
Celltransmissions - Sigma-Aldrich Sigma-RBIp. 11 ... receptor antagonist p. 16 Hydroxyfasudil:...
24
New Products pp. 12-15 Antibodies to Potassium Channels p. 10 SR12813: PXR agonist and HMG-CoA reductase inhibitor p. 11 Exo 1: Reversible inhibitor of exocytosis p. 11 DAPT (LY-374973): γ-secretase inhibitor p. 11 YM-53601: Squalene synthase inhibitor p. 11 sPLA 2 inhibitor Available First from Sigma-RBI p. 11 Monoclonal Anti-PML p. 16 Cyclofenil: ERβ estrogen receptor antagonist p. 16 Hydroxyfasudil: Selective rho kinase inhibitor p. 16 Monoclonal Anti-hnRNP-Q p. 17 GGTI-298 and FTI-276: Geranylgeranyl- and farnesyl- transferase inhibitors p. 17 Anti-Radixin p. 17 ICI 63197: Phosphodiesterase IV (PDE IV) inhibitor p. 17 Assay Kits p. 18-19 Topiramate: Novel anticonvulsant and kainate GluR5 receptor antagonist First Available from Sigma-RBI p. 20 SB-408124: Selective non- peptide OX 1 orexin receptor antagonist p. 20 YIC-C8-434: Acyl-CoA: Cholesterol O-Acyltransferase (ACAT) inhibitor p. 20 GW311616A: Human neutrophil elastase (HNE) inhibitor p. 20 Celltransmissions The Newsletter for Cell Signaling and Neuroscience Research Ligands Interacting with Allosteric Sites on G Protein-Coupled Receptors Nigel J.M. Birdsall 2004 Nobel Prize in Chemistry Awarded to Celltransmissions author for the Discovery of Ubiquitin-Mediated Protein Degradation In this Issue... G protein-coupled receptors (GPCRs) are the largest class of cell surface signaling pro- teins in the human genome. Each receptor responds to a specific stimulus, usually the binding of an endogenous ligand, resulting in the generation of intracellular signals that are characteristic of both the nature of the receptor and of the cell. A high percentage of clinically used drugs generate their therapeutic effects by binding to GPCRs. Historically, such drugs have produced their actions by binding to the orthosteric site, i.e. the same site that binds the endogenous signaling molecule. They compete with the endogenous ligand and, depending on their structure, can act as agonists, antagonists or inverse agonists to modulate receptor function. The characteristic of these competitive inter- actions is that a receptor molecule, while occupied by the ligand, will be in a chronically active or inactive state and insensitive to fluctua- tions in the level of the endogenous signaling ligand. For example, a receptor molecule with an agonist bound is chronically activated and gener- ates a continuous signal that may eventually become attenuated by desensitization and down-regulation of the receptor. This mecha- nism means that the spatial and temporal signaling characteristics of naturally operating receptors are lost. This may explain some of the unwanted effects of even highly receptor- selective ligands, especially agonists, caused by their binding to their specific receptor at the 'wrong' time or place. sigma-aldrich.com/cellsignaling continued on page 3 Vol 20, No 3 • January 2005 S igma-RBI, a division of Sigma-Aldrich (NAS- DAQ: SIAL), congratulates the 2004 Nobel laureates in Chemistry, Dr. Avram Hershko, Dr. Irwin Rose and Celltransmissions author Dr. Aaron Ciechanover. Their contributions have led to the realization that protein degradation is a highly controlled process involving the attach- ment of the molecule ubiquitin to unwanted proteins to mark them for disposal. Failure of this system has been implicated in many diseases incuding cervical cancer and cystic fibrosis. continued on page 8
-
Upload
vuongkhanh -
Category
Documents
-
view
213 -
download
0
Transcript of Celltransmissions - Sigma-Aldrich Sigma-RBIp. 11 ... receptor antagonist p. 16 Hydroxyfasudil:...

