetic assessment of hydrogen production from bioethanol via steam
reforming, partial oxidation and auto-thermal reforming processes. Fuel
Process Technol 2013; 112:19-27.
Kim J, Suh D, Park T, Kim K,. Effect of metal particle size on coking during CO2
reforming of CH4 over Ni-alumina aerogel catalysts. Appl Catal A-Gen. 2000;
197:191-00.

310
Capítulo 10
Koo K, Roh H, Seo Y, Seo D, Yoon W, Park S. Coke study on MgO-promoted
Ni/Al2O3 catalyst in combined H2O and CO2 reforming of methane for gas to
liquid (GTL) process. Appl Catal A-Gen 2008; 340:183-90.
Koo K, Lee S, Jung U, Roh H, Yoon W. Syngas production via combined steam and
carbon dioxide reforming of methane over Ni-Ce/MgAl2O4 catalysts with
enhanced coke resistance. Fuel Process Technol 2014; 119:151-57.
Kraleva E, Sokolov S, Schneider M, Ehrich H. Support effects on the properties of Co
and Ni catalysts for the hydrogen production from bio-ethanol partial oxidation.
Int J Hydrogen Energ 2013; 38:4380-88.
Kraleva E, Sokolov S, Nasillo G, Bentrup U, Ehrich H. Catalytic performance of
CoAlZn and NiAlZn mixed oxides in hydrogen production by bio-ethanol
partial oxidation. Int J Hydrogen Energ 2014; 39:209-20.
Kudakasseril J, Raveendran G, Hussain A, Vijaya GS. Feedstocks, logistics and pre-
treatment processes for sustainable lignocellulosic biorefineries: A
comprehensive review. Renew Sust Energ Rev 2013; 25:205-19.
Kumar A, Prasad R, Sharma Y. Steam Reforming of Ethanol: Production of
Renewable Hydrogen. Int J Environ Res Dev 2014; 3: 203-12.
Lagarias J, Reeds J, Wright M, Wright P. Convergence properties of the Nelder-Mead
simplex method in low dimensions. SIAM J Optim 1999, 9: 112-47.
Li C, Chen Y. Temperature-programmed-reduction studies of nickel oxide/alumina
catalysts: effects of the preparation method. Thermochim Acta 1995; 256:457-
65.
Li S, Li M, Zhang C, Wang S, Ma X, Gong J. Steam reforming of ethanol over
Ni/ZrO2 catalysts: Effect of support on product distribution. Int J Hydrogen
Energ 2012; 37:2940-49.
Li W, Wang H, Ren Z, Wang G, Bai J. Co-production of hydrogen and multi-wall
carbon nanotubes from ethanol decomposition over Fe/Al2O3 catalysts. Appl
Catal B-Environ 2008; 84:433-39.
Lima da Silva A, Müller IL. Hydrogen production by sorption enhanced steam
reforming of oxygenated hydrocarbons (ethanol, glycerol, n-butanol and
methanol): Thermodynamic modelling. Int J Hydrogen Energ 2011; 36:2057-
75.
Liu Q, Liu Z, Zhou X, Li C, Ding J. Hydrogen production by steam reforming of
ethanol over copper doped Ni/CeO2 catalysts. J Rare Earths 2011; 29:872-77.
Llera I, Mas V, Bergamini ML, Laborde M, Amadeo N. Bio-ethanol steam reforming
on Ni based catalyst. Kinetic study. Chem Eng Sci 2012; 71:356-66.
Lobo LS, Figueiredo JL, Bernardo CA. Carbon formation and gasification on metals.
Bulk diffusion mechanism: A reassessment. Catal Today 2011; 178:110-16.
Lovón A, Lovón-Quintana J, Almerindo G, Valença G, Bernardi M, Araújo V y
cols.Preparation, structural characterization and catalytic properties of Co/CeO2

10. Bibliografía 311
Carolina Montero Calderón
catalysts for the steam reforming of ethanol and hydrogen production. J Power
Sources 2012; 216:281-89.
Lu P, Chen T, Chern J. Reaction network and kinetic analysis of ethanol steam
reforming over a Ru/Al2O3 catalyst. Catal Today 2011; 174:17-24.
Lucredio A, Bellido J, Zawadzki A, Assaf E. Co catalysts supported on SiO2 and γ-
Al2O3 applied to ethanol steam reforming: Effect of the solvent used in the
catalyst preparation method. Fuel 2011; 90:1424-30.
Maia T, Assaf J, Assaf E. Steam reforming of ethanol for hydrogen production on
Co/CeO2-ZrO2 catalysts prepared by polymerization method. Mater Chem Phys
2012; 132:1029-34.
Mas V, Bergamini M, Baronetti G, Amadeo N, Laborde M. A kinetic study of ethanol
steam reforming using a nickel based catalyst. Top Catal 2008a; 51:39-48.
Mas V, Dieuzeide ML, Jobbágy M, Baronetti G, Amadeo N, Laborde M. Ni(II)-
Al(III) layered double hydroxide as catalyst precursor for ethanol steam
reforming: Activation treatments and kinetic studies. Catal Today 2008b; 133-
135:319-23.
Matas B, Babich I., Nichols K, Gardeniers J, Lefferts L, Seshan K. Design of a stable
steam reforming catalyst-A promising route to sustainable hydrogen from
biomass oxygenates. Appl Catal B-Environ 2009; 90:38-44.
Matienzo LJ.Caracterización microscópica de superficies en la ciencia de materiales.
Revista de Química 1996;10:47-70.
Mathure PV, Ganguly S, Patwardhan AV, Saha RK. Steam reforming of ethanol using
a commercial nickel-based catalyst. Ind Eng Chem Res 2007; 46:8471-79.
Melchor-Hernández C, Gómez-Cortés A, Díaz G. Hydrogen production by steam
reforming of ethanol over nickel supported on La-modified alumina catalysts
prepared by sol-gel. Fuel 2013; 107:828-35.
Menon V, Rao M. Trends in bioconversion of lignocellulose: Biofuels, platform
chemicals & biorefinery concept. Prog Energ Combust 2012; 38:522-50.
Mezalira D, Probst L, Pronier S, Batonneau Y, Batiot-Dupeyrat C. Decomposition of
ethanol over Ni/Al2O3 catalysts to produce hydrogen and carbon nanostructured
materials. J Mol Catal A-Chem 2011; 340:15-23.
Mier D, Aguayo AT, Atutxa A, Gayubo AG, Bilbao J. Study of Complex Reactions
under Rapid Deactivation - Improvements in the Reaction Equipment and in the
Methodology for Kinetic Calculation. Int J Chem Reac Eng 2007; 5:Article
A66.
Mier, D. Obtención de olefinas por transformación catalítica de parafinas y metanol en
un proceso integrado. Tesis Doctoral 2009. Universidad del País Vasco, Bilbao.
Mier D, Gayubo AG, Aguayo AT, Olazar M, Bilbao J. Olefin Production by co-
feeding methanol and n-butane. Kinetic modelling considering the deactivation
of HZSM-5 zeolite. AIChE J 2011; 57:2841-53.

312
Capítulo 10
Morgenstern D, Fornango J. Low-temperature reforming of ethanol over copper-
plated raney nickel: A new route to sustainable hydrogen for transportation.
Energy Fuels 2005; 19:1708-16.
Moura S, Souza M, Bellido J, Assaf E, Opportus M, Reyes P y cols.Ethanol steam
reforming over rhodium and cobalt-based catalysts: Effect of the support. Int J
Hydrogen Energ 2012; 37:3213-24.
Mousavi SM, Chan SH. Techno-Economic Study of Hydrogen Production via Steam
Reforming of Methanol, Ethanol, and Diesel. Energ Tech Pol 2014; 1:15-22.
Nahar G, Dupont V. Hydrogen via steam reforming of liquid biofeedstock. Biofuels
2012; 3:167-91.
Nasir Uddin M, Wan Daud W, Abbas H. Kinetics and deactivation mechanisms of the
thermal decomposition of methane in hydrogen and carbon nanofiber Co-
production over Ni-supported Y zeolite-based catalysts. Energ Convers Manage
2014; 87:796-09.
Natesakhawat S, Oktar O, Ozkan US. Effect of lanthanide promotion on catalytic
performance of sol-gel Ni/Al2O3 catalysts in steam reforming of propane. J Mol
Catal A-Chem 2005a; 241:133-46.
Natesakhawat S, Watson R, Wang X, Ozkan U. Deactivation characteristics of
lanthanide-promoted sol-gel Ni/Al2O3 catalysts in propane steam reforming. J
Catal 2005b; 234:496-08.
Ni M, Leung D, Leung M. A review on reforming bio-ethanol for hydrogen
production. Int J Hydrogen Energ 2007; 32:3228-47.
Ni L, Kuroda K, Zhou L, Ohta K, Matsuishi K, Nakamura J. Decomposition of metal
carbides as an elementary step of carbon nanotube synthesis. Carbon 2009;
47:3054-62.
Nichele V, Signoretto M, Pinna F, Menegazzo F, Rossetti I, Cruciani G y cols.Ni/ZrO2
catalysts in ethanol steam reforming: Inhibition of coke formation by CaO-
doping. Appl Catal B-Environ 2014; 150-151:12-20.
Oar-Arteta, L. Desarrollo del catalizador y modelado cinético del reformado con vapor
de dimetil éter. Tesis Doctoral 2014. Universidad del País Vasco, Bilbao.
Oar-Arteta L, Remiro A, Vicente J, Aguayo AT, Bilbao J, Gayubo AG. Stability of
CuZnOAl2O3/HZSM-5 and CuFe2O4/HZSM-5 catalysts in dimethyl ether steam
reforming operating in reaction-regeneration cycles. Fuel Process Technol
2014; 126:145-54.
Ochoa A, Aramburu B, Ibáñez M, Valle B, Bilbao J, Gayubo AG y cols.
Compositional insights and valorization pathways for carbonaceous material
deposited during bio-oil thermal treatment. Chem Sus Chem 2014; 7:2597-08.
Organisation for Economic Cooperation and Development (OECD) and the Food and
Agriculture Organization (FAO) of the United Nations. Agricultural Outlook
2014-2023. 2014.

10. Bibliografía 313
Carolina Montero Calderón
Oswald S, Brückner W. XPS depth profile analysis of non-stoichiometric NiO films.
Surf Interface Anal 2004; 36:17-22.
Palma V, Castaldo F, Ciambelli P, Iaquaniello G, Capitani G. On the activity of
bimetallic catalysts for ethanol steam reforming. Int J Hydrogen Energ 2013;
38:6633-45.
Palma V, Castaldo F, Ciambelli P, Iaquaniello G. CeO2-supported Pt/Ni catalyst for
the renewable and clean H2 production via ethanol steam reforming. Appl Catal
B-Environ 2014; 145:73-84.
Panagiotopoulou P, Verykios XE. Mechanistic aspects of the low temperature steam
reforming of ethanol over supported Pt catalysts. Int J Hydrogen Energ 2012;
37:16333-45.
Pang X, Chen Y, Dai R, Cui P. Co/CeO2 catalysts prepared using citric acid
complexing for ethanol steam reforming. Chinese J Catal 2012; 33:281-89.
Parizotto N, Rocha K, Damyanova S, Passos F, Zanchet D, Marques C y
cols.Alumina-supported Ni catalysts modified with silver for the steam
reforming of methane: Effect of Ag on the control of coke formation. Appl
Catal A-Gen 2007; 330:12-22.
Passos AR, Martins L, Pulcinelli SH, Santilli CV, Briois V. Effect of the balance
between Co(II) and Co(0) oxidation states on the catalytic activity of cobalt
catalysts for Ethanol Steam Reforming. Catal Today 2014; 229:88-94.
Patel M, Jindal T, Pant K. Kinetic study of steam reforming of ethanol on Ni-based
ceria-zirconia catalyst. Ind Eng Chem Res 2013; 52:15763-71.
Pelaez-Samaniego MR, Riveros-Godoy G, Torres-Contreras S, Garcia-Perez T,
Albornoz-Vintimilla E. Production and use of electrolytic hydrogen in Ecuador
towards a low carbon economy. Energy 2014; 64:626-31.
PHyrenees association hydrogen, Small-scale reforming systems for hydrogen
refuelling stations. 2010.
Podolski WF, Young GK. Modeling the Water-Gas Shift Reaction. Ind Eng Chem
Process Des Dev 1974; 13:415-42.
Profeti L, Ticianelli E, Assaf E. Ethanol steam reforming for production of hydrogen
on magnesium aluminate-supported cobalt catalysts promoted by noble metals.
Appl Catal A-Gen 2009; 360:17-25.
Qin F, Anderegg JW, Jenks CJ, Gleeson B, Sordelet DJ, Thiel PA. X-ray
photoelectron spectroscopy studies of the early-stage oxidation behavior of (Pt,
Ni)3Al (1 1 1) surfaces in air. Surf Sci 2008; 602:205-15.
Rabenstein G, Hacker V. Hydrogen for fuel cells from ethanol by steam-reforming,
partial-oxidation and combined auto-thermal reforming: A thermodynamic
analysis. J Power Sources 2008; 185:1293-304.
Raele R, Boaventura JMG, Fischmann AA, Sarturi G. Scenarios for the second
generation ethanol in Brazil. Technol Forecast Soc 2014; 87:205-23.

