Chronic loss of noradrenergic tone produces -arrestin2 ...
14
Chronic loss of noradrenergic tone produces β-arrestin2-mediated cocaine hypersensitivity and alters cellular D2 responses in the nucleus accumbens Meriem Gaval-Cruz 1 *, Richard B. Goertz 2 *, Daniel J. Puttick 1 , Dawn E. Bowles 3 , Rebecca C. Meyer 4 , Randy A. Hall 4 , Daijin Ko 5 , Carlos A. Paladini 2 & David Weinshenker 1 Department of Human Genetics, Emory University School of Medicine, Atlanta, GA, USA 1 , Department of Biology, Neurosciences Institute, University of Texas at San Antonio, San Antonio, TX, USA 2 , Department of Surgery, Duke University School of Medicine, Durham, NC, USA 3 , Department of Pharmacology, Emory University School of Medicine, Atlanta, GA, USA 4 and Department of Management Science and Statistics, University of Texas at San Antonio, San Antonio, TX, USA 5 ABSTRACT Cocaine blocks plasma membrane monoamine transporters and increases extracellular levels of dopamine (DA), norepinephrine (NE) and serotonin (5-HT).The addictive properties of cocaine are mediated primarily by DA, while NE and 5-HT play modulatory roles. Chronic inhibition of dopamine β-hydroxylase (DBH), which converts DA to NE, increases the aversive effects of cocaine and reduces cocaine use in humans, and produces behavioral hypersensitivity to cocaine and D2 agonism in rodents, but the underlying mechanism is unknown. We found a decrease in β-arrestin2 (βArr2) in the nucleus accumbens (NAc) following chronic genetic or pharmacological DBH inhibition, and overexpression of βArr2 in the NAc normalized cocaine-induced locomotion in DBH knockout (Dbh −/−) mice. The D2/3 agonist quinpirole decreased excitability in NAc medium spiny neurons (MSNs) from control, but not Dbh −/− animals, where instead there was a trend for an excitatory effect. The Gαi inhibitor NF023 abolished the quinpirole- induced decrease in excitability in control MSNs, but had no effect in Dbh −/− MSNs, whereas the Gαs inhibitor NF449 restored the ability of quinpirole to decrease excitability in Dbh −/− MSNs, but had no effect in control MSNs. These results suggest that chronic loss of noradrenergic tone alters behavioral responses to cocaine via decreases in βArr2 and cellular responses to D2/D3 activation, potentially via changes in D2-like receptor G-protein coupling in NAc MSNs. Keywords Cocaine, D2 receptor, dopamine, dopamine β-hydroxylase, mice, norepinephrine. Correspondence to: David Weinshenker, Department of Human Genetics, Emory University School of Medicine, Whitehead 301, 615 Michael St., Atlanta, GA 30322, USA. E-mail: [email protected] INTRODUCTION Dopamine β-hydroxylase (DBH) is the enzyme that converts dopamine (DA) to norepinephrine (NE) in noradrenergic neurons, thereby controlling NE produc- tion and the DA/NE ratio (Weinshilboum 1978). DBH is of clinical interest in cocaine dependence because: (1) polymorphisms in the human DBH gene that are associ- ated with reduced serum DBH enzymatic activity lead to greater cocaine-induced paranoia (Cubells et al. 2000; Kalayasiri et al. 2007); and (2) inhibition of DBH by the alcoholism medication, disulfiram, or the selective DBH inhibitor, nepicastat (Stanley et al. 1997; Kapoor, Shandilya & Kundu 2011), alters the subjective effects of cocaine and reduces cocaine use in humans (Stanley et al. 1997; Gaval-Cruz & Weinshenker 2009) (K. Cunningham, pers. comm.). Genetic (DBH knockout; Dbh −/−) or pharmacological (disulfiram, nepicastat) DBH inhibition produces hypersensitivity to cocaine-induced locomotion, stereotypy, place preference and place aver- sion in mice; it also enhances the discriminative stimulus effects of cocaine and attenuates cocaine-, cue- and stress-induced reinstatement of cocaine seeking in rats (Schank et al. 2006; Schroeder et al. 2010, 2013; Gaval-Cruz et al. 2012; Manvich, Depoy & Weinshenker 2013). *MGC and RBG contributed equally to this work. Addiction Biology doi:10.1111/adb.12174 © 2014 Society for the Study of Addiction Addiction Biology, 21, 35 48 – PRECLINICAL STUDY
Transcript of Chronic loss of noradrenergic tone produces -arrestin2 ...

Chronic loss of noradrenergic tone producesβ-arrestin2-mediated cocaine hypersensitivity andalters cellular D2 responses in the nucleus accumbens
Meriem Gaval-Cruz1*, Richard B. Goertz2*, Daniel J. Puttick1, Dawn E. Bowles3,Rebecca C. Meyer4, Randy A. Hall4, Daijin Ko5, Carlos A. Paladini2 & David Weinshenker1
Department of Human Genetics, Emory University School of Medicine, Atlanta, GA, USA1, Department of Biology, Neurosciences Institute, University of Texasat San Antonio, San Antonio, TX, USA2, Department of Surgery, Duke University School of Medicine, Durham, NC, USA3, Department of Pharmacology,Emory University School of Medicine, Atlanta, GA, USA4 and Department of Management Science and Statistics, University ofTexas at San Antonio, San Antonio,TX, USA5
ABSTRACT
Cocaine blocks plasma membrane monoamine transporters and increases extracellular levels of dopamine (DA),norepinephrine (NE) and serotonin (5-HT). The addictive properties of cocaine are mediated primarily by DA, while NEand 5-HT play modulatory roles. Chronic inhibition of dopamine β-hydroxylase (DBH), which converts DA to NE,increases the aversive effects of cocaine and reduces cocaine use in humans, and produces behavioral hypersensitivityto cocaine and D2 agonism in rodents, but the underlying mechanism is unknown. We found a decrease in β-arrestin2(βArr2) in the nucleus accumbens (NAc) following chronic genetic or pharmacological DBH inhibition, andoverexpression of βArr2 in the NAc normalized cocaine-induced locomotion in DBH knockout (Dbh −/−) mice. TheD2/3 agonist quinpirole decreased excitability in NAc medium spiny neurons (MSNs) from control, but not Dbh −/−animals, where instead there was a trend for an excitatory effect. The Gαi inhibitor NF023 abolished the quinpirole-induced decrease in excitability in control MSNs, but had no effect in Dbh −/− MSNs, whereas the Gαs inhibitor NF449restored the ability of quinpirole to decrease excitability in Dbh −/− MSNs, but had no effect in control MSNs. Theseresults suggest that chronic loss of noradrenergic tone alters behavioral responses to cocaine via decreases inβArr2 and cellular responses to D2/D3 activation, potentially via changes in D2-like receptor G-protein couplingin NAc MSNs.
Keywords Cocaine, D2 receptor, dopamine, dopamine β-hydroxylase, mice, norepinephrine.
Correspondence to: David Weinshenker, Department of Human Genetics, Emory University School of Medicine, Whitehead 301, 615 Michael St., Atlanta,GA 30322, USA. E-mail: [email protected]
INTRODUCTION
Dopamine β-hydroxylase (DBH) is the enzyme thatconverts dopamine (DA) to norepinephrine (NE) innoradrenergic neurons, thereby controlling NE produc-tion and the DA/NE ratio (Weinshilboum 1978). DBH isof clinical interest in cocaine dependence because: (1)polymorphisms in the human DBH gene that are associ-ated with reduced serum DBH enzymatic activity lead togreater cocaine-induced paranoia (Cubells et al. 2000;Kalayasiri et al. 2007); and (2) inhibition of DBH by thealcoholism medication, disulfiram, or the selective DBHinhibitor, nepicastat (Stanley et al. 1997; Kapoor,
Shandilya & Kundu 2011), alters the subjective effects ofcocaine and reduces cocaine use in humans (Stanleyet al. 1997; Gaval-Cruz & Weinshenker 2009) (K.Cunningham, pers. comm.). Genetic (DBH knockout; Dbh−/−) or pharmacological (disulfiram, nepicastat) DBHinhibition produces hypersensitivity to cocaine-inducedlocomotion, stereotypy, place preference and place aver-sion in mice; it also enhances the discriminative stimuluseffects of cocaine and attenuates cocaine-, cue- andstress-induced reinstatement of cocaine seeking inrats (Schank et al. 2006; Schroeder et al. 2010, 2013;Gaval-Cruz et al. 2012; Manvich, Depoy & Weinshenker2013).
*MGC and RBG contributed equally to this work.
bs_bs_bannerAddiction Biologydoi:10.1111/adb.12174
© 2014 Society for the Study of Addiction Addiction Biology, 21, 35 48–
PRECLINICAL STUD Y

Because Dbh −/− mice are hypersensitive to the D2/3agonist, quinpirole, but not the D1 agonist, SKF81297,cocaine hypersensitivity would appear to be mediated byalterations in the D2 pathway (Weinshenker et al. 2002;Schank et al. 2006). These phenotypes are likely drivenby compensatory responses in DA signaling following thechronic decrease in extracellular DA availability whennoradrenergic excitatory drive on the mesocorticolimbicsystem is missing. We initially reported an increase in theabundance of high-affinity state D2 receptors in thestriatum of Dbh −/− mice, which could explain thecocaine and D2 hypersensitivity (Schank et al. 2006).However, subsequent work failed to confirm this finding(Skinbjerg et al. 2010), suggesting a contribution fromdownstream signaling molecules. Indeed, the behavioralalterations in Dbh −/− mice were accompanied by a rise instriatal pERK and ΔFosB protein levels (Rommelfangeret al. 2007).
The goals of the present study were to determine themolecular and cellular mechanisms behind the D2- andpsychostimulant-induced hypersensitivity that followchronic DBH inhibition. First, we found a decrease ofβ-arrestin2 (βArr2), a protein involved in D2 desensitiza-tion and signaling (Beaulieu & Gainetdinov 2011), in thenucleus accumbens (NAc) of Dbh −/− mice and micetreated chronically with nepicastat. We next used viral-mediated overexpression to determine whether increas-ing βArr2 levels in the NAc could normalize cocaine-induced behavior in Dbh −/− mice. Finally, we assessedelectrophysiological responses to quinpirole in mediumspiny neurons (MSNs) from the NAc of control and Dbh−/− mice in the presence and absence of Gαi and Gαs
inhibitors.