New Products pp. 12-15
Antibodies to PotassiumChannels p. 10
SR12813: PXR agonist andHMG-CoA reductase inhibitorp. 11
Exo 1: Reversible inhibitor ofexocytosis p. 11
DAPT (LY-374973): γ-secretaseinhibitor p. 11
YM-53601: Squalene synthaseinhibitor p. 11
sPLA2 inhibitor Available Firstfrom Sigma-RBI p. 11
Monoclonal Anti-PML p. 16
Cyclofenil: ERβ estrogenreceptor antagonist p. 16
Hydroxyfasudil: Selective rhokinase inhibitor p. 16
Monoclonal Anti-hnRNP-Q p. 17
GGTI-298 and FTI-276:Geranylgeranyl- and farnesyl-transferase inhibitors p. 17
Anti-Radixin p. 17
ICI 63197: PhosphodiesteraseIV (PDE IV) inhibitor p. 17
Assay Kits p. 18-19
Topiramate: Novelanticonvulsant and kainateGluR5 receptor antagonistFirst Available from Sigma-RBIp. 20
SB-408124: Selective non-peptide OX1 orexin receptorantagonist p. 20
YIC-C8-434: Acyl-CoA:Cholesterol O-Acyltransferase(ACAT) inhibitor p. 20
GW311616A: Humanneutrophil elastase (HNE)inhibitor p. 20
CelltransmissionsThe Newsletter for Cell Signaling and Neuroscience Research
Ligands Interacting with Allosteric Sites on G Protein-Coupled ReceptorsNigel J.M. Birdsall
2004 Nobel Prize in Chemistry Awarded toCelltransmissions author for the Discovery ofUbiquitin-Mediated Protein Degradation
In this Issue...
G protein-coupled receptors (GPCRs) are thelargest class of cell surface signaling pro-
teins in the human genome. Each receptorresponds to a specific stimulus, usually thebinding of an endogenous ligand, resulting inthe generation of intracellular signals that arecharacteristic of both the nature of the receptorand of the cell. A high percentage of clinicallyused drugs generate their therapeutic effectsby binding to GPCRs. Historically, such drugshave produced their actions by binding to theorthosteric site, i.e. the same site that binds theendogenous signaling molecule. They competewith the endogenous ligand and, depending ontheir structure, can act as agonists, antagonistsor inverse agonists to modulate receptorfunction.
The characteristic of these competitive inter-actions is that a receptor molecule, whileoccupied by the ligand, will be in a chronicallyactive or inactive state and insensitive to fluctua-tions in the level of the endogenous signalingligand. For example, a receptor molecule with anagonist bound is chronically activated and gener-ates a continuous signal that may eventuallybecome attenuated by desensitization anddown-regulation of the receptor. This mecha-nism means that the spatial and temporalsignaling characteristics of naturally operatingreceptors are lost. This may explain some of theunwanted effects of even highly receptor-selective ligands, especially agonists, caused bytheir binding to their specific receptor at the'wrong' time or place.
sigma-aldrich.com/cellsignaling
continued on page 3
Vol 20, No 3 • January 2005
Sigma-RBI, a division of Sigma-Aldrich (NAS-DAQ: SIAL), congratulates the 2004 Nobel
laureates in Chemistry, Dr. Avram Hershko, Dr.Irwin Rose and Celltransmissions author Dr.Aaron Ciechanover. Their contributions haveled to the realization that protein degradation isa highly controlled process involving the attach-ment of the molecule ubiquitin to unwantedproteins to mark them for disposal. Failure ofthis system has been implicated in many diseasesincuding cervical cancer and cystic fibrosis.
continued on page 8

The Complete Package Includes:
• Antigen design
• Peptide synthesis and conjugation
• 2 animals (rabbit, chicken, sheep or goat)
• 6 immunizations per animal
• ELISA on production bleed #1
• 4 bleeds per animal
The Partial Package Includes:
• Customer supplied antigen (peptide conjugate, protein, gel slice, etc.)
• 2 animals (rabbit, chicken, sheep or goat)
• 6 immunizations per animal
• ELISA on production bleed #1
• 4 bleeds per animal
The Monospecific/Characterized Package Includes:
• All elements of the Complete Package
• Exsanguination of both animals
• Affinity purification on bleed #3– Typically 25 ml of sera yields
2-3 mg of purified antibody
• Western blot testing of purified antibody (full panel of 15 lysates)
– An electronic image of the blot is provided
L E A D E R S H I P I N L I F E S C I E N C E , H I G H T E C H N O L O G Y A N D S E R V I C ESIGMA-ALDRICH CORPORATION • BOX 14508 • ST. LOUIS • MISSOURI 63178 • USAsigma-genosys.com
Sigma-Genosys offers comprehensive services for polyclonal antibody production. By offeringthree different service levels, researchers have the ability to customize the packages based ontheir research needs.