314
Capítulo 10
Ramos IAC, Montini T, Lorenzut B, Troiani H, Gennari FC, Graziani M y
cols.Hydrogen production from ethanol steam reforming on M/CeO2/YSZ
(M=Ru, Pd, Ag) nanocomposites. Catal Today 2012; 180:96-104.
Remiro, A. Producción de hidrógeno mediante reformado con vapor del bio-oil:
Integración en el proceso de las etapas térmica, catalítica y de captura de
CO2.Tesis Doctoral 2012, Universidad del País Vasco, Bilbao.
Remiro A, Valle B, Aguayo AT, Bilbao J, Gayubo AG. Steam reforming of raw bio-
oil in a fluidized bed reactor with prior separation of pyrolytic lignin. Energy
Fuels 2013a; 27:7549-59.
Remiro A, Valle B, Aguayo AT, Bilbao J, Gayubo AG. Operating conditions for
attenuating Ni/La2O3–αAl2O3 catalyst deactivation in the steam reforming of
bio-oil aqueous fraction. Fuel Process Technol 2013b; 115:222-32.
Renewables Global Status Report. Renewable Energy Policy Network for the 21st
Century 2014. ISBN 978-3-9815934-2-6.
Robertson J. Diamond-like amorphous carbon. Mater Sci Eng 2002; 37:129-81.
Rodrigues P, Schmal M. Nickel-alumina washcoating on monoliths for the partial
oxidation of ethanol to hydrogen production. Int J Hydrogen Energ 2011;
36:10709-18.
Rossetti I, Lasso J, Nichele V, Signoretto M, Finocchio E, Ramis G y cols. Silica and
zirconia supported catalysts for the low-temperature ethanol steam reforming.
Appl Catal B-Environ 2014; 150-151:257-67.
Rossi C, Alonso C, Antunes O, Guirardello R, Cardozo-Filho L. Thermodynamic
analysis of steam reforming of ethanol and glycerine for hydrogen production.
Int J Hydrogen Energ 2009; 34:323-32.
Roy B, Artyushkova K, Pham H, Li L, Datye A, Leclerc C. Effect of preparation
method on the performance of the Ni/Al2O3 catalysts for aqueous-phase
reforming of ethanol: Part II-characterization. Int J Hydrogen Energ 2012;
37:18815-26.
Saebea D, Authayanun S, Patcharavorachot Y, Paengjuntuek W, Arpornwichanop A.
Use of different renewable fuels in a steam reformer integrated into a solid
oxide fuel cell: Theoretical analysis and performance comparison. Energy 2013;
51:305-13.
Sahoo DR, Vajpai S, Patel S, Pant KK. Kinetic modeling of steam reforming of
ethanol for the production of hydrogen over Co/Al2O3 catalyst. Chem Eng J
2007; 125:139-47.
Sánchez-Sánchez MC, Navarro RM, Fierro JLG. Ethanol steam reforming over Ni/La-
Al2O3 catalysts: Influence of lanthanum loading. Catal Today 2007; 129:336-
45.
Sierra I, Ereña J, Aguayo A.T, Olazar M, Bilbao J. Deactivation kinetics for direct
dimethyel ether syntesis on CuO-ZnO_Al2O3/ -Al2O3 catalyst. Ind Eng Chem
Res 2010; 49:481-89.

10. Bibliografía 315
Carolina Montero Calderón
Shaikjee A, Coville NJ. The role of the hydrocarbon source on the growth of carbon
materials. Carbon 2012; 50:3376-98.
Shampine L, Reichelt M. The MATLAB ode suite. SIAM. J Sci Comput 1997; 18:1-
22.
Shampine L, Reichelt M, Kierzenka J. Solving index-I DAEs in MATLAB and
Simulink. SIAM Rev 1999; 41:538-552.
Silveira J, Braga L, de Souza A, Antunes J, Zanzi R. The benefits of ethanol use for
hydrogen production in urban transportation. Renew Sust Energ Rev 2009;
13:2525-34.
Simson A, Farrauto R, Castaldi M. Steam reforming of ethanol/gasoline mixtures:
Deactivation, regeneration and stable performance. Appl Catal B-Environ 2011;
106:295-303.
Smith W, Missen R. Chemical reaction equilibrium analysis: theory and
algorithms.Wiley 1982.
Smith J.M., Introducción a la termodinámica en ingeniería química, McGraw-Hill -
Interamericana de Mexico, 2007.
Song H, Ozkan U. The role of impregnation medium on the activity of ceria-supported
cobalt catalysts for ethanol steam reforming. J Mol Catal A-Chem 2010a;
318:21-29.
Song H, Ozkan U. Economic analysis of hydrogen production through a bio-ethanol
steam reforming process: Sensitivity analyses and cost estimations. Int J
Hydrogen Energ 2010b; 35:127-34.
Song H, Mirkelamoglu B, Ozkan US. Effect of cobalt precursor on the performance of
ceria-supported cobalt catalysts for ethanol steam reforming. Appl Catal A-Gen
2010; 382:58-64.
Soykal II, Bayram B, Sohn H, Gawade P, Miller JT, Ozkan US. Ethanol steam
reforming over Co/CeO2 catalysts: Investigation of the effect of ceria
morphology. Appl Catal A-Gen 2012; 449:47-58.
Sun J, Qiu X, Wu F, Zhu W. H2 from steam reforming of ethanol at low temperature
over and catalysts for fuel-cell application. Int J Hydrogen Energ 2005; 30:437-
45.
Sun S, Yan W, Sun P, Chen J. Thermodynamic analysis of ethanol reforming for
hydrogen production. Energy 2012; 44:911-24.
Szijjártó G, Pászti Z, Sajó I, Erdőhelyi A, Radnóczi G, Tompos A. Nature of the
active sites in Ni/MgAl2O4-based catalysts designed for steam reforming of
ethanol. J Catal 2013; 305:290-306.
Takenaka S, Ogihara H, Otsuka K, Structural change of Ni species in Ni/SiO2 catalyst
during decomposition of methane. J Catal 2002; 208:54-63.

316
Capítulo 10
Takenaka S, Kobayashi S, Ogihara H, Otsuka K. Ni/SiO2 catalyst effective for
methane decomposition into hydrogen and carbon nanofiber. J Catal 2003;
217:79-87.
Teng C, Chiu S. Hydrogen production by partial oxidation of ethanol over Pt/CNT
catalysts. Int J Energy Res 2013; 37:1689-98.
Teng L, Tang T. IR study on surface chemical properties of catalytic grown carbon
nanotubes and nanofibers J Zhejiang Univ Sci A 2008; 9:720-26.
Therdthianwong A, Sakulkoakiet T, Therdthianwong S. Hydrogen Production by
Catalytic Ethanol Steam Reforming. Sci Asia 2001; 27:193-98.
Trane-Restrup R, Dahl S, Jensen A. Steam reforming of ethanol: Effects of support
and additives on Ni-based catalysts. Int J Hydrogen Energ 2013; 38:15105-18.
Trimm DL. Coke formation and minimisation during steam reforming reactions. Catal
Today 1997; 37:233-38.
Trimm DL. Catalysts for the control of coking during steam reforming. Catal Today
1999; 49:3-10.
Uckun Kiran E, Trzcinski AP, Ng WJ, Liu Y. Bioconversion of food waste to energy:
A review. Fuel 2014; 134:389-99.
Vagia E, Lemonidou A. Thermodynamic analysis of hydrogen production via
autothermical steam reforming of selected components of aqueous bio-oil
fraction. Int J Hydrogen Energ 2008; 33:2489-00.
Vaidya PD, Rodrigues AE. Insight into steam reforming of ethanol to produce
hydrogen for fuel cells. Chem Eng J 2006a; 117:39-49.
Vaidya PD, Rodrigues AE. Kinetics of steam reforming of ethanol over a Ru/Al2O3
catalyst. Ind Eng Chem Res 2006b; 45:6614-18.
Valle B, Gayubo A.G, Aguayo A.T, Olazar M, Bilbao J. Selective production of
aromatics by crude bio-oil valorization with a Ni modified HZSM-5 zeolite
catalyst. Energy Fuels 2010; 24:2060-70.
Valle B, Aramburu B, Remiro A, Bilbao J, Gayubo AG. Effect of
calcination/reduction conditions of Ni/La2O3-αAl2O3 catalyst on its activity and
stability for hydrogen production by steam reforming of raw bio-oil/ethanol.
Appl Catal B-Environ 2014a; 147:402-10.
Valle B, Aramburu B, Santiviago C, Bilbao J, Gayubo AG. Upgrading of bio-oil in a
continuous process with dolomite catalyst. Energy Fuels 2014b; 28:6419-28.
Van der Lee M, van Dillen A, Geus J, de Jong K, Bitter J. Catalytic growth of
macroscopic carbon nanofiber bodies with high bulk density and high
mechanical strength. Carbon 2006; 44:529-37.
Vasudeva K., Mitra N., Umasankar P., Dhingra SC. Steam Reforming of Ethanol for
Hydrogen Production: Thermodynamic Analysis. Int J Hydrogen Energ 1996;
21: 13-18.

10. Bibliografía 317
Carolina Montero Calderón
Verykios XE. Catalytic dry reforming of natural gas for the production of chemicals
and hydrogen. Int J Hydrogen Energ 2003; 28:1045-63.
Vicente J. Catalizadores y condiciones de proceso para la producción de hidrógeno
mediante reformado con vapor de dimetil éter y etanol. Universidad del País
Vasco 2012. Tesis Doctoral.
Vicente J, Gayubo AG, Ereña J, Aguayo AT, Olazar M, Bilbao J. Improving the DME
steam reforming catalyst by alkaline treatment of the HZSM-5 zeolite. Appl
Catal B-Environ 2013; 130-131:73-83.
Vicente J, Montero C, Ereña J, Azkoiti MJ, Bilbao J, Gayubo AG. Coke deactivation
of Ni and Co catalysts in ethanol steam reforming at mild temperatures in a
fluidized bed reactor. Int J Hydrogen Energ 2014a; 39:12586-96.
Vicente J, Ereña J, Montero C, Azkoiti MJ, Bilbao J, Gayubo AG. Reaction pathway
for ethanol steam reforming on a Ni/SiO2 catalyst including coke formation. Int
J Hydrogen Energ 2014b; 39:18820-34.
Vivanco, R. Proceso MTO (Metanol a olefinas). Catalizadores alternativos y
modelado cinético sobre SAPO-18. Tesis Doctoral. Universidad del País Vasco
2004, Bilbao.
Wang S, Lu G. A Comprehensive Study on Carbon Dioxide Reforming of Methane
over Ni/γ-Al2O3 Catalysts. Ind Eng Chem Res 1999; 38: 2615-25.
Wang H, Liu Y, Wang L, Qin Y. Study on the carbon deposition in steam reforming
of ethanol over Co/CeO2 catalyst. Chem Eng J 2008; 145:25-31.
Wang W, Wang Y. Dry reforming of ethanol for hydrogen production:
Thermodynamic investigation. Int J Hydrogen Energ 2009; 34:5382-89.
Wang F, Li Y, Cai W, Zhan E, Mu X, Shen W. Ethanol steam reforming over Ni and
Ni-Cu catalysts. Catal Today 2009a; 146:31-36.
Wang H, Wang X, Li M, Li S, Wang S, Ma X. Thermodynamic analysis of hydrogen
production from glycerol autothermal reforming. Int J Hydrogen Energ 2009b;
34:5683-90.
Wang F, Cai W, Provendier H, Schuurman Y, Descorme C, Mirodatos C y
cols.Hydrogen production from ethanol steam reforming over Ir/CeO2 catalysts:
Enhanced stability by PrOx promotion. Int J Hydrogen Energ 2011; 36:13566-
74.
Wang H, Zhang L, Li M, Liu Y, Bai X. Co/CeO2 for ethanol steam reforming: effect
of ceria morphology. J Rare Earths 2013; 31:565-71.
Wragg D, Johnsen R, Balasundaram M, Norby P, Fjellvag H, Gronvold A, y
cols.SAPO-34 methanol-to-olefin catalysts under working conditions: A
combined in situ powder X-ray diffraction, mass spectrometry and Raman
study. J Catal 2009; 268:290-96.
Wu X, Kawi S. Steam reforming of ethanol to H2 over Rh/Y2O3: crucial roles of Y2O3
oxidizing ability, space velocity, and H2/C. Energy Environ Sci 2010; 3:334-42.