MATERIALS AND METHODS
Animals
Adult control (Dbh +/−) and Dbh −/− mice were generatedas previously described (Thomas et al. 1998; Schanket al. 2006). Dbh −/− males were bred to Dbh +/− females.Pregnant Dbh +/− mice were given the AR agonistsisoproterenol and phenylephrine (20 μg/ml each) +vitamin C (2 mg/ml) from E9.5-E14.5, and L-3,4-dihydroxyphenylserine (2 mg/ml) + vitamin C (2 mg/ml)from E14.5 birth in their drinking water to rescue theembryonic lethality associated with the homozygous Dbh−/− mutation. Because of this treatment, NE and epineph-rine were present in Dbh −/− animals before but not afterbirth. They were maintained on a mixed C57BL/6J and129SvEv background and group housed, and food andwater were available ad libitum throughout the course ofthe study. Both sexes were used due to the extreme meas-ures required to breed sufficient numbers of knockout
mice for the experiments (Thomas, Matsumoto & Palmiter1995; Thomas et al. 1998). Comparable numbers of maleand female knockouts were used for each experiment, andsex-matched Dbh +/− littermates were used as controls.Although the studies were not powered sufficiently torigorously detect sex differences, no obvious oneswere observed. The Dbh +/− mice were used as controlsbecause their brain catecholamine levels and behaviorsare indistinguishable from wild-type (Dbh +/+) mice(Thomas et al. 1998; Bourdelat-Parks et al. 2005;Mitchell et al. 2006). Some wild-type C57BL/6J mice(Jackson Laboratory, Bar Harbor, ME, USA) were also usedas controls for the electrophysiology experiments.
All animals were treated in accordance with theNational Institutes of Health Intramural Animal Care andUse Program guidelines.The experiments described in thisarticle followed the UTSA and Emory University Divisionof Animal Resources’ Guide for the Care and Use of Labo-ratory Animals and were approved by the UTSA andEmory Institutional Animal Care and Use Committee.
Chronic nepicastat treatment
Nepicastat was administered to Dbh +/− mice via daily i.p.injections (Western blots) or osmotic minipumps (locomo-tor activity). For the i.p. administration, Dbh +/− micereceived vehicle or nepicastat (50 mg/kg, i.p. × 3, eachinjection spaced 2 hours apart) for 5 consecutive days.This dosing regimen reduces brain NE levels by ∼75 per-cent and produces cocaine hypersensitivity (Gaval-Cruzet al. 2012). Mice were euthanized by CO2 asphyxiation 11days later, and their brains were removed, dissected on iceand stored at −80°C. For the minipump administration,nepicastat was dissolved in 50 percent saline and 50percent dimethyl sulfoxide and loaded into Alzet osmoticminipumps (Model #2004, 0.25 μl/hour, 28 days;Durect, Cupertino, CA, USA) to achieve a dose of 50 mg/kg/day. All pumps were placed in a sterile 37°C salinebath for 1 day before implantation. Mice were anesthe-tized with isoflurane, and minipumps were implanted inthe intraperitoneal cavity. Buprenorphine (2.5 mg/kg,s.c.) was given immediately after surgery. Cocaine-induced locomotion was recorded 21 days after pumpimplantation.
Locomotor recordings
Mice were placed in locomotion recording chambers(transparent Plexiglas cages placed into a rack with seveninfrared photobeams spaced 5 cm apart; San DiegoInstruments Inc., La Jolla, CA, USA) and allowed tohabituate for 30 minutes before receiving a single injec-tion of cocaine (10 or 15 mg/kg, i.p.). Novelty-inducedlocomotion was defined as ambulations during the first10 minutes of the habituation period. Ambulations
Meriem Gaval-Cruz et al.
© 2014 Society for the Study of Addiction Addiction Biology, 21, 35 48
36
–

(consecutive beam breaks) were recorded for an addi-tional 1–2 hours following drug administration.
Western blotting
Mouse brain tissue was homogenized in 500 μlharvest buffer [10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 50 mM NaCl,5 mM ethylenediaminetetraacetic acid, pH 7.4, supple-mented with protease inhibitors] using a sonicator.Laemmli sample buffer containing sodium dodecyl sulfate,β-mercaptoethanol, glycerol, Tris-Cl and bromophenolblue was added to samples after measuring protein con-centrations with a bicinchoninic acid assay (ThermoFisher Scientific, Rockford, IL, USA). Samples wereresolved by sodium dodecyl sulfate–polyacrylamide gelelectrophoresis on 4–20 percent Tris-glycine precast gelsfollowed by transfer to nitrocellulose membranes. Follow-ing transfer, membranes were incubated with Ponceaustaining to assess even protein loading, then rinsed withdistilled water. Membranes were then incubated in block-ing buffer (10 mM HEPES, 50 mM NaCl, 1 percent Tween-20, 2 percent dry milk, pH 7.4, for most antibodies; 1XTBS, 0.1 percent Tween-20 with 5 percent w/v non-fatdry milk, for pAKT, GSK3β and pGSK3β) for 30 minutes,and then incubated with primary antibody overnight at4°C.The primary incubation buffer was the same as block-ing buffer for all antibodies except pAKT, GSK3β andpGSK3β. For these, the primary incubation buffer was 1XTBS, 0.1 percent Tween-20 with 5 percent bovine serumalbumin (BSA). The membranes were washed three timesin blocking buffer and incubated with either a fluorescent(1:10 000) or horseradish peroxidase-conjugated second-ary (1:4000) antibody (Invitrogen, Carlsbad, CA, USA) for30 minutes, washed three more times, and then visualizedusing either the Odyssey imaging system (Li-Cor, Lincoln,NE, USA) or via enhanced chemiluminescence reagent(Thermo Fisher Scientific), followed by exposure to film.Membranes were stripped for 20 minutes at 37°C and 10minutes at room temperature with stripping buffer andre-probed for α-actin to confirm equal loading of samples.Blots were analyzed by densitometry using ImageJ Soft-ware (National Institutes of Health, Bethesda, MD, USA).A mean density value was calculated for the ‘control’group (i.e. Dbh +/− mice were the control for Dbh −/− mice,vehicle was the control for nepicastat), and data wereexpressed as % control.
Antibodies
The antibodies used and their working dilutions were asfollows: βArr2 (anti-rabbit; 1:2500; Cell SignalingTechnology, Danvers, MA, USA, CS3857); α-actin(anti-mouse; 1:1000; Santa Cruz Biotechnology, SantaCruz, CA, USA, SC58671); pAkt-Ser473 (anti-rabbit;
1:1000; Cell Signaling, CS9271); Akt (anti-mouse;1:500; Santa Cruz Biotechnology, SC5298); pGSK3β-Ser9 (anti-rabbit; 1:1000; Cell Signaling, CS9322);pGSK3β (anti-rabbit; 1:1000; Cell Signaling, CS9315);FosB (anti-rabbit; 1:1000; Cell Signaling, CS9890).
βArr2 viral vectors
The original βArr2 plasmid (rat sequence) was obtainedfrom Sudha Shenoy in the laboratory of Dr. RobertLefkowitz. The Duke Neurotransgenic Laboratory thenremoved the βArr2 open reading frame, and the insertwas cloned into a pCMVShuttle plasmid (AdEasy System,Stratagene, Santa Clara, CA, USA). The AdEasy βArr2recombinant plasmid was generated per Stratageneinstructions, and the βArr2 adenoviral vector wasexpanded and purified. The viruses were harvested with atiter of 2 × 1012/μl (βArr2) and 5 × 109/μl [green fluo-rescent protein (GFP) control].
βArr2 viral infusions
Mice (n = 16 for each treatment group: βArr2overexpression adenovirus and GFP adenovirus) wereanesthetized using isoflurane and placed in a stereotaxicframe with a nose bar. The animal’s scalp was opened andbregma and lambda aligned to flat-skull position. Thestereotaxic arm was then lowered to the NAc core.The core subregion was chosen because it has been impli-cated in cocaine-induced locomotion and behavioralsensitization to cocaine. The anteroposterior (AP) andmediolateral (ML) coordinates of the NAc core in relationto bregma were AP = 1.4 mm and ML = ±1.0 mm, anda small hole was drilled in the skull at these coordinates.A 5-μl Hamilton microsyringe was lowered to targetthe NAc core (dorsoventral coordinate = −4.2 mm). The26-gauge beveled tip of the Hamilton needle wasprecoated with 2 percent BSA prior to loading the virus toprevent molecular interactions between the syringe andthe viral vectors. Animals received 1 μl of virus per side,injected at a rate of 0.2 μl/minute, and the needleremained in place for 5 minutes after the injection andremoved slowly. The skin was glued together usingVetbond tissue glue (Henry Schein, Roswell, GA, USA).All animals received meloxicam (0.5 mg/kg) for postop-erative pain and water/liquid ibuprofen (0.1 mg/ml).
Ten days after the infusion of βArr2 overexpres-sion and GFP control vectors, all mice were placed inlocomotor chambers, and their basal locomotion wasrecorded for 30 minutes before receiving an injection ofcocaine (15 mg/kg, i.p.), and cocaine-induced locomo-tion was recorded for 2 hours. Mice were anesthetizedand transcardially perfused with saline and 4 percentparaformaldehyde 24–48 hours later, their brains wereremoved, stored in 4 percent paraformaldehyde for 4
Cocaine response in DBH mice
© 2014 Society for the Study of Addiction Addiction Biology, 21, 35 48
37
–

days, and then transferred to 30 percent sucrose. Brainswere sectioned and stained with antibodies against GFPor βArr2, and expression in the NAc was assessed. Threemice that received the βArr2 virus and two mice thatreceived GFP virus were removed from the analysis due toincorrect placement of viral infusion.