For ordering or more information please contact www.sigma-genosys.com/Anti_CT or call 1-800-234-5362.

®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
3
Allo
ster
ic L
igan
ds
to
G P
rote
in-C
ou
ple
d R
ecep
tors
An alternative approach in principle is to target the drugto a second site that is different from the orthosteric site,but which is conformationally linked to the orthosteric site(i.e. an allosteric site). This provides the opportunity to beable to 'tune up' or 'tune down' a receptor response,rather than 'switching' it on (or off) by the use of anorthosteric agonist or antagonist, respectively. Further-more, the allosteric approach can generate novel forms ofselectivity that are absent in orthosteric ligands.
This approach is well known in the ionotropic receptorfield where for many years drugs such as benzodiazepines,acting allosterically to enhance the actions of γ-aminobutyric acid (GABA) at GABAA receptors, have had clinicalutility. However, even in this well-established field, it hasproven difficult until recently to generate allosteric ligandsthat are selective in their actions on a given subtype ofGABAA receptors [1,2]. In the past few years there havebeen substantial and increasing scientific efforts directedtowards the detection and characterization of allostericsites on a number of GPCRs, as well as the accompanyingdiscovery and development of molecules that begin todefine the pharmacology of these sites. The purpose ofthis article is to provide a compilation of some of theallosteric ligands that act at GPCRs and a flavor of thenovel selectivities associated with this type of ligand. A number of reviews of this field have been publishedrecently [3-8].
Analysis of Allosteric Interactions: TheAllosteric Ternary Complex ModelFigure 1 represents the simplest model that describes anallosteric interaction between two ligands, L and X, at areceptor, R. This is a symmetrical scheme but, for thepurposes of this article, L will be defined as an orthostericligand and X as the allosteric ligand.
In this model there are two topographically separatedbinding sites on the receptor (R) that are conforma-tionally linked. Orthosteric ligands (L) including, and mostimportantly, the endogenous hormone or neurotrans-mitter bind to one site with an affinity constant KL. Thebinding of ligands (X) to the second site allostericallymodulates the affinity of L by a factor α, the coopera-tivity factor. In a complementary manner, the binding of Lto the receptor changes the affinity constant of X from
KX (in the absence of L) to α.KX (in the presence of highconcentrations of L). If α > 1, there is positive hetero-tropic cooperativity, if α < 1, there is a negative coopera-tive interaction between L and X. For the very specialcase where α = 1, there is neutral cooperativity and thebinding of X to R does not affect the equilibrium bindingof L. Therefore, the binding and actions of an allostericligand with any given L and R are defined by two param-eters, its affinity KX (which is independent of L) and itscooperativity α (which depends on L). The selectivity andthe pharmacological activity of X at different receptors istherefore manifest by differences in either or both KL andα. The importance is that there are two very differentindependent parameters (affinity and cooperativity) thatcan generate receptor selectivity.
In equilibrium binding studies it is possible to measure KXand α (and hence α.KX) for an allosteric ligand. Analyticalmethods by which these parameters can be visualized andcalculated have been devised [9-12]. Allosteric ligands arealso generally found to modulate the dissociation rate ofradioligands bound to the orthosteric site because thedissociation rate constants of L from R.L and X.R.L wouldbe expected to be different. This effect on radioligandkinetics generates a second estimate of α.KX which shouldbe the same as that determined in the equilibrium experi-ment if the ternary complex describes the L-R-X interactionand the effects of X on the binding of L are not simply dueto non-specific effects of X, such as membrane perturba-tion. We have used these approaches to confirm thevalidity of the allosteric ternary complex model for thebinding of allosteric ligands to a number of GPCRs [10-19].