318
Capítulo 10
Wu Y, Li P, Yu J, Cunha AF, Rodrigues AE. Sorption-enhanced steam reforming of
ethanol for continuous high-purity hydrogen production: 2D adsorptive reactor
dynamics and process design. Chem Eng Sci 2014a; 118:83-93.
Wu Y, Santos JC, Li P, Yu J, Cunha AF, Rodrigues AE. Simplified kinetic model for
steam reforming of ethanol on a Ni/Al2O3 catalyst. Can J Chem Eng 2014b;
92:116-30.
Xu W, Liu Z, Johnston-Peck AC, Senanayake SD, Zhou G, Stacchiola D y cols. Steam
Reforming of Ethanol on Ni/CeO2: Reaction Pathway and Interaction between
Ni and the CeO2 Support. ACS Catal 2013; 3:975-84.
Yun S, Lim H, Ted Oyama S. Experimental and kinetic studies of the ethanol steam
reforming reaction equipped with ultrathin Pd and Pd-Cu membranes for
improved conversion and hydrogen yield. J Membr Sci 2012; 409-410:222-31.
Zawadzki A, Bellido J, Lucredio A, Assaf E. Dry reforming of ethanol over supported
Ni catalysts prepared by impregnation with methanolic solution. Fuel Process
Technol 2014; 128:432-40.
Zeng G, Gu R, Li Y. The preparation and catalytic behavior of a shell-core Ni/Mg-Al
catalyst for ethanol steam reforming. Int J Hydrogen Energ 2013; 38:11256-67.
Zhang C, Li S, Wu G, Gong J. Synthesis of stable Ni-CeO2 catalysts via ball-milling
for ethanol steam reforming. Catal Today 2014a; 233:53-60.
Zhang C, Li S, Wu G, Huang Z, Han Z, Wang T y cols. Steam reforming of ethanol
over skeletal Ni-based catalysts: A temperature programmed desorption and
kinetic study. AIChE J 2014b; 60:635-44.
Zhou J, Sui Z, Zhu J, Li P, Chen D, Dai Y y cols. Characterization of surface oxygen
complexes on carbon nanofibers by TPD, XPS and FT-IR. Carbon 2007;
45:785-96.

ANEXOS


321
Carolina Montero Calderón
ANEXO A. BALANCE DE MATERIA EN EL REACTOR
Para validar el sistema de análisis cromatográfico y el correcto funcionamiento
de la unidad de reacción, se ha calculado el balance atómico de C, H y O para cada
reacción, así como el balance de materia global tras finalizar cada reacción. En este
Anexo se recogen los cálculos correspondientes a unas condiciones de operación
tomadas como ejemplo: 500 ºC, S/E=6, W/F0=0.18 gcath/gEtOH, PEtOH=0.08 bar, t = 20h.
El procedimiento seguido ha sido el siguiente:
1) A intervalos de tiempo regulares (∼ 7.5 min) se han analizado los productos de
reacción a la salida del reactor en el Micro GC Agilent 3000, cuantificándose de
este modo la composición de todos los productos de reacción, a excepción del
coque depositado sobre la superficie del catalizador. En la Tabla A.1 se muestran
las fracciones molares de los productos de reacción calculadas para diferentes
tiempos de reacción.
2) Una vez cuantificados todos los productos de reacción se ha calculado el balance
de C, H y O a la salida del reactor para cada tiempo de reacción. En la Figura A.1
se muestran las relaciones C/H, C/O y H/O obtenidas, que se mantienen
prácticamente constantes e iguales a las relaciones iniciales correspondientes a los
caudales molares de reactantes alimentados al reactor. Este resultado confirma que
el calibrado del cromatógrafo se ha llevado a cabo correctamente.
3) El cierre del balance de materia se lleva a cabo igualando el termino de salida,
formado por la masa de la corriente de salida gaseosa y de la corriente de salida
líquida y el contenido de coque, al termino de entrada, formado por la masa de
reactantes.
� Masa en la corriente de salida gaseosa: A partir de la composición de la
corriente gaseosa, se calcula su peso molecular promedio, que junto con el
caudal volumétrico medido periódicamente, mediante un medidor de
burbuja, permite calcular la masa total en la corriente gaseosa en cada
intervalo de tiempo. Los resultados se muestran en la Tabla A.2.
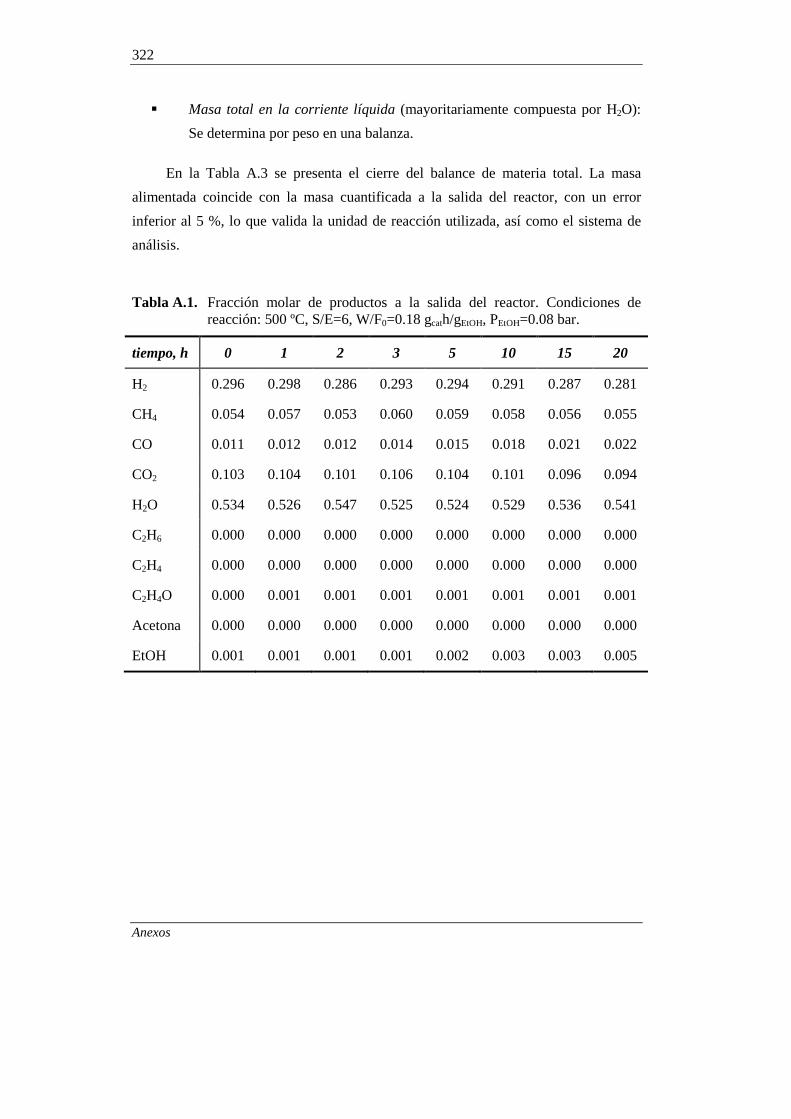
322
Anexos
� Masa total en la corriente líquida (mayoritariamente compuesta por H2O):
Se determina por peso en una balanza.
En la Tabla A.3 se presenta el cierre del balance de materia total. La masa
alimentada coincide con la masa cuantificada a la salida del reactor, con un error
inferior al 5 %, lo que valida la unidad de reacción utilizada, así como el sistema de
análisis.
Tabla A.1. Fracción molar de productos a la salida del reactor. Condiciones de reacción: 500 ºC, S/E=6, W/F0=0.18 gcath/gEtOH, PEtOH=0.08 bar.
tiempo, h 0 1 2 3 5 10 15 20
H2 0.296 0.298 0.286 0.293 0.294 0.291 0.287 0.281
CH4 0.054 0.057 0.053 0.060 0.059 0.058 0.056 0.055
CO 0.011 0.012 0.012 0.014 0.015 0.018 0.021 0.022
CO2 0.103 0.104 0.101 0.106 0.104 0.101 0.096 0.094
H2O 0.534 0.526 0.547 0.525 0.524 0.529 0.536 0.541
C2H6 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000
C2H4 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000
C2H4O 0.000 0.001 0.001 0.001 0.001 0.001 0.001 0.001
Acetona 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000
EtOH 0.001 0.001 0.001 0.001 0.002 0.003 0.003 0.005
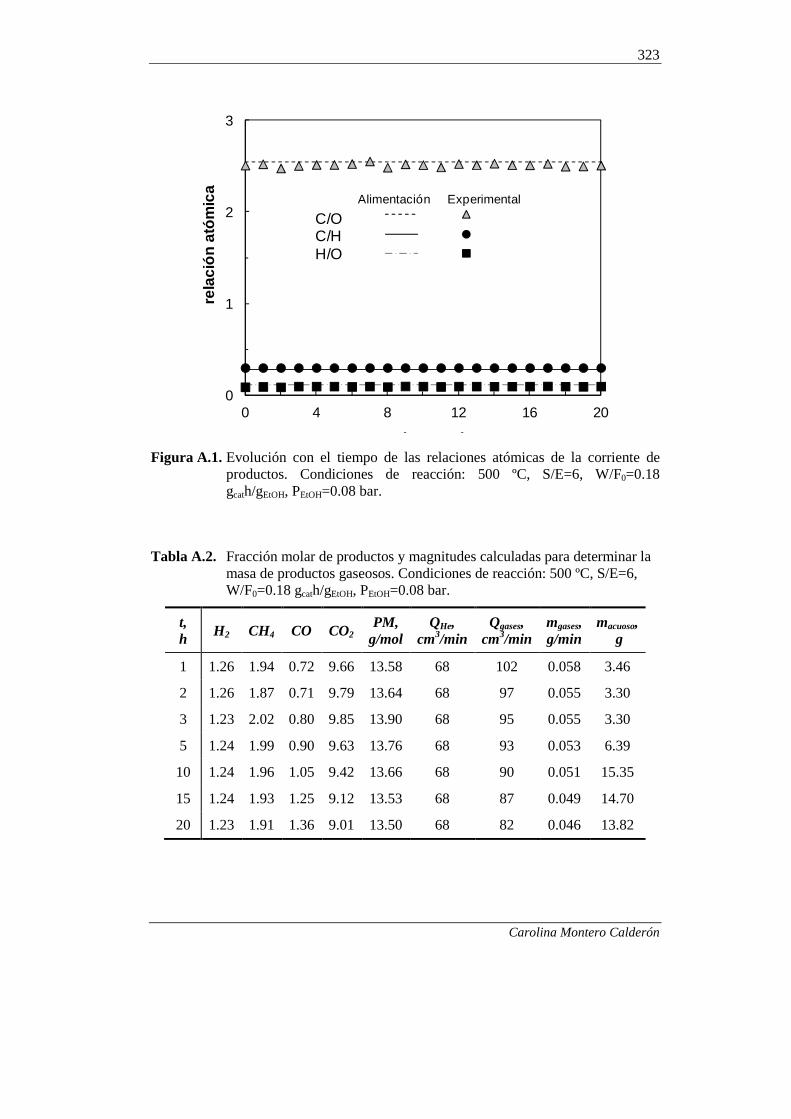
323
Carolina Montero Calderón
Figura A.1. Evolución con el tiempo de las relaciones atómicas de la corriente de productos. Condiciones de reacción: 500 ºC, S/E=6, W/F0=0.18 gcath/gEtOH, PEtOH=0.08 bar.
Tabla A.2. Fracción molar de productos y magnitudes calculadas para determinar la masa de productos gaseosos. Condiciones de reacción: 500 ºC, S/E=6, W/F0=0.18 gcath/gEtOH, PEtOH=0.08 bar.
t, h H2 CH4 CO CO2
PM, g/mol
QHe, cm3/min
Qgases, cm3/min
mgases, g/min
macuoso, g
1 1.26 1.94 0.72 9.66 13.58 68 102 0.058 3.46
2 1.26 1.87 0.71 9.79 13.64 68 97 0.055 3.30
3 1.23 2.02 0.80 9.85 13.90 68 95 0.055 3.30
5 1.24 1.99 0.90 9.63 13.76 68 93 0.053 6.39
10 1.24 1.96 1.05 9.42 13.66 68 90 0.051 15.35
15 1.24 1.93 1.25 9.12 13.53 68 87 0.049 14.70
20 1.23 1.91 1.36 9.01 13.50 68 82 0.046 13.82
0
1
2
3
0 4 8 12 16 20
rela
ción
ató
mic
a
tiempo , h
C/OC/HH/O
Alimentación Experimental
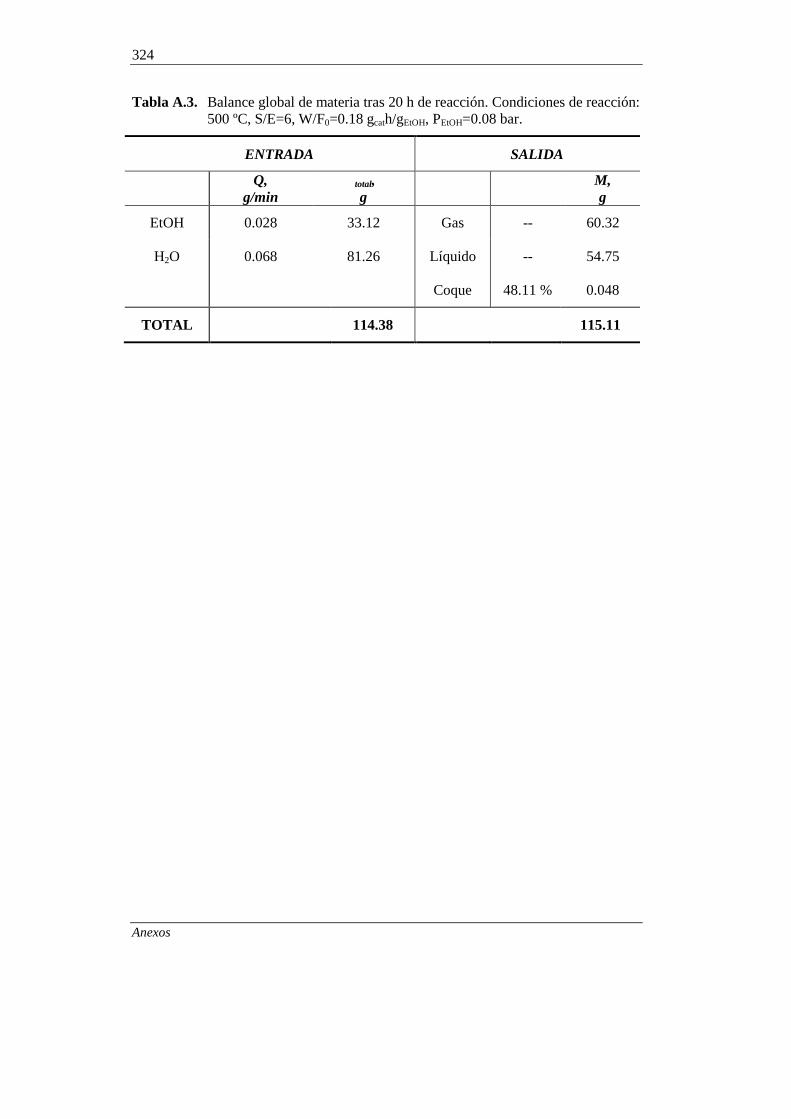
324
Anexos
Tabla A.3. Balance global de materia tras 20 h de reacción. Condiciones de reacción: 500 ºC, S/E=6, W/F0=0.18 gcath/gEtOH, PEtOH=0.08 bar.
ENTRADA SALIDA
Q, g/min
total, g
M, g
EtOH 0.028 33.12 Gas -- 60.32
H2O 0.068 81.26 Líquido -- 54.75
Coque 48.11 % 0.048
TOTAL 114.38 115.11

