deficit alfa-1-antitripsina
14
(alpha-1 antiproteinasa, antitripsina) ALFA 1-ANTITRIPSINA ( AAT/A1AT) o α 1 -antitripsina (α 1 AT) Es una proteína (serina) que actúa como antiproteasa, previniendo a los tejidos del daño causado por las enzimas proteasas (elastasas, catalasas y antitrombinas)
-
Upload
teofanes-vargas-salcedo -
Category
Health & Medicine
-
view
221 -
download
0
Transcript of deficit alfa-1-antitripsina

(alpha-1 antiproteinasa, antitripsina)
ALFA 1-ANTITRIPSINA( AAT/A1AT) o α1-antitripsina (α1AT)
Es una proteína (serina) que actúacomo antiproteasa, previniendo alos tejidos del daño causado porlas enzimas proteasas (elastasas,catalasas y antitrombinas)
• El 90 % de la síntesis de AAT serealiza en el hígado desde dóndeescapa a la circulación general.
• Se expresa en hígado, neutrófilos ymonocitos.
• La AAT sérica constituye el 90 % dela alfa-1 del proteinograma.
(Laurell y Erickson 1963).

• Esta proteína es codificada por un gen,llamado inhibidor de proteasa (Pi).
• La proteína tiene 394 aminoácidos.
• Hay reconocidos aprox. 100 alelos
diferentes.
Protege a los tejidos delas proteasas presentesprincipalmente enlas células inflamatorias,en especial la elastasa.Está presente enla sangre humana de 1,5 -3,5 gramos/litro.
Es una alteración autosómica recesiva fenotípicamente y autosómica codominante
genotípicamente
Mutación puntual o mutación delectura errónea porque altera lalectura de la cadena codificadoradel gen al especificar unaminoácido diferente.
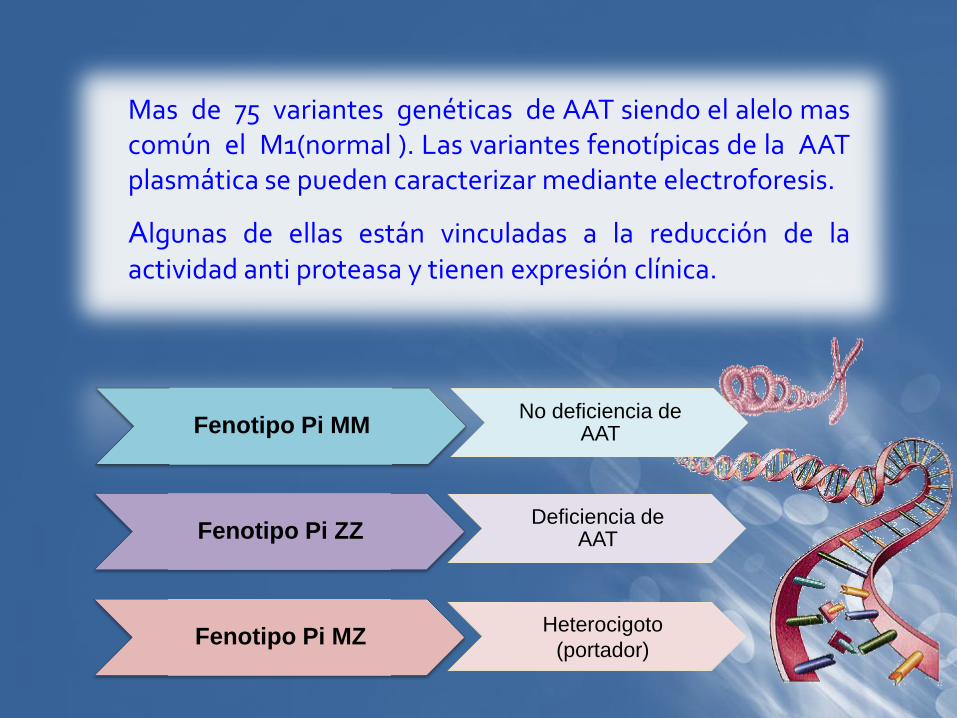
Mas de 75 variantes genéticas de AAT siendo el alelo mascomún el M1(normal ). Las variantes fenotípicas de la AATplasmática se pueden caracterizar mediante electroforesis.
Algunas de ellas están vinculadas a la reducción de laactividad anti proteasa y tienen expresión clínica.
Fenotipo Pi MM
Fenotipo Pi ZZ
Fenotipo Pi MZ
No deficiencia de AAT
Deficiencia de AAT
Heterocigoto
(portador)

El fenotipo Pi ZZ está caracterizado por una sustitución de unaminoácido (ac. glu-lys en posición 342). Se pliega de formaanómala y bloquea su movimiento por vía secretora.
Esto causa polimerización del péptido impidiendo suliberación desde el hígado a la circulación general. Laacumulación en el RER en el 10% de los pacientes, esta
asociado con daño hepático.