Electrophysiological recordings of NAc neurons
C57BL/6J, Dbh +/− and Dbh −/− mice were used forelectrophysiological recordings. C57BL/6J mice wereused to: (1) confirm that Dbh +/− mice NAc MSNs weresimilar to wild-type NAc MSNs; and (2) increase thenumber of cells in a few experiments when not enoughappropriately sex- and age-matched Dbh +/− controlanimals were available. Mice were anesthetized with alethal dose of isoflurane and decapitated. The brains werequickly removed and placed into an ice-cold, oxygenatedcutting solution containing (in mM): 110 choline Cl, 2.5KCl, 1.25 NaH2PO4, 4 MgCl2, 2 CaCl2, 10 dextrose, 25NaHCO3, 1.3 ascorbic acid, 2.4 sodium pyruvate and0.05 glutathione. Parasagittal brain slices containing theNAc (250 μm) were cut using a vibrating tissue slicer(Microm HM 650V, Thermo Fisher Scientific). The sliceswere then transferred to an incubation chamber contain-ing warm (35°C) artificial cerebral spinal fluid (ACSF) for1 hour prior to recordings, and then stored at room tem-perature. The slices were transferred to a recordingchamber for the experiments, where they were sub-merged in oxygenated ACSF. The ACSF was equilibratedwith 95 percent O2-5 percent CO2, had a pH of 7.2, andcontained (in mM): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2MgCl2, 2 CaCl2, 10 dextrose, 25 NaHCO3, 1.3 ascorbicacid and 2.4 sodium pyruvate. The slices were superfusedwith 34–36°C ACSF at a rate of 2 ml/minute.
The cells were visualized using gradient contrast illu-mination through a 40X water-immersion lens attachedto an Olympus BX51 (Olympus, Center Valley, PA, USA)upright microscope. Patch pipettes were pulled fromborosilicate glass (o.d. 1.5 mm, i.d. 0.84 mm) using aP-97 Flaming/Brown electrode puller (Sutter Instru-ments, Novato, CA USA). Pipettes were filled with a solu-tion containing (in mM): 138 K-gluconate, 10 HEPES,0.0001 CaCl2, 0.2 ethylene glycol tetraacetic acid, 4NaATP, 0.4 NaGTP and 2 MgCl2, with an osmolarity of270–275 mOsm and adjusted to a pH of 7.3 with potas-sium hydroxylase (KOH). Recordings were made using aMultiClamp 700B amplifier (Molecular Devices, Sunny-vale, CA, USA). Signals were digitized at 15–30 kHz andsaved to a hard drive for analysis using the softwareprogram AxoGraph X (AxoGraph Scientific, Berkeley, CA,USA).
Spiny neurons in the NAc core were identified ashaving the following properties: a hyperpolarized mem-brane potential (< −70 mV), a low input resistance
(< 350 MΩ), and delayed spiking upon current injection.Drugs were applied to the slice by superfusion at the indi-cated concentration. All experiments were performed inthe presence of 5 μM NBQX (AMPA antagonist), 25 μMD-APV (NMDA antagonist), 100 μM picrotoxin (GABAaantagonist) and 10 μM SCH 23390 (D1 antagonist). Thedrug NF023 (10 μM) was applied internally. For NF 449(1 μM) application, the slices were incubated in the Gαs
antagonist for 1 hour prior to recording, and then con-tinuously exposed to NF449 throughout the recordingprocess. All drugs were obtained from Tocris Bioscience(Bristol, UK) or Sigma-Aldrich (St. Louis, MO, USA). Incurrent-clamp configuration, current was injected for200 ms at 100-pA step intervals (100–500 pA) with 5seconds between each pulse, until the cell was depolarizedand spikes were evoked. An input/output curve wasobtained under baseline conditions before and aftersuperfusing 10 ml of a 5 μM solution of quinpirole forapproximately 5 minutes. Action potentials were detectedusing an amplitude threshold, and spike frequency wascalculated as the reciprocal of the interspike interval.
Statistical analysis
Western blot data were analyzed by t-test using GraphPadPrism 6.0 (La Jolla, CA, USA) for Macintosh (Apple,Cupertino, CA, USA). Behavioral data were analyzed bytwo-way repeated measures ANOVA (RMANOVA), fol-lowed by Bonferroni post hoc tests, where appropriate,using Prism. Electrophysiological data were analyzed byRMANOVA with a generalized estimating equation (GEE).In our data, the number of observations differed betweendifferent current steps across cells. Because of the unbal-anced design, the classic RMANOVA was therefore not anappropriate test. We used the RMANOVA with a GEEapproach with an exchangeable correlation structure totake into account the unbalanced design, as well as cor-related observations. For each test, GEE uses a robust test(Wald χ2 test based on robust variance estimators) foreach effect. These analyses were performed using R(www.r-project.org).
RESULTS
Dbh −/− mice have decreased βArr2 in the NAc
We showed previously that ΔFosB, which is induced inthe NAc by chronic drug exposure and is known topromote psychostimulant-induced behaviors (Kelz et al.1999), is elevated in the striatum of drug-naïve Dbh −/−mice (Rommelfanger et al. 2007). As part of a largersurvey to identify potential upstream mediators of thecocaine hypersensitivity that follows chronic DBH inhibi-tion, we found that Dbh −/− mice had significantly lessβArr2 in the NAc (t6 = 3.493, P < 0.05) (Fig. 1a). Besides
Meriem Gaval-Cruz et al.
© 2014 Society for the Study of Addiction Addiction Biology, 21, 35 48
38
–
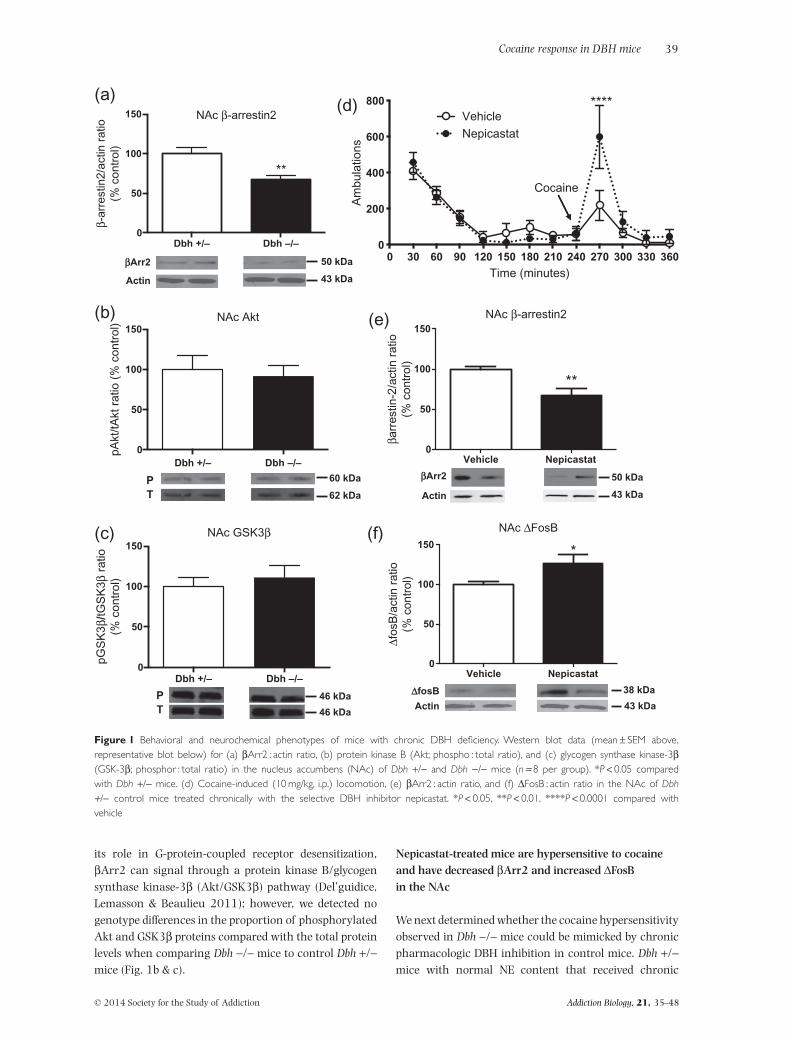
its role in G-protein-coupled receptor desensitization,βArr2 can signal through a protein kinase B/glycogensynthase kinase-3β (Akt/GSK3β) pathway (Del’guidice,Lemasson & Beaulieu 2011); however, we detected nogenotype differences in the proportion of phosphorylatedAkt and GSK3β proteins compared with the total proteinlevels when comparing Dbh −/− mice to control Dbh +/−mice (Fig. 1b & c).