The allosteric ternary complex model can also be used toanalyze functional data [10-12,20]. However, it has to beassumed that X has no intrinsic activity and the binding ofX to the receptor only changes agonist 'affinity', but doesnot affect agonist 'efficacy'. If this is not so, the estimatesof the cooperativity obtained using this simple model toanalyze binding and functional data may differ from eachother. Under such circumstances more complex models,involving receptor isomerization states and/or the ability ofallosteric ligands to couple to and uncouple from G proteins,need to be considered [7,21-23].
Novel Selectivities Associated withAllosteric Ligands (Table 1)It is of prime importance to characterize the interactionsbetween an allosteric ligand and the endogenous signalingmolecule(s) at GPCRs as these are the only allosteric inter-actions that are generally going to be therapeutically use-ful in vivo. As stated earlier, different allosteric ligands actto ‘tune up’ or ‘tune down’ endogenous signaling by thereceptor. If ligand X has no intrinsic activity, it has noaction on its own and therefore, ‘silent’ receptors are notaffected by allosteric ligands. This provides temporal aswell as spatial resolution for the actions of the allosteric
Ligands Interacting with Allosteric Sites on G Protein-Coupled ReceptorsNigel J.M. Birdsall (continued from cover)
αKL
α.KX
X.R.LX.R + L
KX
R + L+ +X XKL
R.L
Figure 1. The allosteric ternary complex model.

ligand. In contrast, agonists provide no such resolution andthis can lead to potential unwanted effects caused by'inappropriate' and chronic stimulation of the target receptor.
Furthermore, allosteric ligands have a limited maximumactivity that is determined by the value of their coopera-tivity. It might be anticipated that an allosteric ligandthat exhibits positive cooperativity with the endogenousligand (an allosteric enhancer) could be of therapeuticuse in diseases where a focal deficit in neurotransmitterlevel gives rise to symptoms of the disease. Examples areAlzheimer’s disease, where there is a cholinergic deficitin the cerebral cortex and hippocampus, and Parkinson’sdisease where there are low levels of dopamine in thebasal ganglia. Allosteric enhancers should produce aselective boost in signaling in those regions of lowneurotransmitter release.
Receptor subtypes are closely homologous proteins that areactivated by the same endogenous ligand and thus arelikely to have a conserved orthosteric site. This can result indifficulties in designing subtype-selective ligands, especiallyagonists. In contrast, an allosteric binding site on a recep-tor may not have been subject to the same evolutionarypressure to conserve its structure and hence there is theincreased possibility of allosteric ligands that exhibitselectivity based on affinity.
A much more subtle, but nevertheless profound, form ofselectivity is that based on a ligand having different coop-erativities for different receptor species, especially receptorsubtypes. In this case, the important feature of an allostericligand is that if it has neutral cooperativity with theendogenous neurotransmitter at one receptor subtype itwill be totally inactive at that subtype even though it bindsto the receptor. It will have no action on that receptor atany concentration. If this allosteric ligand exhibits positiveor negative cooperativity with the same endogenous ligandat another subtype, it will have an action at that subtype.The form of selectivity, that is action at one subtype, noaction at another subtype at any concentration, has beentermed ‘absolute subtype selectivity’ [11,12]. Subtypeselectivity, based on cooperativity, was first described forbrucine analogs that bind to an allosteric site on muscarinicreceptors [11,12]. Subsequently thiochrome, an oxidationproduct and metabolite of thiamine, was shown to demon-strate ‘absolute subtype selectivity’, being an allostericenhancer at M4 muscarinic acetylcholine receptors, buthaving no effect on acetylcholine binding at the other fourmuscarinic receptor subtypes [24].