325
Carolina Montero Calderón
ANEXO B. ESTUDIO TERMODINAMICO DEL PROCESO SRE
B.1. FUNDAMENTOS
Las composiciones en equilibrio pueden calcularse directamente a partir de la
constante de equilibrio de la reacción. Sin embargo, cuando no se conoce este
parámetro, o cuando el proceso involucra una serie de reacciones simultáneas, el
cálculo de las composiciones de equilibrio requiere la minimización de la energía libre
de Gibbs total del sistema (G):
0i
iN
1ii
N
1i
0ii
N
1iii
N
1iii
f
flnnRTGnnGnG ∑+∑=∑ µ=∑=
==== (B.1)
donde: iG es la energía libre de Gibbs parcial molar de la especie i; 0iG es la energía
libre de Gibbs estándar; iµ es el potencial químico; R es la contante universal de los
gases; T y P son la temperatura y presión del sistema; if es la fugacidad de la especie
i en el sistema; 0if es la fugacidad en el estado estacionario y in el numero de moles
de la especie i.
Si la reacción está en equilibrio en fase gas, se considera que:
Pyf iii φ= 00i Pf = y 0
fi0i GG ∆= (B.2)
Aplicando el método de multiplicadores de Lagrange, la energía libre de Gibbs
mínima para cada una de las especies gaseosas y para el sistema total puede calcularse
con las siguientes ecuaciones
0aP
PylnRTG
kikk0
ii0fi =∑λ+
φ+∆ (B.3)
0aP
PylnRTGn
kikk0
ii0fi
N
1ii =
∑λ+
φ+∆∑
= (B.4)
donde: 0fiG∆ es la energía de Gibbs estándar de formación de la especie i; 0P la
presión en condiciones estándar, 101.3 kPa; iy la fracción molar en fase gas; iφ el
coeficiente de fugacidad de la especie i; kλ el multiplicador de Lagrange y ika el
número de átomos del elemento k presente en cada molécula de la especie i.

326
Anexos
El cálculo de las composiciones de equilibrio consiste en determinar los valores
del número de moles de cada especie i, ni, que minimicen G a presión y temperatura
constante, y que además cumpla el balance de masa:
0an ik
N
1ii =∑
= (B:5)
Para el reformado con vapor de etanol los balances de masa pueden realizarse
aplicando el método estequiométrico (que considera todas las reacciones involucradas)
como propusieron Fishtik y cols. (2000), y mediante el método no estequiométrico,
que es independiente de las reacciones involucradas en el proceso y solo es función de
los componentes presentes en el equilibrio. Los dos métodos conducen a resultados
equivalentes (Smith y Missen, 1982), aunque es el método no estequiométrico el de
uso más extendido en la bibliografía.
Para la minimización de la energía libre de Gibbs en el proceso SRE, en la
bibliografía se han utilizado diferentes programas de cálculo como: lenguaje Fortran,
aplicando el método de programación cuadrática secuencial (SQP) (Vasudeva, 1996) o
el algoritmo de hormiga (Sun y cols., 2012); la función Solver de Excel (Lima da
Silva y Müller., 2009); o bien directamente con software comercial de simulación
como AspenTech (Rabenstein y Hacker, 2008), AspenPlus (Khila y cols., 2013),
HySys (Hernández y Kafarov, 2009) o ChemCAD (Wang y cols., 2009).
La bibliografía proporciona información importante sobre la termodinámica. No
obstante, en esta Tesis se ha realizado el estudio termodinámico para disponer de datos
de equilibrio correspondientes a los valores concretos de las variables de operación
(temperatura y relación molar S/E) utilizados, y también para conocer los valores de
las variables de operación que proporcionan el máximo rendimiento de H2 y
cuantificar el requerimiento energético de la operación, utilizando para ello un
software comercial de simulación. Además, se ha considerado oportuno extender
todos los cálculos al proceso de reformado oxidativo con vapor del etanol (OSRE),
para comparar tanto los rendimientos como los requerimientos energéticos de los dos
procesos de reformado del etanol.

327
Carolina Montero Calderón
B.2. PROCEDIMIENTO DE SIMULACIÓN
Se ha realizado la simulación del proceso utilizando el software PRO II 8.3 de
SIMSCI, que cuenta con una extensa base de datos y métodos termodinámicos. En la
simulación se han considerando los siguientes parámetros:
� Reactivos: etanol, vapor de agua (SRE) y oxígeno (OSRE).
� Productos: H2, CO, CO2, CH4 y coque (considerado como carbón en estado
sólido). No se han considerado en la simulación la acetona, acetaldehído, etileno
y etano, dado que son productos intermedios en el esquema de reacción, cuya
ausencia en las condiciones de equilibrio se ha comprobado experimentalmente.
� Presión del sistema: 1 atmósfera.
� Temperatura de reacción: desde 300 a 1000 ºC.
� Relación molar vapor de agua/etanol (S/E): entre 1 y 12
� Relación molar oxígeno etanol (O2/E)= entre 0.0525 y 0.84, que equivalen a 0.25
y 4 moles de aire por mol de etanol, respectivamente.
� Paquete de propiedades termodinámicas: SRKS, paquete propio de SIMSCI
basado en el método de Soave-Redlich-Kwong, que incluye una variación en la
correlación la cual permite predecir con mayor precisión la presión de vapor.
� Tipo de Reactor para el reformado: Reactor Gibbs, que permite resolver los
balances de masa y energía por medio de la minimización de la energía libre de
los componentes de la reacción aplicando el método no estequiométrico.
En la Figura B.1 se muestra un esquema del proceso simulado de SRE U OSRE,
que incluye: un mezclador de la alimentación de etanol y de agua (M1), un evaporador
para el calentamiento de la mezcla (E1) y el reactor de reformado (R1).
Como resultado de la simulación se obtiene el flujo molar de hidrógeno
formado en el equilibrio termodinámico, por flujo molar de etanol alimentado, así
como los flujos molares de los compuestos de carbono y la entalpía de la reacción en
condiciones estándar (∆Hrº), con la cual se han podido determinar los requerimientos
energéticos.

328
Anexos
Figura B.1. Esquema de proceso global utilizado para la simulación de los procesos SRE y OSR.
A partir de los flujos molares calculados se han determinado los rendimientos
de los productos en el equilibrio. El rendimiento de hidrógeno se ha definido referido
al máximo estequiométrico correspondiente a la reacción de reformado, Ec. (1.8):
alimentado etanol demolar flujo 6obtenidoH demolar flujo
R 2H2 ⋅
= (B.6)
El rendimiento de cada componente con carbono (CO, CO2, CH4 y Csólido ), se
cuantifica con la expresión:
alimentado etanol demolar flujo 2
obtenido )C,CH,CO(CO, i componente delmolar flujoRi S42
⋅= (B.7)
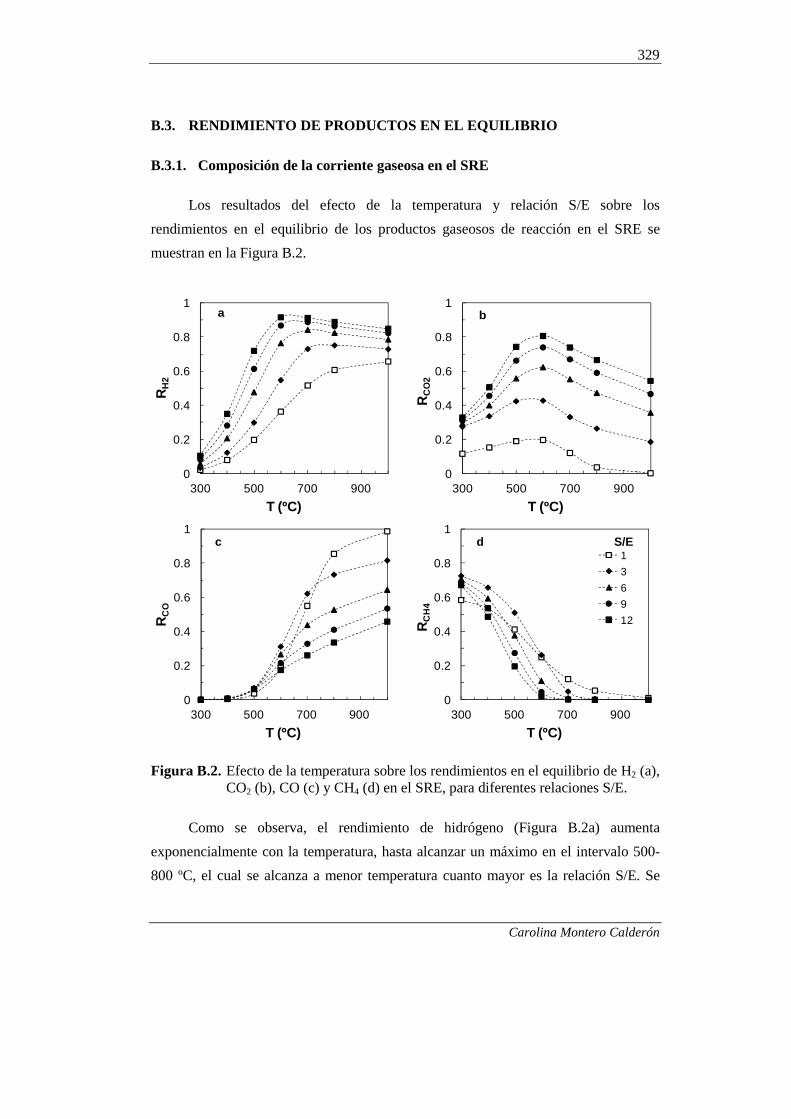
329
Carolina Montero Calderón
B.3. RENDIMIENTO DE PRODUCTOS EN EL EQUILIBRIO
B.3.1. Composición de la corriente gaseosa en el SRE
Los resultados del efecto de la temperatura y relación S/E sobre los
rendimientos en el equilibrio de los productos gaseosos de reacción en el SRE se
muestran en la Figura B.2.
0
0.2
0.4
0.6
0.8
1
300 500 700 900
RH
2
T (ºC)
a
0
0.2
0.4
0.6
0.8
1
300 500 700 900
RC
O2
T (ºC)
b
0
0.2
0.4
0.6
0.8
1
300 500 700 900
RC
O
T (ºC)
c
0
0.2
0.4
0.6
0.8
1
300 500 700 900
RC
H4
T (ºC)
1
3
6
9
12
S/Ed
Figura B.2. Efecto de la temperatura sobre los rendimientos en el equilibrio de H2 (a), CO2 (b), CO (c) y CH4 (d) en el SRE, para diferentes relaciones S/E.
Como se observa, el rendimiento de hidrógeno (Figura B.2a) aumenta
exponencialmente con la temperatura, hasta alcanzar un máximo en el intervalo 500-
800 ºC, el cual se alcanza a menor temperatura cuanto mayor es la relación S/E. Se