La deficiencia de AAT es un trastorno genético que puedeocasionar enfermedad pulmonar obstructiva crónica,principalmente enfisema precoz, en la tercera y cuarta décadade vida. La otra manifestación más frecuente es la enfermedadcrónica del hígado, la cual puede afectar a recién nacidos, niños yadultos.
• El déficit de AAT es la segunda causa de enfermedadpulmonar de naturaleza genética en orden deimportancia.
• La prevalencia en población caucásica se estima en1/1600 a 1/4500. rara en personas asiáticos y negras.
• Población del norte de Europa o península ibérica

Niveles plasmáticos de AAT
• Los niveles normales son 20 a 48 µM/L, (150-350 mg/dl)
• Niveles por encima de 11 µM/L son consideradosprotectores.
• Niveles por debajo de 2.5 a 7 µM/L (20-45 mg/dl), estánvinculados a una actividad deficiente
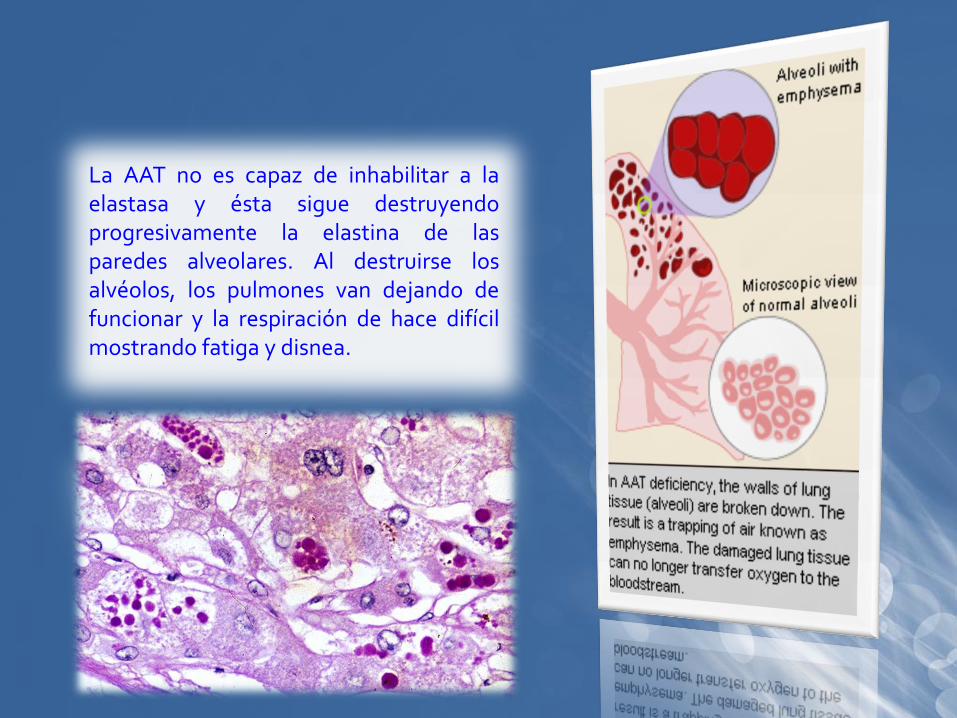
La AAT no es capaz de inhabilitar a laelastasa y ésta sigue destruyendoprogresivamente la elastina de lasparedes alveolares. Al destruirse losalvéolos, los pulmones van dejando defuncionar y la respiración de hace difícilmostrando fatiga y disnea.
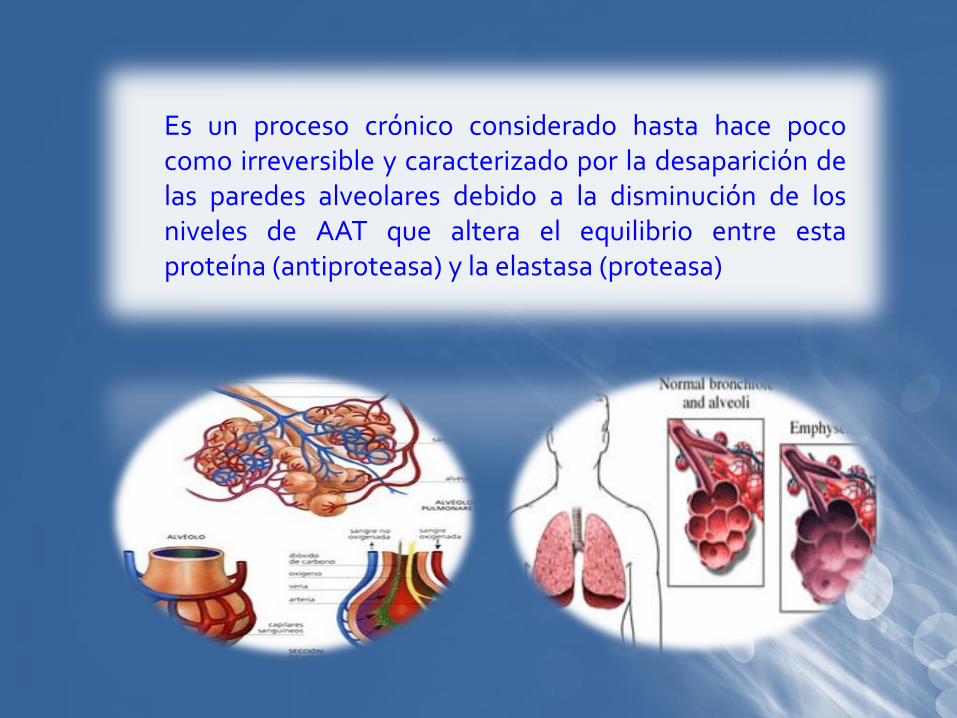
Es un proceso crónico considerado hasta hace pococomo irreversible y caracterizado por la desaparición delas paredes alveolares debido a la disminución de losniveles de AAT que altera el equilibrio entre estaproteína (antiproteasa) y la elastasa (proteasa)