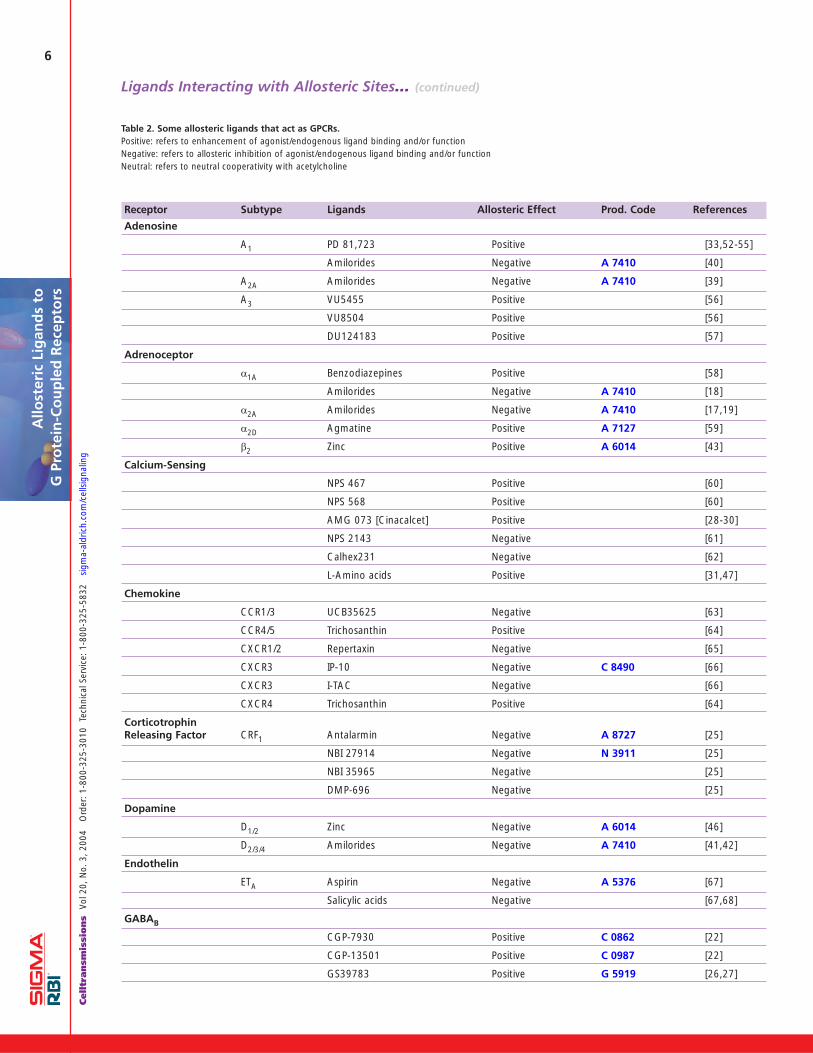
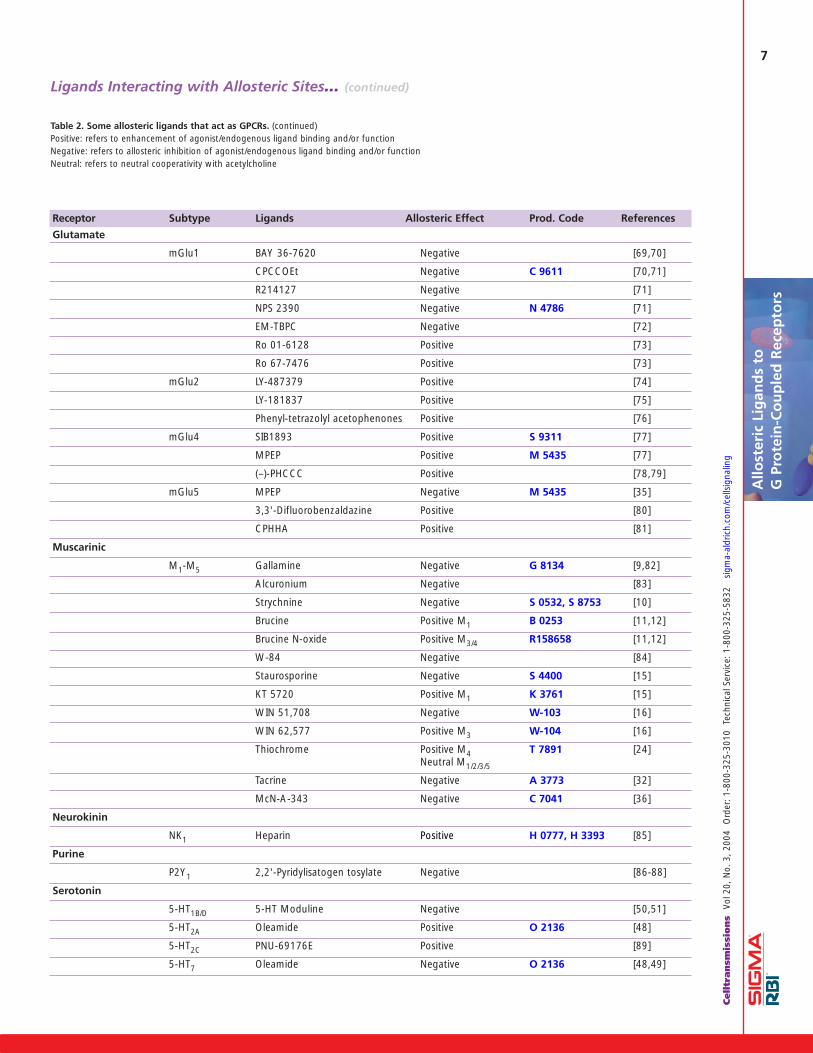
Allosteric LigandsTable 2 (pages 6 and 7) presents a list of GPCRs that havebeen claimed to possess allosteric sites together withselected ligands that have been used to characterize thesesites. In a number of examples, the data are limited and/ornot capable of simple interpretation in terms of allosterism.Further characterization of these ligand-receptor interac-tions would be beneficial.
The receptors listed in Table 2 are primarily members oftwo of the three major subclasses of GPCRs: namely class-es A and C. The only class B receptor reported to have awell-defined allosteric site is the CRF1 corticotrophin releas-ing factor receptor [25]. In the case of class C receptors thefocus, driven primarily by the pharmaceutical industry, hasbeen on the GABAB receptor, the metabotropic glutamatereceptor subtypes and the calcium-sensing receptor.Allosteric enhancers of binding and function at GABABreceptors, CGP-7930 (Prod. Code C 0862) [22], CGP-13501(Prod. Code C 0987) [22] and GS39783 (Prod Code G 5919)[26], have been reported. The latter compound has ananxiolytic effect in a rodent model without displaying someof the undesirable side effects produced by either baclofenor the benzodiazepines [27]. Selective allosteric enhancersat each of subtypes 1,2,4 and 5 of metabotropic glutamatemGlu receptors have also been described.
The calcium-sensing receptor represents the only GPCR forwhich an allosteric ligand (Cinacalcet, an allostericenhancer) has been approved by the FDA for the treatmentof secondary hyperparathyroidism in patients with chronickidney disease on dialysis, and hypercalcemia in patientswith parathyroid cancer [28-30]. It acts by increasing thesensitivity of the receptor to circulating calcium and conse-quently reducing parathyroid hormone release thereby nor-malizing serum calcium levels. Of the family A (rhodopsin-like) receptors, most investigations have involved the dis-covery of allosteric muscarinic ligands and the investigationof their mechanisms of action. In the case of muscarinicreceptors, it has been demonstrated that there are twoallosteric sites with very different pharmacologies [15,16].A similar spatial separation of the L-amino acid and thecalcimimetic binding sites on the calcium-sensing receptorhas also been demonstrated [31]. For some allostericmuscarinic ligands, e.g. tacrine (Prod. Code A 3773) andsome pentacyclic carbazolones, the allosteric interactionsexhibit slope factors greater than 1 and less than 2, indi-cating a positively homotropic cooperative interaction. Ithas been suggested that these ligands may be modulatingreceptor-receptor interactions in a receptor dimer [13,32].