330
Anexos
observa que el rendimiento de H2 aumenta notablemente al aumentar la relación S/E
en el intervalo 1-9, si bien para mayor relación S/E el aumento del rendimiento es
poco apreciable. Este es el resultado que cabe esperar, dado que el exceso de agua
desplaza hacia la derecha las reacciones de formación de H2 (reacciones de reformado
y WGS).
El rendimiento de CO2 (Figura B.2b), aumenta en el intervalo de temperatura de
300 - 600 ºC para todas las relaciones S/E, dado que la reacción de reformado de
etanol está favorecida por la temperatura al ser altamente endotérmica. Pero disminuye
a partir de 600 ºC, dado que la reacción WGS, exotérmica, es desfavorecida a altas
temperaturas, lo que conduce a un aumento progresivo del rendimiento de CO con la
temperatura a partir de 600 ºC (Figura B.2c). El aumento de la relación S/E disminuye
el rendimiento de CO y aumenta el de CO2, así como el de H2, dado que se desplaza el
equilibrio de la reacción WGS hacia formación de CO2 + H2.
El rendimiento de CH4 (Figura B.2d) es muy elevado a temperatura baja (< 500
ºC), porque la reacción de metanación está muy favorecida termodinámicamente, y
disminuye notablemente a medida que aumenta temperatura, siendo prácticamente
nulo por encima de 800 ºC para todas las relaciones S/E estudiadas. La rápida
disminución del rendimiento de CH4 está acompañada por un rápido aumento del
rendimiento de CO en el intervalo 400-600 ºC, dado que el aumento de temperatura
desfavorece las reacciones de metanación. Para valores de S/E más altos que el
estequiométrico (S/E=3), el rendimiento de CH4 en el equilibrio disminuye al
aumentar la relación S/E, dado que el exceso de agua desplaza la reacción de
metanación hacia la izquierda (esto es, tiene lugar el reformado del metano).
B.3.2. Composición de la corriente gaseosa en el OSRE
Los resultados del efecto de la temperatura sobre los rendimientos en el
equilibrio de los productos gaseosos en el proceso de reformado oxidativo con vapor
de etanol, para una relación S/E=3 y para diferentes proporciones de oxígeno en la
alimentación, se muestran en la Figura B.3, correspondiendo cada gráfica a un
producto de reacción. Como se observa, el efecto de la temperatura sobre los
rendimientos de H2, CO2, CO y CH4 es el mismo anteriormente comentado para el
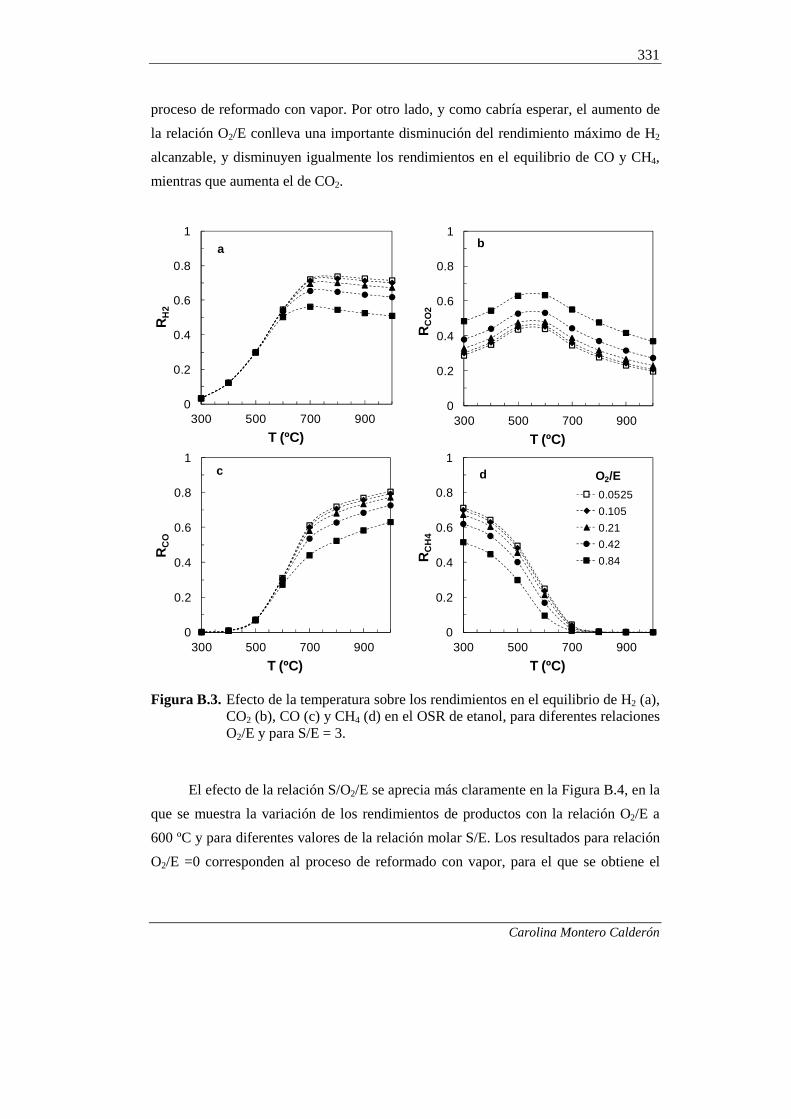
331
Carolina Montero Calderón
proceso de reformado con vapor. Por otro lado, y como cabría esperar, el aumento de
la relación O2/E conlleva una importante disminución del rendimiento máximo de H2
alcanzable, y disminuyen igualmente los rendimientos en el equilibrio de CO y CH4,
mientras que aumenta el de CO2.
0
0.2
0.4
0.6
0.8
1
300 500 700 900
RH
2
T (ºC)
a
0
0.2
0.4
0.6
0.8
1
300 500 700 900R
CO
2
T (ºC)
b
0
0.2
0.4
0.6
0.8
1
300 500 700 900
RC
O
T (ºC)
c
0
0.2
0.4
0.6
0.8
1
300 500 700 900
RC
H4
T (ºC)
0.0525
0.105
0.21
0.42
0.84
O2/Ed
Figura B.3. Efecto de la temperatura sobre los rendimientos en el equilibrio de H2 (a), CO2 (b), CO (c) y CH4 (d) en el OSR de etanol, para diferentes relaciones O2/E y para S/E = 3.
El efecto de la relación S/O2/E se aprecia más claramente en la Figura B.4, en la
que se muestra la variación de los rendimientos de productos con la relación O2/E a
600 ºC y para diferentes valores de la relación molar S/E. Los resultados para relación
O2/E =0 corresponden al proceso de reformado con vapor, para el que se obtiene el
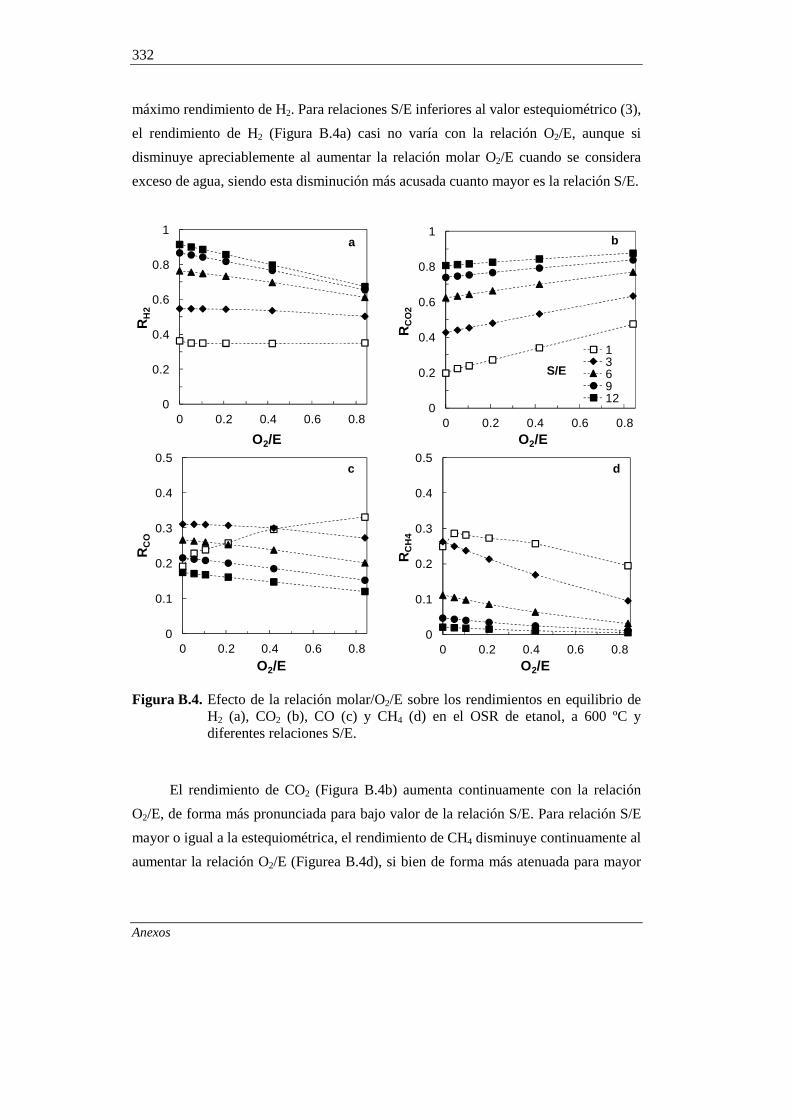
332
Anexos
máximo rendimiento de H2. Para relaciones S/E inferiores al valor estequiométrico (3),
el rendimiento de H2 (Figura B.4a) casi no varía con la relación O2/E, aunque si
disminuye apreciablemente al aumentar la relación molar O2/E cuando se considera
exceso de agua, siendo esta disminución más acusada cuanto mayor es la relación S/E.
0
0.2
0.4
0.6
0.8
1
0 0.2 0.4 0.6 0.8
RH
2
O2/E
a
0
0.2
0.4
0.6
0.8
1
0 0.2 0.4 0.6 0.8
RC
O2
O2/E
136912
b
S/E
0
0.1
0.2
0.3
0.4
0.5
0 0.2 0.4 0.6 0.8
RC
O
O2/E
c
0
0.1
0.2
0.3
0.4
0.5
0 0.2 0.4 0.6 0.8
RC
H4
O2/E
d
Figura B.4. Efecto de la relación molar/O2/E sobre los rendimientos en equilibrio de H2 (a), CO2 (b), CO (c) y CH4 (d) en el OSR de etanol, a 600 ºC y diferentes relaciones S/E.
El rendimiento de CO2 (Figura B.4b) aumenta continuamente con la relación
O2/E, de forma más pronunciada para bajo valor de la relación S/E. Para relación S/E
mayor o igual a la estequiométrica, el rendimiento de CH4 disminuye continuamente al
aumentar la relación O2/E (Figurea B.4d), si bien de forma más atenuada para mayor