Nepicastat-treated mice are hypersensitive to cocaineand have decreased βArr2 and increased ΔFosBin the NAc
We next determined whether the cocaine hypersensitivityobserved in Dbh −/− mice could be mimicked by chronicpharmacologic DBH inhibition in control mice. Dbh +/−mice with normal NE content that received chronic
0 30 60 90 120 150 180 210 240 270 300 330 3600
200
400
600
800Vehicle
Nepicastat
Time (minutes)
Am
bula
tions
Cocaine
****
(b)
bArr2
Actin
NAc ΔFosB
Vehicle Nepicastat0
50
100
150
Δfos
B/a
ctin
rat
io(%
con
trol
)
*
*
DfosB
Actin
NAc β-arrestin2
Vehicle Nepicastat0
50
100
150
βarr
estin
-2/a
ctin
rat
io(%
con
trol
)
**
Actin
P T
(c)
(e)
(f)
NAc β-arrestin2
Dbh +/– Dbh –/–
Dbh +/– Dbh –/–
Dbh +/– Dbh –/–
0
50
100
150β-
arre
stin
2/ac
tin r
atio
(% c
ontr
ol)
**
(a)
NAc Akt
0
50
100
150
pAkt
/tAkt
rat
io (
% c
ontr
ol)
NAc GSK3β
50
100
150
0
pGS
K3β
/tGS
K3β
rat
io(%
con
trol
)
P T
50 kDa
43 kDa
60 kDa
62 kDa
46 kDa
46 kDa
50 kDa
43 kDa
43 kDa
38 kDa
(d)
bArr2
Figure 1 Behavioral and neurochemical phenotypes of mice with chronic DBH deficiency. Western blot data (mean ± SEM above,representative blot below) for (a) βArr2 : actin ratio, (b) protein kinase B (Akt; phospho : total ratio), and (c) glycogen synthase kinase-3β(GSK-3β; phosphor : total ratio) in the nucleus accumbens (NAc) of Dbh +/− and Dbh −/− mice (n = 8 per group). *P < 0.05 comparedwith Dbh +/− mice. (d) Cocaine-induced (10 mg/kg, i.p.) locomotion, (e) βArr2 : actin ratio, and (f) ΔFosB : actin ratio in the NAc of Dbh+/− control mice treated chronically with the selective DBH inhibitor nepicastat. *P < 0.05, **P < 0.01, ****P < 0.0001 compared withvehicle
Cocaine response in DBH mice
© 2014 Society for the Study of Addiction Addiction Biology, 21, 35 48
39
–

nepicastat (via osmotic minipump or daily i.p. injections)had no change in locomotion induced by a novel environ-ment, but displayed increased cocaine-induced stereo-typy (Gaval-Cruz et al. 2012) and/or locomotion(Fig. 1d), reminiscent of Dbh −/− mice (Weinshenkeret al. 2002; Schank et al. 2006; Gaval-Cruz et al. 2012).Two-way ANOVA revealed a main effect of time(F11,110 = 17.55, P < 0.0001) and a treatment × timeinteraction (F11,110 = 2.64, P < 0.01). Post hoc testsshowed that peak cocaine-induced locomotion was sig-nificantly enhanced by chronic nepicastat administra-tion. Acute DBH inhibition, in contrast, does notaugment cocaine responses and can even inhibit them(Maj, Przegalinski & Wielosz 1968; Haile et al. 2003;Schroeder et al. 2013). These results indicate that thehypersensitivity to psychostimulants seen in Dbh −/−mice cannot be attributed to developmental alterationsproduced solely by DBH knockout, but likely results fromdownstream changes in the signaling pathways thatoccur following prolonged deficits in NE.
Because Dbh −/− mice have decreased βArr2 in theNAc and increased ΔFosB in the striatum, we measuredthe relative levels of these proteins in the NAc of controlmice following chronic treatment with nepicastat. Wefound that nepicastat-treated mice had decreased βArr2(t14 = 3.49, P < 0.01; Fig. 1e) and increased ΔFosB(t14 = 2.69, P < 0.05; Fig. 1f), confirming that geneticand pharmacological inhibition of NE synthesis producessimilar alterations in DA signaling proteins in the ventralstriatum.
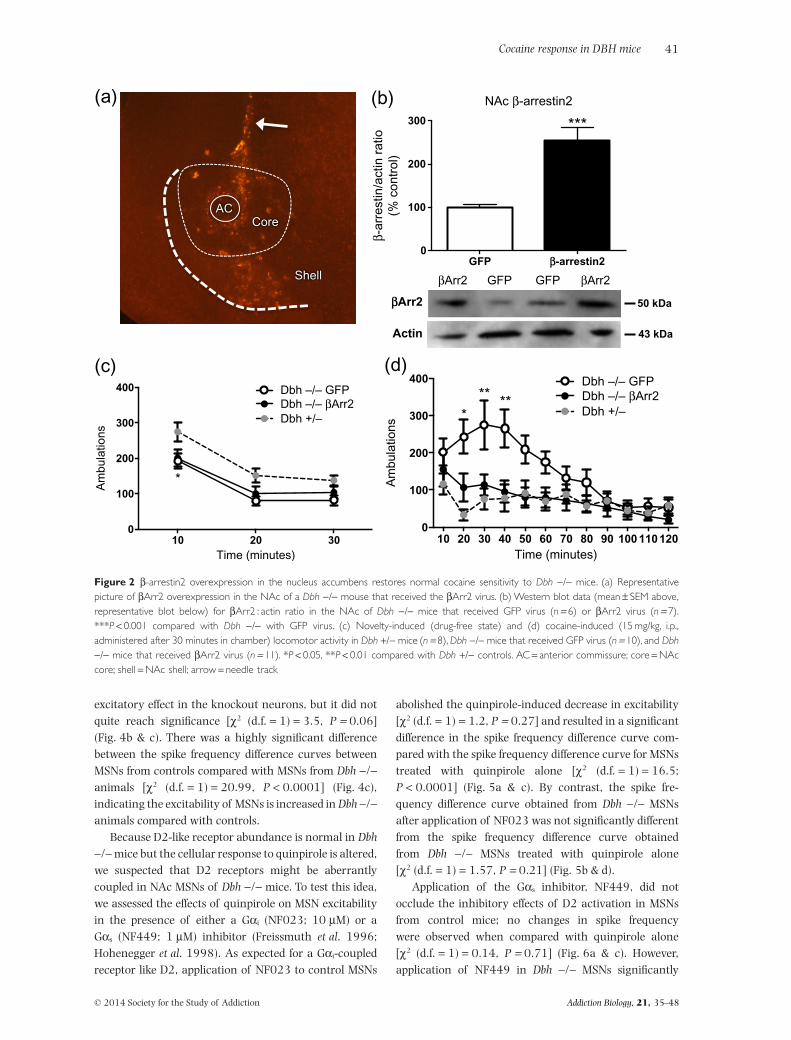
Overexpression of βArr2 in the NAc reverses cocainehypersensitivity in Dbh −/− mice
While ΔFosB is induced in the NAc by chronic drug expo-sure and is known to promote psychostimulant-inducedbehaviors (Kelz et al. 1999), the role of βArr2 is less clear.To determine whether the decreased βArr2 in the NAc ofmice with chronic NE deficiency contributes to theirbehavioral hypersensitivity to cocaine, we overexpressedGFP or βArr2 in the NAc of Dbh −/− mice usingadenoviral vectors and assessed novelty- and cocaine-induced locomotor activity. High levels of βArr2immunoreactivity were evident along the needle trackand in both the core and shell subregions (Fig. 2a), andβArr2 protein levels were doubled in the Dbh −/− NAcas assessed by Western blot (Fig. 2b) 7–10 days fol-lowing viral vector injection, indicating that βArr2overexpression was achieved and that the antibody weused was detecting βArr2. βArr2 overexpression had noeffect on the reduced novelty-induced locomotor activityof Dbh −/− mice (Fig. 2c). Two-way ANOVA revealed amain effect of time (F2,66 = 99.44, P < 0.0001) and geno-type (F2,33 = 4.46, P < 0.05). Post hoc tests showed that
Dbh −/− mice overexpressing GFP or βArr2 had decreasednovelty-induced locomotion compared with Dbh +/− con-trols with normal NE content at the 10-minute timepoint. As expected, Dbh −/− mice that were infused withthe GFP virus were hypersensitive to cocaine-inducedlocomotion compared with mice with normal NEcontent. By contrast, overexpression of βArr2 in the NAccore of Dbh −/− mice completely normalized their cocaineresponse (Fig. 2d). Two-way ANOVA revealed a maineffect of time (F11,286 = 9.62, P < 0.0001), genotype(F2,26 = 5.20, P < 0.05), and a time × genotype interac-tion (F22,286 = 2.88, P < 0.0001). Post hoc tests showedthat Dbh −/− mice overexpressing GFP displayedincreased locomotion compared with Dbh +/− mice andDbh −/− overexpressing βArr2 at the 20-, 30- and40-minute time points following cocaine administration,whereas there were no apparent differences between Dbh+/− mice and Dbh −/− mice overexpressing βArr2 in theNAc at any time point. These results suggest that thecocaine hypersensitivity conferred by chronic DBH inhi-bition is mediated, at least in part, by reduced βArr2levels in the NAc.
Dbh −/− NAc MSNs have aberrant responsesto quinpirole
To uncover the cellular underpinnings of the D2 andcocaine hypersensitivity following chronic DBH inhibi-tion, we measured the spike frequency of trains of actionpotentials elicited by the injection of current steps in NAcMSNs both at baseline and following bath application ofquinpirole (5 μM) from control and Dbh −/− mice. Thecontrol group consisted of both Dbh +/− and wild-typeC57Bl/6J mice because results comparing baseline andquinpirole responses in these groups were not signifi-cantly different [n = 22 Dbh −/−, 24 C57Bl/6J; χ2
(d.f. = 1) = 0.1, P = 0.75] (Fig. 3). We found no genotypedifferences between control and Dbh −/− mice in baselinefiring rate in untreated MSNs (F4,160 = 0.43; P = 0.79;data not shown), and activation of D2 receptors byquinpirole did not significantly change the input resist-ance or the resting membrane potential in either controlor Dbh −/− mice (control ΔRin = 5.84 ± 4.28 MΩ, Dbh −/−ΔRin = 6.57 ± 8.55 MΩ, P = 0.54; control ΔVrest = −0.71± 1.80 mV, Dbh −/− ΔVrest = −1.36 ± 1.04 mV, P = 0.43).As reported previously and expected for a Gαi/o-coupledreceptor (Surmeier & Kitai 1993; Zamponi & Snutch1998; Yasumoto et al. 2002; Perez, White & Hu 2006),activation of D2 receptors by quinpirole (5 μM) reducedevoked mean spike frequency in MSNs from controlanimals [χ2 (d.f. = 1) = 5.2, P = 0.02] (Fig. 4a & c).However, the D2-mediated reduction in MSN excitabilityseen in control mice was absent in MSNs recorded fromDbh −/− mice. Instead, quinpirole tended to have an
Meriem Gaval-Cruz et al.