La Deficiencia de Alfa 1-Antitripsina es la principal causagenética de enfermedad hepática, misma que genera cercade la mitad de los trasplantes de hígado que se realizan en lapoblación pediátrica; además de ser considerada laenfermedad genética más frecuente para la que se requieretransplantes de hígado.
La proporción de la AAT como expresión desu mutación, impide agregarse en el RERde los hepatocitos ( enfermedad hepática).
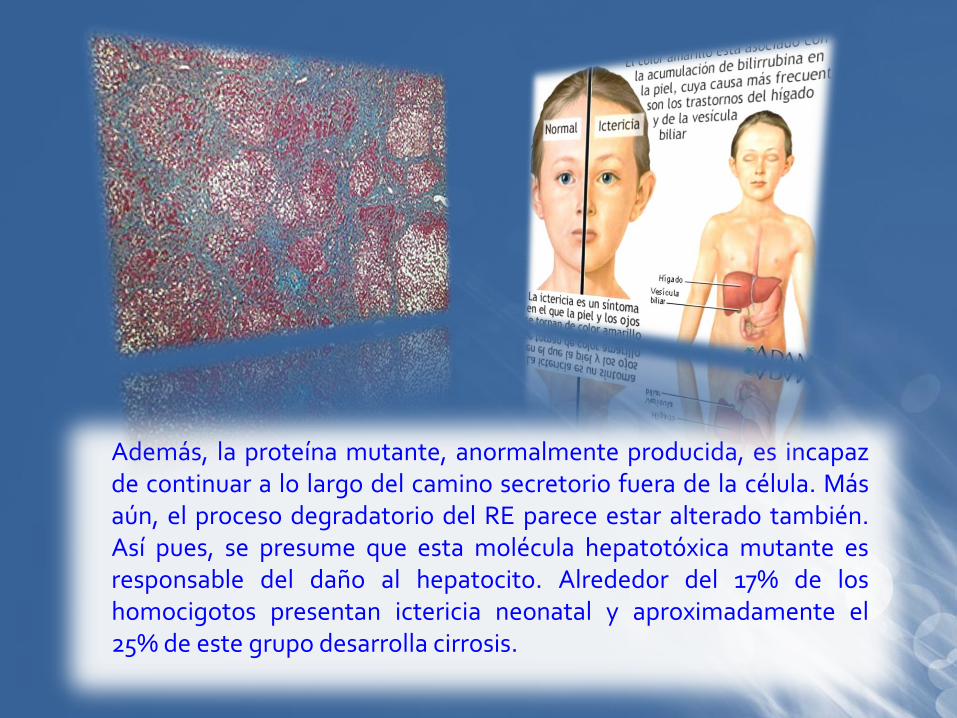
Además, la proteína mutante, anormalmente producida, es incapazde continuar a lo largo del camino secretorio fuera de la célula. Másaún, el proceso degradatorio del RE parece estar alterado también.Así pues, se presume que esta molécula hepatotóxica mutante esresponsable del daño al hepatocito. Alrededor del 17% de loshomocigotos presentan ictericia neonatal y aproximadamente el25% de este grupo desarrolla cirrosis.

• Eric A. Wulfs. Diane E. Hoffmann JD. Maimon M. Déficit de alfa 1 antitripsina.Impacto del descubrimiento genético sobre la medicina y la sociedad. JAMA1994; 271: 217-222.
• Byth BC. Billingsley GD. Cox DW. Physical and genetic mapping of the serpingene cluster at 14q32.1.: allelic association and a unique haplotype associatedwith a 1-antitrypsin deficiency. Am J Hum Genet 1994; 55: 126-133.