333
Carolina Montero Calderón
relación S/E. El rendimiento de CO (Figura B.4c) también disminuye con la relación
O2/E, aunque más atenuadamente que el rendimiento de CH4, y aumenta con la
relación O2/E para relación S/E inferior a la estequiométrica. Esta diferente evolución
de los rendimientos de CO y de CH4 para muy bajo valor de la relación S/E debe
atribuirse a que en tales condiciones se promueven las reacciones de formación de
coque, como se analiza a continuación.
B.3.3. Rendimientos de coque en el SRE y OSRE
En las simulaciones se ha considerado que el coque que se forma es carbón en
estado grafito, ya que el programa de simulación no proporciona otras opciones de
tipos de carbón. En la Figura B.5, que muestra el rendimiento simulado de coque en
los procesos SRE (Figura B.5a) y OSRE (Figura B.5b).
En la Figura B.5a se observa que en el proceso SRE solo se predice la
formación de coque para relaciones S/E menores al valor estequiométrico (S/E=3), y
que en estos casos hay un ligero máximo de rendimiento de coque en el intervalo 500 -
600 ºC. El aumento de la temperatura por encima de 600 ºC conlleva una notable
disminución del rendimiento de coque predicho. En el proceso OSRE (Figura B.5b) la
formación de coque se atenúa, como cabe esperar por la presencia de O2 en la
alimentación, siendo la disminución del rendimiento de coque con la relación O2/E
más pronunciada cuanto mayor es la temperatura de reformado.
Debe indicarse que estos resultados simulados de rendimientos de coque
difieren notablemente de los valores experimentales determinados para el contenido de
coque (Capítulo 5), lo que indica que no es adecuada la suposición de carbono grafito
para cuantificar la formación de coque en el equilibrio, aunque el efecto cualitativo de
las variables de operación sí es predicho adecuadamente por la simulación con el
software utilizado.

334
Anexos
0
0.2
0.4
0.6
0.8
1
300 500 700 900
RC
T (ºC)
1236912
S/Ea
0
0.2
0.4
0.6
0.8
1
300 500 700 900
RC
T (ºC)
0 (SR)0.05250.1050.210.420.84
O2/Eb
Figura B.5. Efecto de la temperatura sobre el rendimiento de coque en el equilibrio en el SRE, con diferentes relaciones S/E (a) y OSRE, con diferentes relaciones O2/E y para relación S/E=1(b).
B.3.4. Comparación con resultados de la bibliografía
Para validar el método de minimización de la energía libre de Gibbs utilizado,
se han comparado los resultados de número de moles de H2 en el equilibrio obtenidos
para el SRE con resultados previos de la bibliografía, para 800, 1000 y 1100 K y
diferentes relaciones S/E. Los resultados se muestran en la Tabla B.1. Se observa una
gran similitud entre los resultados obtenidos en esta Tesis y los obtenidos por otros
autores, para las diferentes relaciones S/E y a las tres temperaturas, lo que da validez
al procedimiento de cálculo utilizado. Las pequeñas diferencias pueden ser atribuibles
a los diferentes métodos utilizados para la minimización de la energía libre de Gibbs.
Tabla B.1. Comparación del número de moles de H2 en el equilibrio calculados en esta Tesis con resultados de la bibliografía para el SRE.
T (K) 800 1000 1100
S/E 1 3 6 9 3 6 9 3 6 9
Sun y cols.(2012) 1.5 2.5 3.8 4.2 4.5 5.1 5.4 4.6 4.9 5.1
Lima da Silva y cols. (2012) 1.2 2.2 3.2 4.2 4.5 4.9 5.3 4.5 4.8 5.1
PRO II (esta Tesis) 1.4 2.2 3.4 4.2 4.5 5 5.3 4.5 4.9 5.1

335
Carolina Montero Calderón
B.4. REQUERIMIENTOS ENERGÉTICOS EN LOS PROCESOS DE
REFORMADO DE ETANOL
Para determinar los requerimientos energéticos globales de los procesos, tanto
en el reformado con vapor como en el reformado oxidativo con vapor, se ha realizado
un balance energético del proceso global. En dichos procesos, el etanol, agua y
oxígeno (para el OSR) entran a una unidad de mezclado y se unen en una misma
corriente, que se precalienta hasta la temperatura de reformado, para luego reaccionar
en el reactor isotermo de Gibbs hasta alcanzar el equilibrio termodinámico y obtener
una corriente de productos que se enfrían de nuevo a la temperatura de referencia,
según se esquematiza en la Figura B.6.
Figura B.6. Esquema del balance energético global en el SRE y OSRE.
Por tanto, el balance de energía global del sistema incluye la energía para el
calentamiento, para el reformado y para el enfriamiento:
Qtotal = Qcalentamiento + Qreformado + Q enfriamiento (B.8)
Se ha asumido que la alimentación y los productos están en condiciones
estándar y que no hay pérdidas de calor en ninguno de los procesos, de modo que la
energía del sistema se puede expresar de la siguiente forma:
Qtotal = -∆Hr0 = np∆Hc,p
0 - nr∆Hc,r0 (B.9)
donde: ∆Hrº es la entalpía de reacción a condiciones estándar (dato que suministra
directamente la simulación del reactor Gibbs de reformado con el software PRO II); n
es el número de moles y ∆Hc0 es la entalpía de combustión, refiriéndose los subíndices
r y p a reactivos y productos, respectivamente.
Calentamiento Reformado Enfriamiento
Qcalentamiento QSREQenfriamiento
Productosde reformado298.15 K1 atm
EtOH+H2O+O2
(OSRE)298.15 K1 atm
QOSRE

336
Anexos
Por conveniencia, para la discusión de los resultados se ha expresado la energía
total del sistema de forma adimensional, mediante la siguiente relación (Sun y cols.,
2012):
0EtOHc,
total
∆H
Q(B.10)
siendo 0EtOHc,∆H el calor de combustión del etanol = (-1235 kJ/mol). De esta forma,
cuando la relación Qtotal/∆cHEtOH es mayor que cero, el sistema global es endotérmico,
mientras que cuando es menor de cero el sistema es exotérmico.
La Figura B.7 muestra los resultados de requerimiento energético neto
adimensional para el reformado con vapor de etanol, calculado para diferentes valores
de temperatura y de la relación S/E. Esta gráfica muestra que el proceso global puede
ser exotérmico a baja temperatura de reformado y baja relación S/E, condiciones en
las que el rendimiento de H2 obtenido en el equilibrio es muy bajo (Figura B.2a),
mientras que para cualquier relación S/E es un proceso claramente endotérmico
operando a altas temperaturas de reformado (> 540 ºC).
Por tanto, al aumentar la temperatura y la relación S/E se requiere mayor aporte
de energía para el reformado con vapor de etanol. Teóricamente se podría alcanzar el
régimen autotérmico (Qtotal/0
EtOHc,∆H =0) para este proceso a 530 °C con S/E=1, y a
menor temperatura a medida que se aumenta la relación S/E.
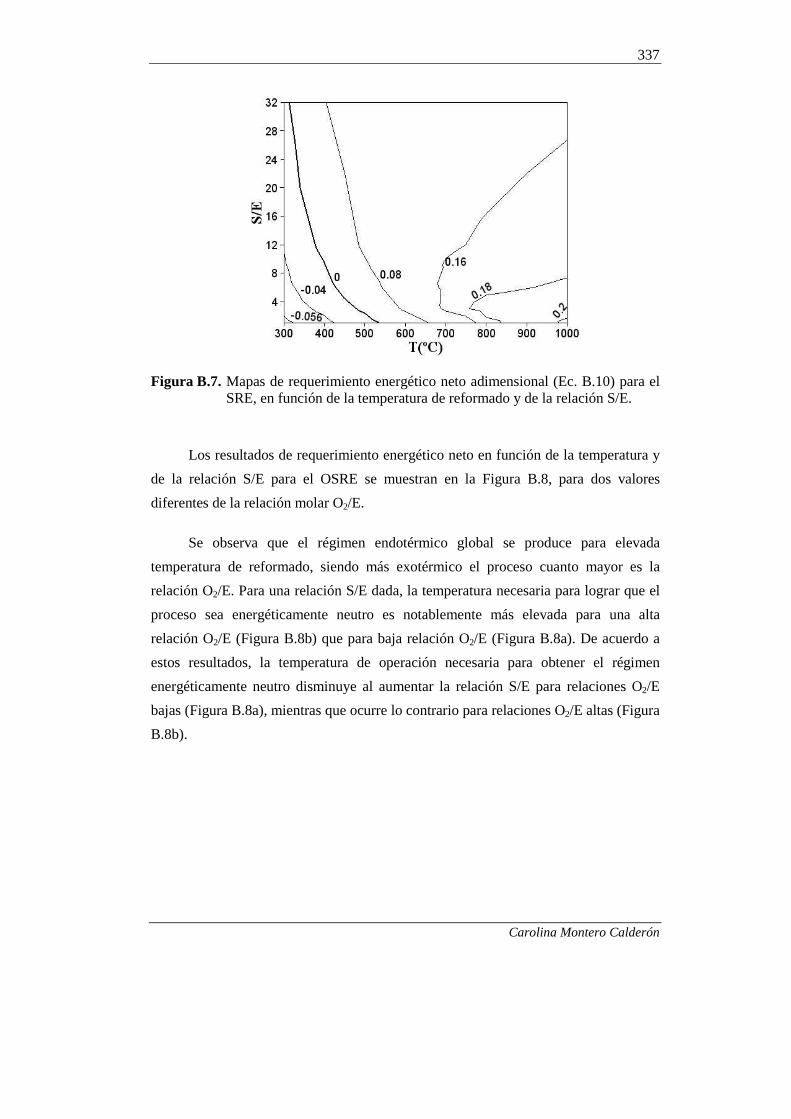
337
Carolina Montero Calderón
Figura B.7. Mapas de requerimiento energético neto adimensional (Ec. B.10) para el SRE, en función de la temperatura de reformado y de la relación S/E.
Los resultados de requerimiento energético neto en función de la temperatura y
de la relación S/E para el OSRE se muestran en la Figura B.8, para dos valores
diferentes de la relación molar O2/E.
Se observa que el régimen endotérmico global se produce para elevada
temperatura de reformado, siendo más exotérmico el proceso cuanto mayor es la
relación O2/E. Para una relación S/E dada, la temperatura necesaria para lograr que el
proceso sea energéticamente neutro es notablemente más elevada para una alta
relación O2/E (Figura B.8b) que para baja relación O2/E (Figura B.8a). De acuerdo a
estos resultados, la temperatura de operación necesaria para obtener el régimen
energéticamente neutro disminuye al aumentar la relación S/E para relaciones O2/E
bajas (Figura B.8a), mientras que ocurre lo contrario para relaciones O2/E altas (Figura
B.8b).
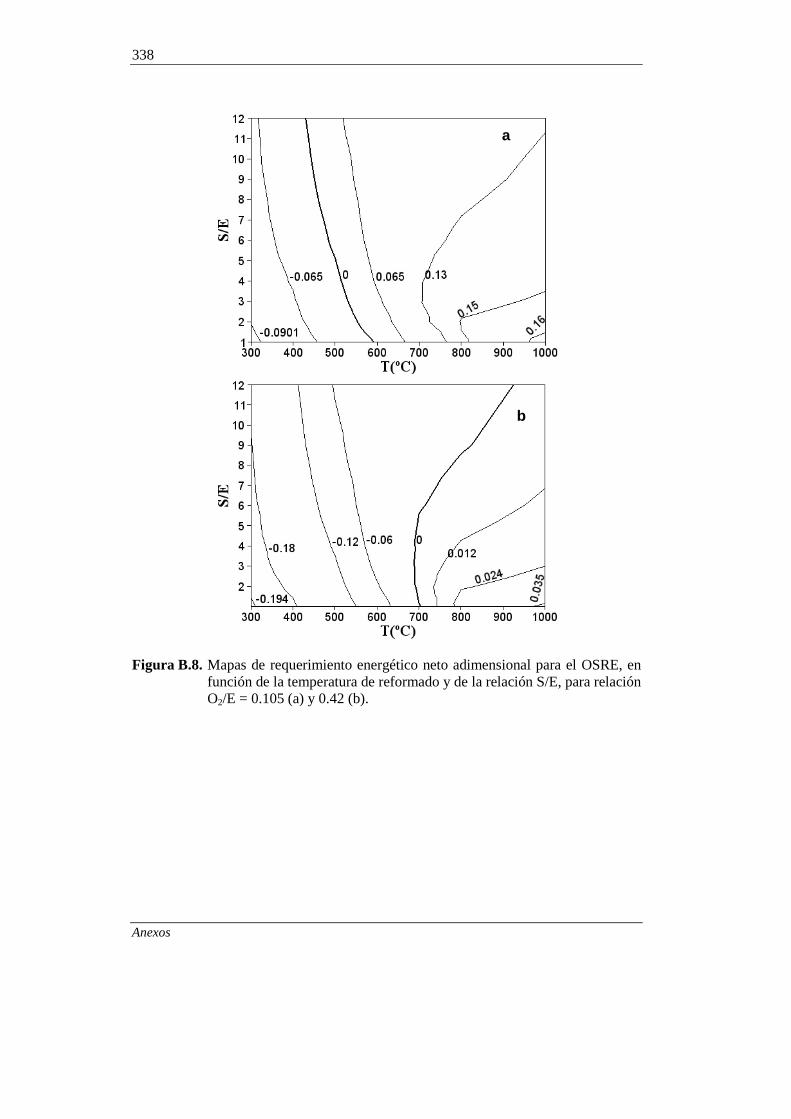
338
Anexos
a
b
Figura B.8. Mapas de requerimiento energético neto adimensional para el OSRE, en función de la temperatura de reformado y de la relación S/E, para relación O2/E = 0.105 (a) y 0.42 (b).