© 2014 Society for the Study of Addiction Addiction Biology, 21, 35 48
40
–
excitatory effect in the knockout neurons, but it did notquite reach significance [χ2 (d.f. = 1) = 3.5, P = 0.06](Fig. 4b & c). There was a highly significant differencebetween the spike frequency difference curves betweenMSNs from controls compared with MSNs from Dbh −/−animals [χ2 (d.f. = 1) = 20.99, P < 0.0001] (Fig. 4c),indicating the excitability of MSNs is increased in Dbh −/−animals compared with controls.
Because D2-like receptor abundance is normal in Dbh−/− mice but the cellular response to quinpirole is altered,we suspected that D2 receptors might be aberrantlycoupled in NAc MSNs of Dbh −/− mice. To test this idea,we assessed the effects of quinpirole on MSN excitabilityin the presence of either a Gαi (NF023; 10 μM) or aGαs (NF449; 1 μM) inhibitor (Freissmuth et al. 1996;Hohenegger et al. 1998). As expected for a Gαi-coupledreceptor like D2, application of NF023 to control MSNs
abolished the quinpirole-induced decrease in excitability[χ2 (d.f. = 1) = 1.2, P = 0.27] and resulted in a significantdifference in the spike frequency difference curve com-pared with the spike frequency difference curve for MSNstreated with quinpirole alone [χ2 (d.f. = 1) = 16.5;P < 0.0001] (Fig. 5a & c). By contrast, the spike fre-quency difference curve obtained from Dbh −/− MSNsafter application of NF023 was not significantly differentfrom the spike frequency difference curve obtainedfrom Dbh −/− MSNs treated with quinpirole alone[χ2 (d.f. = 1) = 1.57, P = 0.21] (Fig. 5b & d).
Application of the Gαs inhibitor, NF449, did notocclude the inhibitory effects of D2 activation in MSNsfrom control mice; no changes in spike frequencywere observed when compared with quinpirole alone[χ2 (d.f. = 1) = 0.14, P = 0.71] (Fig. 6a & c). However,application of NF449 in Dbh −/− MSNs significantly
NAc β-arrestin2
GFP b-arrestin20
100
200
300
β-ar
rest
in/a
ctin
rat
io(%
con
trol
)
***
10 20 300
100
200
300
400
Time (minutes)
Am
bula
tions
Dbh –/– βArr2Dbh –/– βArr2Dbh –/– GFP
ACCore
Shell
*
Dbh +/–
(a) (b)
(c)
10 20 30 40 50 60 70 80 90 100 1101200
100
200
300
400
Time (minutes)
Am
bula
tions
Dbh –/– GFP
*
** **Dbh +/–
(d)
βArr2 βArr2GFP GFP
bArr2
Actin
50 kDa
43 kDa
Figure 2 β-arrestin2 overexpression in the nucleus accumbens restores normal cocaine sensitivity to Dbh −/− mice. (a) Representativepicture of βArr2 overexpression in the NAc of a Dbh −/− mouse that received the βArr2 virus. (b) Western blot data (mean ± SEM above,representative blot below) for βArr2 : actin ratio in the NAc of Dbh −/− mice that received GFP virus (n = 6) or βArr2 virus (n = 7).***P < 0.001 compared with Dbh −/− with GFP virus. (c) Novelty-induced (drug-free state) and (d) cocaine-induced (15 mg/kg, i.p.,administered after 30 minutes in chamber) locomotor activity in Dbh +/− mice (n = 8), Dbh −/− mice that received GFP virus (n = 10), and Dbh−/− mice that received βArr2 virus (n = 11). *P < 0.05, **P < 0.01 compared with Dbh +/− controls. AC = anterior commissure; core = NAccore; shell = NAc shell; arrow = needle track
Cocaine response in DBH mice
© 2014 Society for the Study of Addiction Addiction Biology, 21, 35 48
41
–
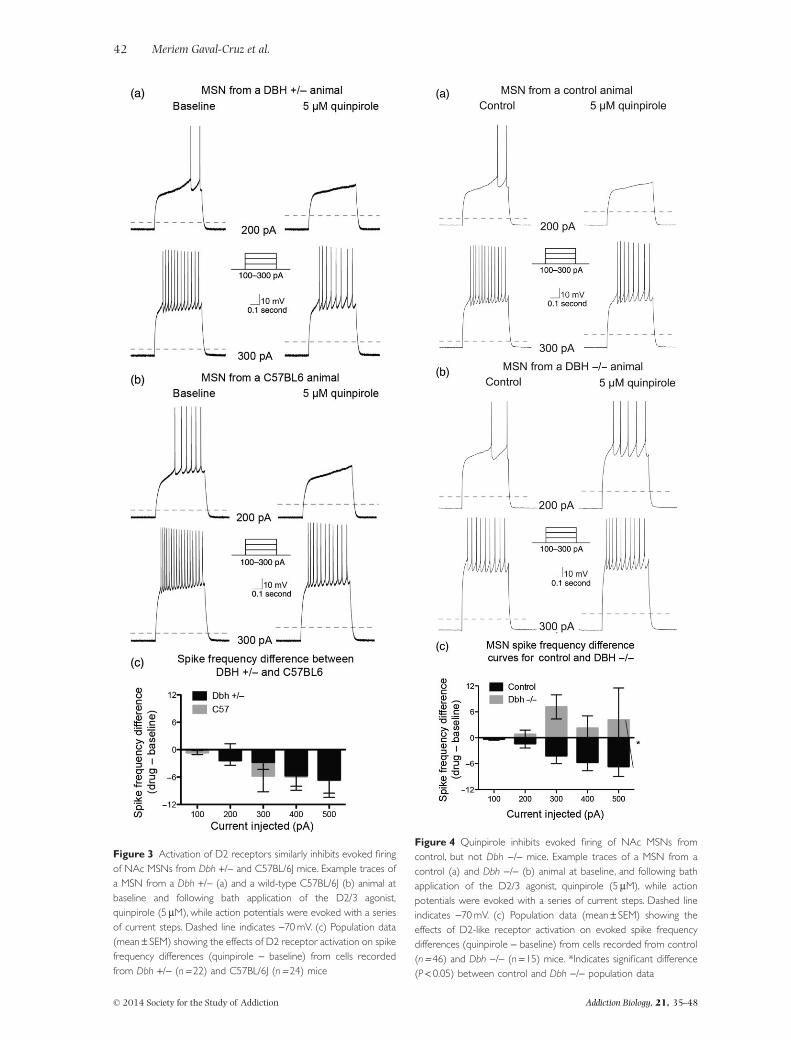
Figure 3 Activation of D2 receptors similarly inhibits evoked firingof NAc MSNs from Dbh +/− and C57BL/6J mice. Example traces ofa MSN from a Dbh +/− (a) and a wild-type C57BL/6J (b) animal atbaseline and following bath application of the D2/3 agonist,quinpirole (5 μM), while action potentials were evoked with a seriesof current steps. Dashed line indicates −70 mV. (c) Population data(mean ± SEM) showing the effects of D2 receptor activation on spikefrequency differences (quinpirole − baseline) from cells recordedfrom Dbh +/− (n = 22) and C57BL/6J (n = 24) mice
Figure 4 Quinpirole inhibits evoked firing of NAc MSNs fromcontrol, but not Dbh −/− mice. Example traces of a MSN from acontrol (a) and Dbh −/− (b) animal at baseline, and following bathapplication of the D2/3 agonist, quinpirole (5 μM), while actionpotentials were evoked with a series of current steps. Dashed lineindicates −70 mV. (c) Population data (mean ± SEM) showing theeffects of D2-like receptor activation on evoked spike frequencydifferences (quinpirole − baseline) from cells recorded from control(n = 46) and Dbh −/− (n = 15) mice. *Indicates significant difference(P < 0.05) between control and Dbh −/− population data
Meriem Gaval-Cruz et al.
© 2014 Society for the Study of Addiction Addiction Biology, 21, 35 48
42
–
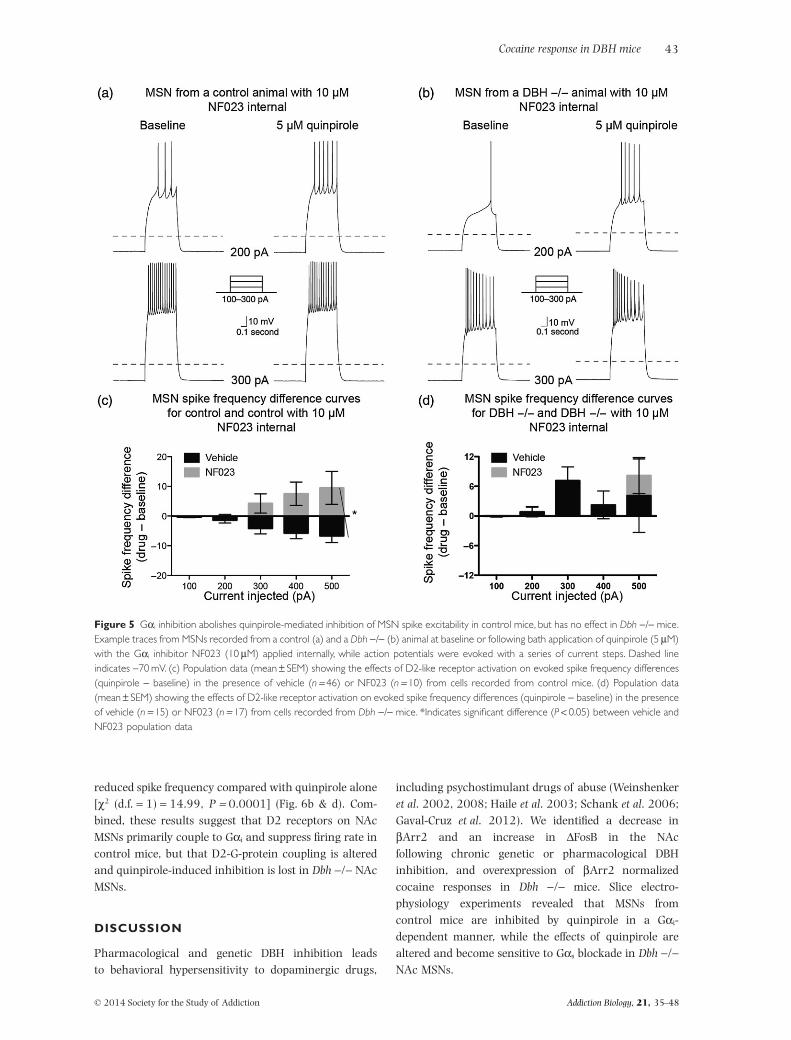
reduced spike frequency compared with quinpirole alone[χ2 (d.f. = 1) = 14.99, P = 0.0001] (Fig. 6b & d). Com-bined, these results suggest that D2 receptors on NAcMSNs primarily couple to Gαi and suppress firing rate incontrol mice, but that D2-G-protein coupling is alteredand quinpirole-induced inhibition is lost in Dbh −/− NAcMSNs.
DISCUSSION
Pharmacological and genetic DBH inhibition leadsto behavioral hypersensitivity to dopaminergic drugs,
including psychostimulant drugs of abuse (Weinshenkeret al. 2002, 2008; Haile et al. 2003; Schank et al. 2006;Gaval-Cruz et al. 2012). We identified a decrease inβArr2 and an increase in ΔFosB in the NAcfollowing chronic genetic or pharmacological DBHinhibition, and overexpression of βArr2 normalizedcocaine responses in Dbh −/− mice. Slice electro-physiology experiments revealed that MSNs fromcontrol mice are inhibited by quinpirole in a Gαi-dependent manner, while the effects of quinpirole arealtered and become sensitive to Gαs blockade in Dbh −/−NAc MSNs.
Figure 5 Gαi inhibition abolishes quinpirole-mediated inhibition of MSN spike excitability in control mice, but has no effect in Dbh −/− mice.Example traces from MSNs recorded from a control (a) and a Dbh −/− (b) animal at baseline or following bath application of quinpirole (5 μM)with the Gαi inhibitor NF023 (10 μM) applied internally, while action potentials were evoked with a series of current steps. Dashed lineindicates −70 mV. (c) Population data (mean ± SEM) showing the effects of D2-like receptor activation on evoked spike frequency differences(quinpirole − baseline) in the presence of vehicle (n = 46) or NF023 (n = 10) from cells recorded from control mice. (d) Population data(mean ± SEM) showing the effects of D2-like receptor activation on evoked spike frequency differences (quinpirole − baseline) in the presenceof vehicle (n = 15) or NF023 (n = 17) from cells recorded from Dbh −/− mice. *Indicates significant difference (P < 0.05) between vehicle andNF023 population data
Cocaine response in DBH mice
© 2014 Society for the Study of Addiction Addiction Biology, 21, 35 48
43
–
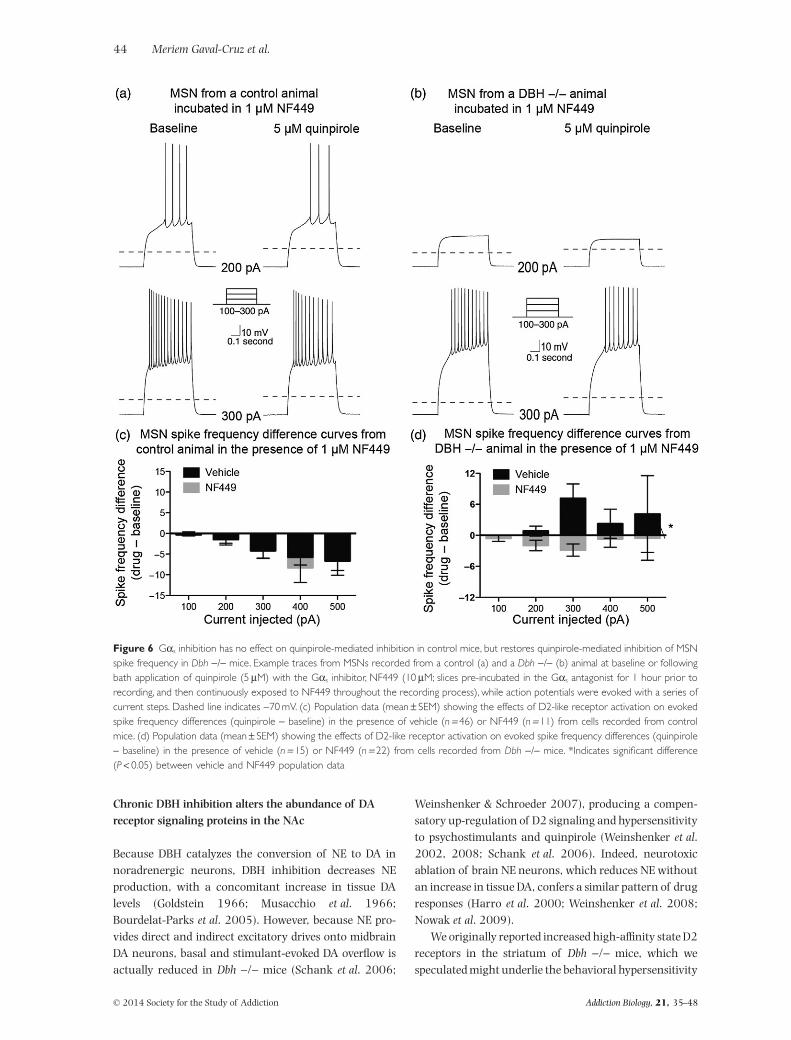
Chronic DBH inhibition alters the abundance of DAreceptor signaling proteins in the NAc
Because DBH catalyzes the conversion of NE to DA innoradrenergic neurons, DBH inhibition decreases NEproduction, with a concomitant increase in tissue DAlevels (Goldstein 1966; Musacchio et al. 1966;Bourdelat-Parks et al. 2005). However, because NE pro-vides direct and indirect excitatory drives onto midbrainDA neurons, basal and stimulant-evoked DA overflow isactually reduced in Dbh −/− mice (Schank et al. 2006;
Weinshenker & Schroeder 2007), producing a compen-satory up-regulation of D2 signaling and hypersensitivityto psychostimulants and quinpirole (Weinshenker et al.2002, 2008; Schank et al. 2006). Indeed, neurotoxicablation of brain NE neurons, which reduces NE withoutan increase in tissue DA, confers a similar pattern of drugresponses (Harro et al. 2000; Weinshenker et al. 2008;Nowak et al. 2009).
We originally reported increased high-affinity state D2receptors in the striatum of Dbh −/− mice, which wespeculated might underlie the behavioral hypersensitivity
Figure 6 Gαs inhibition has no effect on quinpirole-mediated inhibition in control mice, but restores quinpirole-mediated inhibition of MSNspike frequency in Dbh −/− mice. Example traces from MSNs recorded from a control (a) and a Dbh −/− (b) animal at baseline or followingbath application of quinpirole (5 μM) with the Gαs inhibitor, NF449 (10 μM; slices pre-incubated in the Gαs antagonist for 1 hour prior torecording, and then continuously exposed to NF449 throughout the recording process), while action potentials were evoked with a series ofcurrent steps. Dashed line indicates −70 mV. (c) Population data (mean ± SEM) showing the effects of D2-like receptor activation on evokedspike frequency differences (quinpirole − baseline) in the presence of vehicle (n = 46) or NF449 (n = 11) from cells recorded from controlmice. (d) Population data (mean ± SEM) showing the effects of D2-like receptor activation on evoked spike frequency differences (quinpirole− baseline) in the presence of vehicle (n = 15) or NF449 (n = 22) from cells recorded from Dbh −/− mice. *Indicates significant difference(P < 0.05) between vehicle and NF449 population data
Meriem Gaval-Cruz et al.
© 2014 Society for the Study of Addiction Addiction Biology, 21, 35 48
44
–

of the knockouts to psychostimulants (Schank et al.2006). However, subsequent in vitro radioligand compe-tition experiments failed to confirm these results(Skinbjerg et al. 2010) (our unpublished data). The dis-crepancy between studies measuring D2 affinity statesmay be due to some differences in the radioligandsand approaches employed, but more concerning wasour failure to observe a genotype difference in the abun-dance of high-affinity state D2 receptors in vivo usingpositron emission tomography imaging (Skinbjerg et al.2010).
Because of these issues and inconsistencies, we sus-pected that changes in downstream signaling molecules,rather than D2 receptor affinity state, were responsible forcocaine hypersensitivity following chronic NE deficiency.Both genetic and pharmacological DBH inhibition pro-duced a decrease of βArr2 and an increase of ΔFosB in theNAc. ΔFosB is a transcription factor that is induced bychronic exposure to drugs or other environmental stimuliand enhances behavioral responses to cocaine (Kelz et al.1999). We chose to pursue the contribution of βArr2because it is upstream of ΔFosB in the DA receptorsignaling pathway, and because it had not been implicatedin cocaine-induced behaviors; locomotor activity andconditioned place preference following cocaine adminis-tration are unchanged in βArr2 knockout mice (Bohnet al. 2003). We found that viral-mediated overexpressionof βArr2 in the NAc suppressed the cocaine hypersensi-tivity in Dbh −/− mice. This effect was not due to a generalsuppression of motor activity because ambulatorybehavior in a novel environment was unaffected byβArr2 overexpression, and cocaine-induced locomotionwas normalized to, but not below, control levels. Becausewe examined and used a non-selective CMV promoter todrive overexpression of βArr2, we cannot attribute itseffects on cocaine-induced locomotion specifically tochanges in D2 signaling. Future experiments using D1-and D2-specific promoters will be required to delineate theimportance of βArr2 in direct versus indirect pathwayMSNs. The discrepancies between the βArr2-mediatedphenotypes in Dbh −/− and βArr2 knockout mice may bedue to the complete lack of global βArr2 in the βArr2knockout mice versus the partial reduction of βArr2specifically in the NAc of Dbh −/− mice.