339
Carolina Montero Calderón
ANEXO C. SELECCIÓN DE LA EXPRESIÓN PARA LA FUNCIÓN θθθθ
(ATENUACIÓN POR ADSORCIÓN DE COMPONENTES
DEL MEDIO DE REACCIÓN)
Para seleccionar la expresión o expresiones más adecuadas para cuantificar la
posible atenuación de la velocidad de reacción debido a la adsorción de componentes
del medio de reacción en los centros activos, considerando las etapas del modelo
cinético M1 propuesto (Tabla 6.5), se han considerado varias posibilidades que se
muestran en la Tabla C.1.
El modelo M1A no considera la adsorción de ningún componente del medio de
reacción en la ecuación cinética (es decir, asume reacciones elementales).
El modelo M1B se ha propuesto basándose en el importante efecto observado
de atenuación de la velocidad de reacción al aumentar la relación S/E de la
alimentación, para comprobar si este efecto atenuante es mayor que el debido a la
propia dilución (disminución de Pi) que conlleva el exceso de agua.
El modelo M1C es una modificación del modelo M1B, que incluye para la
reacción WGS el término de adsorción que propusieron Palma y cols. (2014) para esta
reacción, si bien en esta Tesis no se han considerado valores fijos de las constantes de
adsorción, sino variables con la temperatura.
El resto de expresiones para el/los término/s de atenuación θ consisten en
asumir que algunas o todas las etapas son atenuadas por la adsorción de sus
correspondientes reactantes.
El resultados de los ajustes obtenidos con cada modelo se recogen en la Tabla
C.2, y posteriormente se incluye el análisis de significación de las varianzas.
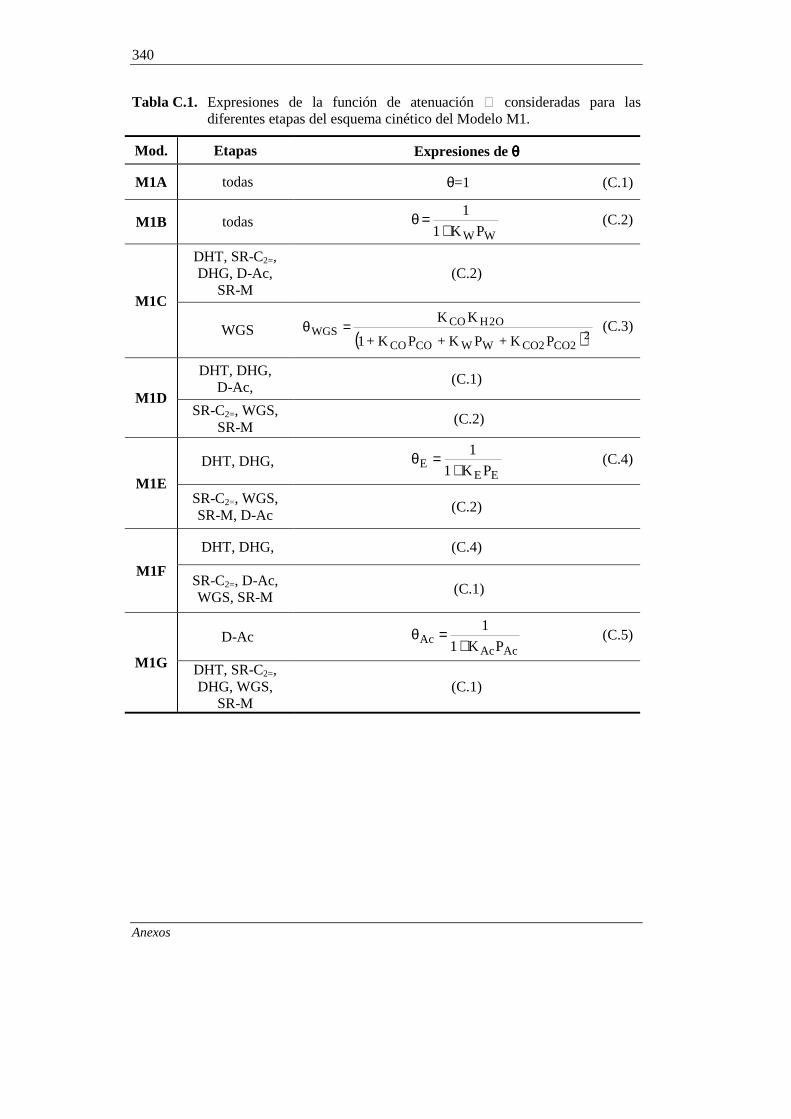
340
Anexos
Tabla C.1. Expresiones de la función de atenuación � consideradas para las diferentes etapas del esquema cinético del Modelo M1.
Mod. Etapas Expresiones de θθθθ
M1A todas θ=1 (C.1)
M1B todas WW PK1
1
+=θ (C.2)
M1C
DHT, SR-C2=, DHG, D-Ac,
SR-M (C.2)
WGS ( )2CO2CO2WWCOCO
O2HCOWGS
PK + PK + PK + 1
KK =θ (C.3)
M1D
DHT, DHG, D-Ac,
(C.1)
SR-C2=, WGS, SR-M
(C.2)
M1E
DHT, DHG, EE
E PK1
1
+=θ (C.4)
SR-C2=, WGS, SR-M, D-Ac
(C.2)
M1F
DHT, DHG, (C.4)
SR-C2=, D-Ac, WGS, SR-M
(C.1)
M1G
D-Ac AcAc
Ac PK1
1
+=θ (C.5)
DHT, SR-C2=, DHG, WGS,
SR-M (C.1)
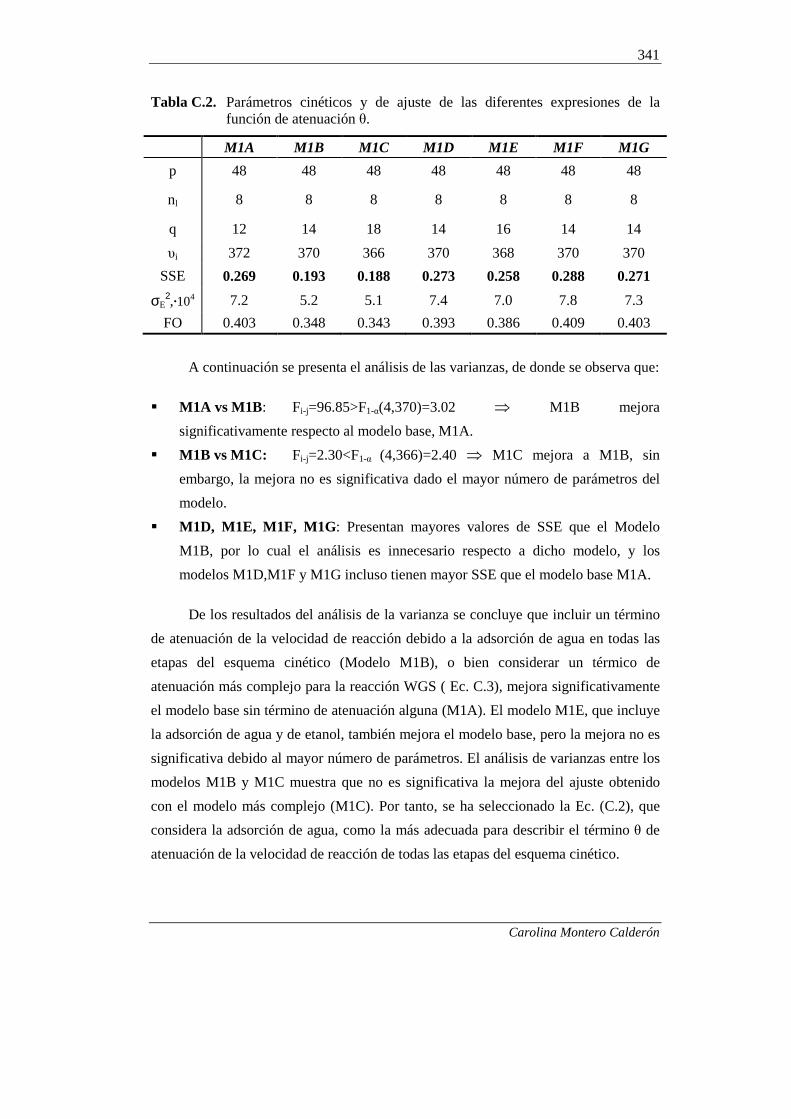
341
Carolina Montero Calderón
Tabla C.2. Parámetros cinéticos y de ajuste de las diferentes expresiones de la función de atenuación θ.
M1A M1B M1C M1D M1E M1F M1G
p 48 48 48 48 48 48 48
nl 8 8 8 8 8 8 8
q 12 14 18 14 16 14 14
υi 372 370 366 370 368 370 370
SSE 0.269 0.193 0.188 0.273 0.258 0.288 0.271
σE2,·104 7.2 5.2 5.1 7.4 7.0 7.8 7.3
FO 0.403 0.348 0.343 0.393 0.386 0.409 0.403
A continuación se presenta el análisis de las varianzas, de donde se observa que:
� M1A vs M1B: Fi-j=96.85>F1-α(4,370)=3.02 ⇒ M1B mejora
significativamente respecto al modelo base, M1A.
� M1B vs M1C: Fi-j=2.30<F1-α (4,366)=2.40 ⇒ M1C mejora a M1B, sin
embargo, la mejora no es significativa dado el mayor número de parámetros del
modelo.
� M1D, M1E, M1F, M1G: Presentan mayores valores de SSE que el Modelo
M1B, por lo cual el análisis es innecesario respecto a dicho modelo, y los
modelos M1D,M1F y M1G incluso tienen mayor SSE que el modelo base M1A.
De los resultados del análisis de la varianza se concluye que incluir un término
de atenuación de la velocidad de reacción debido a la adsorción de agua en todas las
etapas del esquema cinético (Modelo M1B), o bien considerar un térmico de
atenuación más complejo para la reacción WGS ( Ec. C.3), mejora significativamente
el modelo base sin término de atenuación alguna (M1A). El modelo M1E, que incluye
la adsorción de agua y de etanol, también mejora el modelo base, pero la mejora no es
significativa debido al mayor número de parámetros. El análisis de varianzas entre los
modelos M1B y M1C muestra que no es significativa la mejora del ajuste obtenido
con el modelo más complejo (M1C). Por tanto, se ha seleccionado la Ec. (C.2), que
considera la adsorción de agua, como la más adecuada para describir el término θ de
atenuación de la velocidad de reacción de todas las etapas del esquema cinético.