Aberrant cellular responses to quinpirole in NAc MSNsof Dbh −/− mice
Because normal D2 autoreceptor function is preserved inDbh −/− mice (Paladini, Beckstead & Weinshenker 2007),we focused our attention on potential changes in D2receptor signaling in accumbal MSNs. Activation of D2and other Gαi/o-coupled receptors typically inhibitsevoked action potentials, reducing firing and spike
frequency in MSNs. By contrast, Gαs-coupled receptors,such as D1, have a facilitatory effect on MSN responses(Hu & Wang 1988; West & Grace 2002; Surmeier et al.2010). Our electrophysiological recordings from NAcneurons confirmed that the D2/3 agonist, quinpirole,suppressed evoked MSN firing in slices from control mice.By contrast, the inhibitory effects of quinpirole were abol-ished in Dbh −/− MSNs, and in fact there was a trend forquinpirole to be excitatory.
There are several potential explanations for thischange in cellular response to D2/3 activation. Forexample, the involvement of βArr2 in receptor desensiti-zation and endocytosis suggests a possible contribution ofaltered D2 trafficking and localization. In addition,because βArr2 recruits cAMP-degrading phospho-diesterase to the membrane upon receptor binding (Perryet al. 2002; Kendall & Luttrell 2009), reduction of βArr2could dampen inhibition and promote excitation bypotentiating cAMP abundance. The reduction in βArr2could also affect neuronal firing by altering G-protein-independent GSK3β/Akt signaling, although we did notdetect any differences in these proteins in the NAc of Dbh−/− mice. It is also possible that the decrease in βArr2 andthe altered quinpirole response are unrelated. Futureexperiments to determine whether loss of βArr2 directlycauses aberrant D2/3 signaling will help identify theunderlying mechanisms.
Given that NF023 abolished quinpirole-induced inhi-bition in MSNs from control mice, but had no effect onquinpirole response in Dbh −/− MSNs, while NF449 hadno effect on quinpirole-induced inhibition in controlMSNs, but suppressed firing in the presence of quinpirolein Dbh −/− mice, it is possible that at least some D2 recep-tors are coupled to Gαs instead of Gαi in Dbh −/− MSNs. Invivo Gαi-to-Gαs switching has been reported for μ-opioidreceptors and CB1 cannabinoid receptors followingchronic agonist exposure (Wang et al. 2005; Paquetteet al. 2007), and a reduction in βArr2 promotes Gαs-to-Gαi switching of β-adrenergic receptors (Baillie et al.2003). However, these data are associated with severallimitations and must be interpreted with caution.Quinpirole is an agonist of both D2 and D3 DA receptors,and both subtypes are present in the NAc. Thus, the Gαs-to-Gαi switch could be solely or preferentially affectingone of these two subtypes. D3 receptors and D2-D3heterodimer receptors have been reported to couple toGαq, and thus promote neuronal excitation. However,increased D3-Gαq or D2/D3-Gαq heterodimer signaling isunlikely to underlie quinpirole-induced excitation in Dbh−/− MSNs for two reasons. First, Dbh −/− mice are hyper-sensitive to quinpirole but not the preferential D2/D3heterodimer agonist SKF83959 (our unpublished data).Second, the altered response of Dbh −/− MSNs toquinpirole is blocked by the Gαs inhibitor NF449, which
Cocaine response in DBH mice
© 2014 Society for the Study of Addiction Addiction Biology, 21, 35 48
45
–

does not interfere with Gαq signaling. NF023 and NF449are not totally selective for Gαi and Gαs; e.g. both com-pounds are also P2X and P2Y receptor antagonists(Lambrecht 1996; Braun et al. 2001). A contribution ofpurine/pyrimidine receptors is nevertheless unlikelygiven our results because, as a ligand-gated ion channel,would have a direct effect on membrane potential andfiring rate, which we did not observe. NF023 and NF449also had different effects in our different groups ofanimals, their inhibition which is inconsistent with thesedrugs acting as purinergic/pyrimidinergic antagonists.Finally, our methods did not allow us to distinguishbetween D1 and D2 MSNs, which precludes assigningdirect effects of quinpirole and could also account for thehigh variability in some of our experiments, particularlythe ones involving Dbh −/− recordings. In addition, itwill be important to test alterations in D2-G-proteinassociations using other techniques such as co-immunoprecipitation. Unfortunately, D2 antibodies ofsufficient quality and specificity for this approach are notcurrently available.
Clinical implications
The consequences of chronic reduction in DBH functionmay be relevant to drug addiction. Non-selective DBHinhibitors, like disulfiram, have shown promise in humanlaboratory studies and clinical trials for the treatment ofstimulant dependence (Gaval-Cruz & Weinshenker2009), and a large phase II trial of nepicastat for cocaineaddiction is ongoing (http://clinicaltrials.gov/show/NCT01704196). Genetic or chronic pharmacologicalreduction of DBH activity enhances some interoceptiveproperties of cocaine, particularly its aversive effects,such as paranoia and anxiety (Hameedi et al. 1995;McCance-Katz, Kosten & Jatlow 1998a,b; Schank et al.2006; Kalayasiri et al. 2007; Sofuoglu et al. 2008;Gaval-Cruz & Weinshenker 2009; Mutschler, Diehl &Kiefer 2009), suggesting that the clinical efficacy of DBHinhibitors is related to an increase in cocaine aversion.Interestingly, optogenetic inhibition of D2 MSNs drives,while excitation of D2 MSNs inhibits, compulsive cocaineseeking in mice (Bock et al. 2013). Thus, under normalconditions, cocaine may facilitate its own use by promot-ing DA signaling via the D2 receptor and suppressing D2MSN activity; however, our data suggest that, under con-ditions of chronic DBH inhibition, cocaine may actuallylimit its own use because D2-mediated inhibition isabsent, leading to increased cell excitability.
Acknowledgements
We thank Dainippon-Sumitomo Pharmaceuticals Inc.(Osaka, Japan) for providing the DOPS needed tomaintain our Dbh mouse colony, Synosia Therapeutics for
providing the nepicastat, and C. Strauss for helpfulediting of the manuscript. This work was supported bythe National Institute of Drug Abuse, National Instituteof Mental Health, and National Institute of NeurologicalDisorders and Stroke (NIDA grants DA017963 andDA027535 to D.W., DA25040 and DA015040 to M.G.C.,and DA030530 to C.A.P.; NIMH grant MH079276 toC.A.P.; and NINDS grant NS060658 to C.A.P.).
Disclosure/Conflict of Interest
DW is coinventor on a patent concerning the use of selec-tive DBH inhibitors for the treatment of cocaine depend-ence (US-201-0274303-A1; ‘Methods and Compositionsfor Treatment of Drug Addiction’). MGC, RBG, DJP, DEB,RCM, RAH, DJ and CAP declare no conflict of interest.
Authors Contribution
MGC, RBG, CAP and DW participated in the researchdesign. MGC, DJP and RCM conducted the Western blotexperiments. MGC and DJP conducted the behavioralexperiments. RBG conducted the electrophysiologyexperiments. DK conducted the statistical analysis of theelectrophysiology experiments. RAH and DEB contrib-uted analytic tools. MGC, RBG, CAP and DW wrote themanuscript.
References
Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ,Houslay MD (2003) Beta-arrestin-mediated PDE4 cAMPphosphodiesterase recruitment regulates beta-adrenoceptorswitching from Gs to Gi. Proc Natl Acad Sci U S A 100:940–945.
Beaulieu JM, Gainetdinov RR (2011) The physiology, signaling,and pharmacology of dopamine receptors. Pharmacol Rev63:182–217.
Bock R, Shin JH, Kaplan AR, Dobi A, Markey E, Kramer PF,Gremel CM, Christensen CH, Adrover MF, Alvarez VA (2013)Strengthening the accumbal indirect pathway promotes resil-ience to compulsive cocaine use. Nat Neurosci 16:632–638.
Bohn LM, Gainetdinov RR, Sotnikova TD, Medvedev IO,Lefkowitz RJ, Dykstra LA, Caron MG (2003) Enhancedrewarding properties of morphine, but not cocaine, inbeta(arrestin)-2 knock-out mice. J Neurosci 23:10265–10273.
Bourdelat-Parks BN, Anderson GM, Donaldson ZR, Weiss JM,Bonsall RW, Emery MS, Liles LC, Weinshenker D (2005)Effects of dopamine beta-hydroxylase genotype and disulfiraminhibition on catecholamine homeostasis in mice.Psychopharmacology (Berl) 183:72–80.
Braun K, Rettinger J, Ganso M, Kassack M, Hildebrandt C,Ullmann H, Nickel P, Schmalzing G, Lambrecht G (2001)NF449: a subnanomolar potency antagonist at recombinantrat P2X1 receptors. Naunyn Schmiedebergs Arch Pharmacol364:285–290.
Cubells JF, Kranzler HR, McCance-Katz E, Anderson GM,Malison RT, Price LH, Gelernter J (2000) A haplotype at the
Meriem Gaval-Cruz et al.
© 2014 Society for the Study of Addiction Addiction Biology, 21, 35 48
46
–

DBH locus, associated with low plasma dopamine beta-hydroxylase activity, also associates with cocaine-inducedparanoia. Mol Psychiatry 5:56–63.
Del’guidice T, Lemasson M, Beaulieu JM (2011) Role of beta-arrestin 2 downstream of dopamine receptors in the BasalGanglia. Front Neuroanat 5:58.
Freissmuth M, Boehm S, Beindl W, Nickel P, Ijzerman AP,Hohenegger M, Nanoff C (1996) Suramin analogues assubtype-selective G protein inhibitors. Mol Pharmacol49:602–611.
Gaval-Cruz M, Weinshenker D (2009) Mechanisms ofdisulfiram-induced cocaine abstinence: antabuse and cocainerelapse. Mol Interv 9:175–187.
Gaval-Cruz M, Liles LC, Iuvone PM, Weinshenker D (2012)Chronic inhibition of dopamine beta-hydroxylase facilitatesbehavioral responses to cocaine in mice. PLoS ONE 7:e50583.
Goldstein M (1966) Inhibition of norepinephrine biosynthesis atthe dopamine-beta-hydroxylation stage. Pharmacol Rev18:77–82.
Haile CN, During MJ, Jatlow PI, Kosten TR, Kosten TA (2003)Disulfiram facilitates the development and expression of loco-motor sensitization to cocaine in rats. Biol Psychiatry 54:915–921.
Hameedi FA, Rosen MI, McCance-Katz EF, McMahon TJ, PriceLH, Jatlow PI, Woods SW, Kosten TR (1995) Behavioral,physiological, and pharmacological interaction of cocaineand disulfiram in humans. Biol Psychiatry 37:560–563.
Harro J, Merikula A, Lepiku M, Modiri AR, Rinken A, Oreland L(2000) Lesioning of locus coeruleus projections by DSP-4neurotoxin treatment: effect on amphetamine-inducedhyperlocomotion and dopamine D2 receptor binding in rats.Pharmacology & Toxicology 86:197–202.
Hohenegger M, Waldhoer M, Beindl W, Boing B, Kreimeyer A,Nickel P, Nanoff C, Freissmuth M (1998) Gsalpha-selective Gprotein antagonists. Proc Natl Acad Sci U S A 95:346–351.
Hu XT, Wang RY (1988) Comparison of effects of D-1 and D-2dopamine receptor agonists on neurons in the rat caudateputamen: an electrophysiological study. J Neurosci 8:4340–4348.
Kalayasiri R, Sughondhabirom A, Gueorguieva R, Coric V,Lynch WJ, Lappalainen J, Gelernter J, Cubells JF, Malison RT(2007) Dopamine beta-hydroxylase gene (DbetaH) -1021C–>:T influences self-reported paranoia during cocaine self-administration. Biol Psychiatry 61:1310–1313.
Kapoor A, Shandilya M, Kundu S (2011) Structural insight ofdopamine beta-hydroxylase, a drug target for complex traits,and functional significance of exonic single nucleotidepolymorphisms. PLoS ONE 6:e26509.
Kelz MB, Chen J, Carlezon WA Jr., Whisler K, Gilden L, BeckmannAM, Steffen C, Zhang YJ, Marotti L, Self DW, Tkatch T,Baranauskas G, Surmeier DJ, Neve RL, Duman RS, PicciottoMR, Nestler EJ (1999) Expression of the transcription factordeltaFosB in the brain controls sensitivity to cocaine. Nature401:272–276.
Kendall RT, Luttrell LM (2009) Diversity in arrestin function.Cell Mol Life Sci 66:2953–2973.
Lambrecht G (1996) Design and pharmacology of selectiveP2-purinoceptor antagonists. J Auton Pharmacol 16:341–344.
Maj J, Przegalinski E, Wielosz M (1968) Disulfiram and the drug-induced effects on motility. J Pharm Pharmacol 20:247–248.
Manvich DF, Depoy L, Weinshenker D (2013) Dopamine beta-hydroxylase inhibitors enhance the discriminative stimuluseffects of cocaine in rats. J Pharmacol Exp Ther 347:564–573.
McCance-Katz EF, Kosten TR, Jatlow P (1998a) Chronic disulfi-ram treatment effects on intranasal cocaine administration:initial results. Biol Psychiatry 43:540–543.
McCance-Katz EF, Kosten TR, Jatlow P (1998b) Disulfirameffects on acute cocaine administration. Drug Alcohol Depend52:27–39.
Mitchell HA, Ahern TH, Liles LC, Javors MA, Weinshenker D(2006) The effects of norepinephrine transporter inactivationon locomotor activity in mice. Biol Psychiatry 60:1046–1052.
Musacchio JM, Goldstein M, Anagnoste B, Poch G, Kopin IJ(1966) Inhibition of dopamine-beta-hydroxylase by disulfi-ram in vivo. J Pharmacol Exp Ther 152:56–61.
Mutschler J, Diehl A, Kiefer F (2009) Pronounced paranoia as aresult of cocaine-disulfiram interaction: case report and modeof action. J Clin Psychopharmacol 29:99–101.
Nowak P, Nitka D, Kwiecinski A, Josko J, Drab J, Pojda-Wilczek D,Kasperski J, Kostrzewa RM, Brus R (2009) Neonatal co-lesionby DSP-4 and 5,7-DHT produces adulthood behavioralsensitization to dopamine D(2) receptor agonists. PharmacolRep 61:311–318.
Paladini CA, Beckstead MJ, Weinshenker D (2007)Electrophysiological properties of catecholaminergic neuronsin the norepinephrine-deficient mouse. Neuroscience144:1067–1074.
Paquette JJ, Wang HY, Bakshi K, Olmstead MC (2007)Cannabinoid-induced tolerance is associated with a CB1receptor G protein coupling switch that is prevented byultra-low dose rimonabant. Behav Pharmacol 18:767–776.
Perez MF, White FJ, Hu XT (2006) Dopamine D(2) receptormodulation of K(+) channel activity regulates excitability ofnucleus accumbens neurons at different membrane poten-tials. J Neurophysiol 96:2217–2228.
Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL,Miller WE, McLean AJ, Conti M, Houslay MD, Lefkowitz RJ(2002) Targeting of cyclic AMP degradation to beta2-adrenergic receptors by beta-arrestins. Science 298:834–836.
Rommelfanger KS, Edwards GL, Freeman KG, Liles LC, MillerGW, Weinshenker D (2007) Norepinephrine loss producesmore profound motor deficits than MPTP treatment in mice.Proc Natl Acad Sci U S A 104:13804–13809.
Schank JR, Ventura R, Puglisi-Allegra S, Alcaro A, Cole CD,Liles LC, Seeman P, Weinshenker D (2006) Dopaminebeta-hydroxylase knockout mice have alterations in dopaminesignaling and are hypersensitive to cocaine. Neuropsycho-pharmacology 31:2221–2230.
Schroeder JP, Cooper DA, Schank JR, Lyle MA, Gaval-Cruz M,Ogbonmwan YE, Pozdeyev N, Freeman KG, Iuvone PM,Edwards GL, Holmes PV, Weinshenker D (2010) Disulfiramattenuates drug-primed reinstatement of cocaine seeking viainhibition of dopamine beta-hydroxylase. Neuropsycho-pharmacology 35:2440–2449.
Schroeder JP, Epps SA, Grice TW, Weinshenker D (2013) Theselective dopamine beta-hydroxylase inhibitor nepicastatattenuates multiple aspects of cocaine-seeking behavior.Neuropsychopharmacology 38:1032–1038.
Skinbjerg M, Seneca N, Liow JS, Hong J, Weinshenker D, PikeVW, Halldin C, Sibley DR, Innis RB (2010) Dopamine beta-hydroxylase-deficient mice have normal densities of D(2)dopamine receptors in the high-affinity state based on in vivoPET imaging and in vitro radioligand binding. Synapse64:699–703.
Cocaine response in DBH mice
© 2014 Society for the Study of Addiction Addiction Biology, 21, 35 48
47
–

Sofuoglu M, Poling J, Waters A, Sewell A, Hill K, Kosten T (2008)Disulfiram enhances subjective effects of dextroamphetaminein humans. Pharmacol Biochem Behav 90:394–398.
Stanley WC, Li B, Bonhaus DW, Johnson LG, Lee K, Porter S,Walker K, Martinez G, Eglen RM, Whiting RL, Hegde SS (1997)Catecholamine modulatory effects of nepicastat (RS-25560-197), a novel, potent and selective inhibitor of dopamine-beta-hydroxylase. Br J Pharmacol 121:1803–1809.
Surmeier DJ, Kitai ST (1993) D1 and D2 dopamine receptormodulation of sodium and potassium currents in ratneostriatal neurons. Prog Brain Res 99:309–324.
Surmeier DJ, Shen W, Day M, Gertler T, Chan S, Tian X, PlotkinJL (2010) The role of dopamine in modulating the structureand function of striatal circuits. Prog Brain Res 183:149–167.
Thomas SA, Matsumoto AM, Palmiter RD (1995) Noradrenalineis essential for mouse fetal development. Nature 374:643–646.
Thomas SA, Marck BT, Palmiter RD, Matsumoto AM (1998)Restoration of norepinephrine and reversal of phenotypes inmice lacking dopamine beta-hydroxylase. J Neurochem70:2468–2476.
Wang HY, Friedman E, Olmstead MC, Burns LH (2005) Ultra-low-dose naloxone suppresses opioid tolerance, dependenceand associated changes in mu opioid receptor-G protein cou-pling and Gbetagamma signaling. Neuroscience 135:247–261.
Weinshenker D, Schroeder JP (2007) There and back again: atale of norepinephrine and drug addiction. Neuropsycho-pharmacology 32:1433–1451.
Weinshenker D, Miller NS, Blizinsky K, Laughlin ML, PalmiterRD (2002) Mice with chronic norepinephrine deficiencyresemble amphetamine-sensitized animals. Proc Natl Acad SciU S A 99:13873–13877.
Weinshenker D, Ferrucci M, Busceti CL, Biagioni F, Lazzeri G,Liles LC, Lenzi P, Pasquali L, Murri L, Paparelli A, Fornai F(2008) Genetic or pharmacological blockade of noradre-naline synthesis enhances the neurochemical, behavioral,and neurotoxic effects of methamphetamine. J Neurochem105:471–483.
Weinshilboum RM (1978) Serum dopamine beta-hydroxylase.Pharmacol Rev 30:133–166.
West AR, Grace AA (2002) Opposite influences of endogenousdopamine D1 and D2 receptor activation on activity states andelectrophysiological properties of striatal neurons: studiescombining in vivo intracellular recordings and reversemicrodialysis. J Neurosci 22:294–304.
Yasumoto S, Tanaka E, Hattori G, Maeda H, Higashi H (2002)Direct and indirect actions of dopamine on the membranepotential in medium spiny neurons of the mouse neostriatum.J Neurophysiol 87:1234–1243.
Zamponi GW, Snutch TP (1998) Modulation of voltage-dependent calcium channels by G proteins. Curr OpinNeurobiol 8:351–356.
Meriem Gaval-Cruz et al.
© 2014 Society for the Study of Addiction Addiction Biology, 21, 35 48
48
–