342
Anexos
ANEXO D. SELECCIÓN DE UN MODELO CINÉTICO PARA
CUANTIFICAR LA CONTRIBUCIÓN DE LAS RUTAS
TÉRMICAS
A tenor de los resultados mostrados en el Apartado 4.1, se ha considerado en
principio que todas las reacciones del esquema cinético del modelo de mejor ajuste,
M7, son susceptibles de estar promovidas por rutas térmicas con excepción de la
reacción de reformado de etileno (que contribuye poco al mecanismo global dada la
poca concentración de etileno en el medio). La velocidad de las reacciones
individuales promovidas por el efecto térmico se ha asumido que corresponde a la de
una reacción elemental. Se han planteado varios modelos (que se muestran en la Tabla
D.1) que difieren en el número de reacciones que se considera que están promovidas
por rutas térmicas y en cuales de ellas se considera una misma constante cinética (para
simplificarlo). Los modelos denominados MT1x no consideran que la reacción de
reformado de etanol esté promovida por el efecto térmico, mientras que los modelos
denominados MT2x sí consideran las rutas térmicas para esta reacción.
En Tabla D.2 se presentan los resultados de los ajustes obtenidos entre los
valores calculados con cada modelo y los valores experimentales de la composición
del medio de reacción obtenida en ausencia de catalizador (para diferentes
temperaturas y relaciones S/E utilizadas en los ensayos). Se muestra también el
análisis de varianzas entre pares de modelos, referido al modelo MT1a. Como se
cuenta con el número de grados de libertad < 120, se ha realizado el test de
significación dado por la Ec. (6.57).
Los resultados del análisis de varianzas reflejan que la mejora lograda con los
otros modelos respecto al MT1a no es significativa, principalmente debido a las bajas
concentraciones detectadas para cada uno de los componentes. Sin embargo, el
modelo MT2f es el que presenta menor varianza (σE2=4.08·10-05), y en la Tabla D.3 se
compara la suma de cuadrados residuales para cada componente individual (SSEi) y
total (SSET) de los modelos MT1a y MT2f (de peor y mejor ajuste).
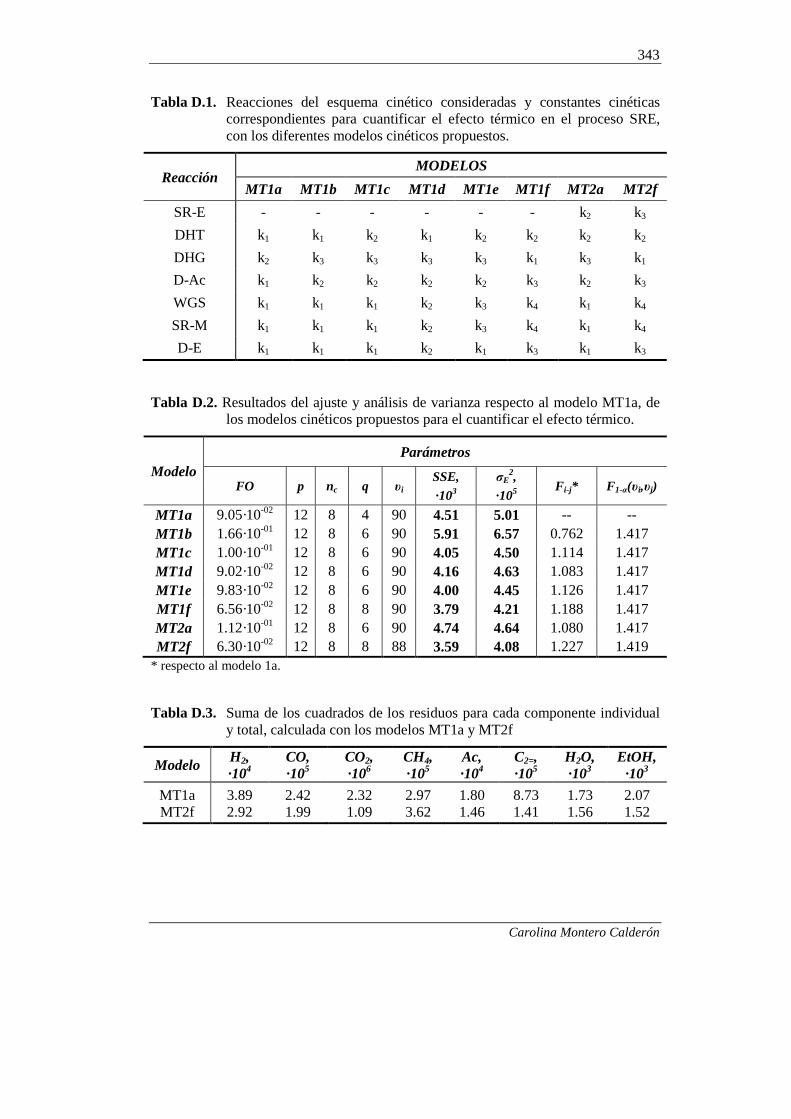
343
Carolina Montero Calderón
Tabla D.1. Reacciones del esquema cinético consideradas y constantes cinéticas correspondientes para cuantificar el efecto térmico en el proceso SRE, con los diferentes modelos cinéticos propuestos.
Reacción MODELOS
MT1a MT1b MT1c MT1d MT1e MT1f MT2a MT2f
SR-E - - - - - - k2 k3
DHT k1 k1 k2 k1 k2 k2 k2 k2
DHG k2 k3 k3 k3 k3 k1 k3 k1
D-Ac k1 k2 k2 k2 k2 k3 k2 k3
WGS k1 k1 k1 k2 k3 k4 k1 k4
SR-M k1 k1 k1 k2 k3 k4 k1 k4
D-E k1 k1 k1 k2 k1 k3 k1 k3
Tabla D.2. Resultados del ajuste y análisis de varianza respecto al modelo MT1a, de los modelos cinéticos propuestos para el cuantificar el efecto térmico.
Modelo Parámetros
FO p nc q υi SSE, σE
2, F i-j* F 1-α(υi,υj) ·103 ·105
MT1a 9.05·10-02 12 8 4 90 4.51 5.01 -- -- MT1b 1.66·10-01 12 8 6 90 5.91 6.57 0.762 1.417 MT1c 1.00·10-01 12 8 6 90 4.05 4.50 1.114 1.417 MT1d 9.02·10-02 12 8 6 90 4.16 4.63 1.083 1.417 MT1e 9.83·10-02 12 8 6 90 4.00 4.45 1.126 1.417 MT1f 6.56·10-02 12 8 8 90 3.79 4.21 1.188 1.417 MT2a 1.12·10-01 12 8 6 90 4.74 4.64 1.080 1.417 MT2f 6.30·10-02 12 8 8 88 3.59 4.08 1.227 1.419
* respecto al modelo 1a.
Tabla D.3. Suma de los cuadrados de los residuos para cada componente individual y total, calculada con los modelos MT1a y MT2f
Modelo H2, ·104
CO, ·105
CO2, ·106
CH4, ·105
Ac, ·104
C2=, ·105
H2O, ·103
EtOH, ·103
MT1a 3.89 2.42 2.32 2.97 1.80 8.73 1.73 2.07 MT2f 2.92 1.99 1.09 3.62 1.46 1.41 1.56 1.52

344
Anexos
Como se observa en la Tabla D.3, el modelo MT2f mejora el ajuste de las
concentraciones de los componentes mayoritarios del medio de reacción (H2 y H2O),
además de mejorar el ajuste del etanol, que es el compuesto que reacciona en el
proceso térmico. También se observa mejora del ajuste de la concentración de
acetaldehído y etileno, que son productos de la deshidrogenación y deshidratación de
etanol, respectivamente. Como se discutió en el Apartado 4.1, estas reacciones se
promueven desde bajas temperaturas.
La comparación de los ajustes para los modelos MT1a y MT2f (peor y mejor
ajuste), se puede observar en la Figura D.1, en la que se han representado los gráficos
de paridad entre valores experimentales y calculados con ambos modelos.
Los parámetros cinéticos de mejor ajuste para las rutas térmicas obtenido con el
modelo MT2f y los intervalos de confianza se recogen en la Tabla D.4, en la cual las
constantes cinéticas k i* corresponden a la temperatura de referencia (600 ºC).
Tabla D.4. Parámetros cinéticos de mejor ajuste del modelo MT2f para cuantificar el efecto térmico en el proceso SRE.
Parámetros MODELO T2f
k1*, mol(gcat·h)-1·atm-1 3.02·10-03 ±1.00·10-04
k2*, mol(gcat·h)-1·atm-1 9.37·10-04 ±8.67·10-05
k3*,mol(gcat·h)-1·atm-1 3.03·10-04 ±4.67·10-05
k4*, mol(gcat·h)-1·atm-2 9.22·10-04 ±3.24·10-04
E1, kcal·mol-1 20.50 ±0.65
E2, kcal·mol-1 32.00 ±8.66
E3, kcal·mol-1 38.90 ±2.23
E4, kcal·mol-1 3.52 ±0.82
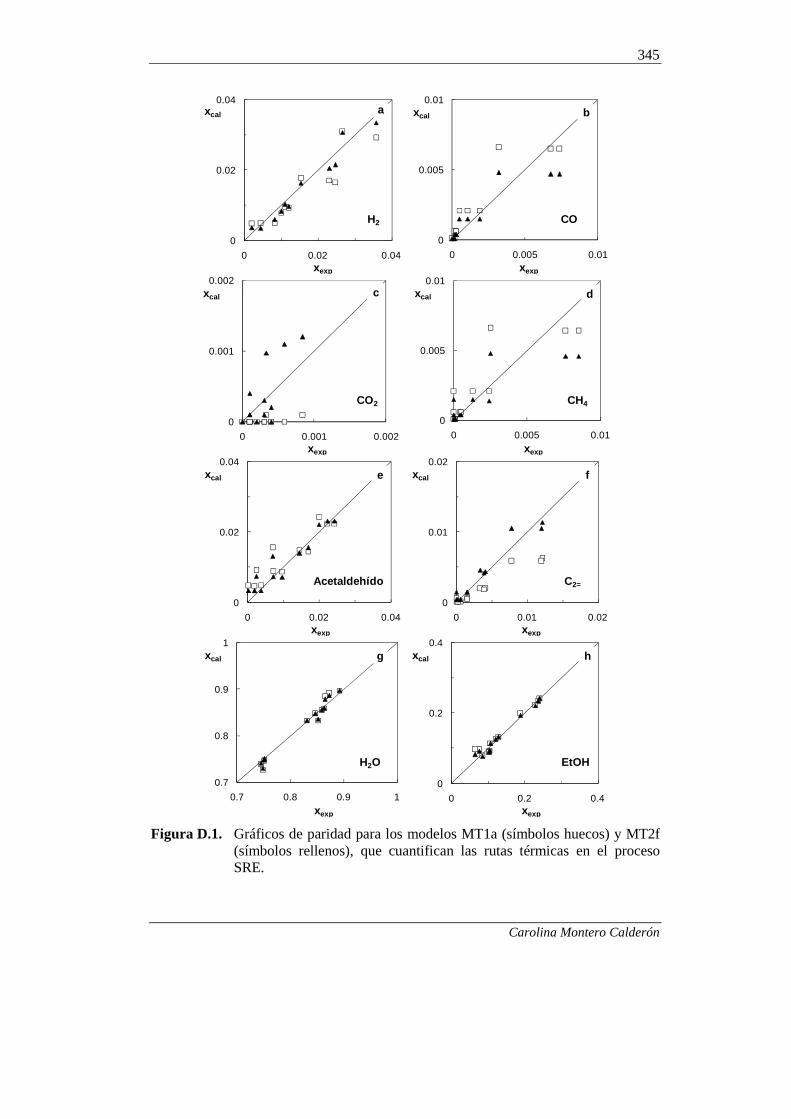
345
Carolina Montero Calderón
Figura D.1. Gráficos de paridad para los modelos MT1a (símbolos huecos) y MT2f (símbolos rellenos), que cuantifican las rutas térmicas en el proceso SRE.
0
0.02
0.04
0 0.02 0.04
xcal
xexp
H2
a
0
0.005
0.01
0 0.005 0.01
xcal
xexp
CO
b
0
0.001
0.002
0 0.001 0.002
xcal
xexp
CO2
c
0
0.005
0.01
0 0.005 0.01
xcal
xexp
CH4
d
0
0.02
0.04
0 0.02 0.04
xcal
xexp
Acetaldehído
e
0
0.01
0.02
0 0.01 0.02
xcal
xexp
C2=
f
0.7
0.8
0.9
1
0.7 0.8 0.9 1
xcal
xexp
H2O
g
0
0.2
0.4
0 0.2 0.4
xcal
xexp
EtOH
h



