Studies on muscarinic receptors and A1 adenosine recep-tors have indicated that certain allosteric ligands may haveintrinsic activity. The best example is PD 81,723, the firstallosteric enhancer reported to act at a GPCR (A1 adeno-sine receptors) [33]. This ligand also displays agonist activityin the absence of an orthosteric ligand. Therefore, it has a
®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
4A
llost
eric
Lig
and
s to
G
Pro
tein
-Co
up
led
Rec
epto
rsLigands Interacting with Allosteric Sites... (continued)
Table 1. Potential novel selectivities associated with allosteric ligands
• Maximum effect limited by the cooperativity factor α• No activation of ‘silent’ receptors-spatial and temporal selectivity of action• Potential for greater subtype selectivity based on affinity• Subtype selectivity based on cooperativity• Ability to modulate receptor-protein and receptor-receptor interactions by
a small molecule binding to an allosteric site

dual action as allosteric enhancer and allosteric agonist. Incontrast, alcuronium acts as an inverse agonist on functionat M2 muscarinic receptors [34]. It decreases the efficacyof pilocarpine to such an extent that this partial agonistbecomes an antagonist. This is an example of an allostericligand changing the pharmacology of an orthostericligand. Some other allosteric ligands, e.g. 6-methyl-2-(phenylethynyl)pyridine (MPEP; Prod. Code M 5435)acting at mGlu5 receptors, have also inverse agonist activi-ty and decrease agonist efficacy [35]. Another cholinergicagonist, McN-A-343 (Prod. Code C 7041) has been shownto interact allosterically at M2 muscarinic receptors and itwas speculated at that time that it might also be able toactivate the receptor from the allosteric site [36]. Morerecently, the atypical muscarinic agonists, AC-42 [37],clozapine (Prod. Code C 6305) and N-desmethylclozap-ine (Prod. Code D 5676) [38] have been reported toexhibit a subtype selectivity that is greater than thatusually observed for muscarinic agonists. They appear tobind in a different mode to acetylcholine and, on thatbasis, it has been hypothesized that they also may activatethe muscarinic receptor from an allosteric site. However,direct evidence for such a mechanism is lacking at present.Some allosteric ligands seem to act allosterically at many classA GPCRs. The two notable examples are amilorides thatinteract at adenosine, dopamine and α-adrenoceptor sub-types [17-19, 39-42]. An even greater promiscuity and com-plexity is shown by SCH-202676 (Prod. Code S 4063) whichinteracts with adrenergic, dopamine, muscarinic and opioidreceptor subtypes [43,44]. The findings suggest that theseligands may interact with a binding site (or sites) that is con-served between at least some members of class A GPCRs.
A question that is often asked is whether there should bean endogenous ligand if there is a well-defined allostericsite on a GPCR. At present there is very limited evidence tosupport such a hypothesis, although the actions of zinc (atβ2-adrenoceptors [45] and D1/D2 dopamine receptors [46]), L-amino acids (calcium sensing receptor [47]), oleamide(Prod. Code O 2136; 5-HT2A [48] and 5-HT7 serotoninreceptors [49]) and 5-HT-moduline, an endogenoustetrapeptide LSAL, (at 5-HT1B/1D serotonin receptors[50,51]) are suggestive that this may be so.
SummaryAllosteric ligands, especially allosteric enhancers, have thepotential for exhibiting different selectivities at GPCRs fromthose of orthosteric ligands. The difficulty is that, at theinitiation of a project, the information about the pharma-cology of an allosteric site on a GPCR may be very limitedor non-existent. In addition, as the effects of an allostericligand on receptor binding or function may be small, anyscreening assay must be very sensitive and robust in orderto detect 'hits'. Equally, the actions of allosteric ligands invivo may be more subtle and different from those oforthosteric ligands. Nevertheless the large number ofrecent publications provides strong evidence of an increas-ing interest from both academia, and especially the
pharmaceutical industry. The recent approval of Cinacalcetfor clinical use supports allosteric modulation at GPCRs asa valid and novel therapeutic approach.
References1. Street, L.J., et al., J. Med. Chem., 47, 3642-3657 (2004).2. Whiting, P.J., Drug Discov. Today, 8, 445-450 (2003).3. Christopoulos, A., Nat. Rev. Drug Discov., 1, 198-209 (2002).4. Soudijn, W.S., et al., Drug Discov. Today, 9, 754-758 (2002).5. Christopoulos, A. and Kenakin, T., Pharmacol. Rev., 54, 323-374 (2002).6. Jensen, A.A. and Spalding, T.A., Eur. J. Pharmacol. Sci., 21, 407-420 (2004).7. May, L.T., et al., Curr. Pharmaceut. Design, 10, 2003-2013 (2004).8. Pin, J-P., et al., Mol. Pharmacol., 60, 881-884 (2001).9. Stockton, J.M., et al., Mol. Pharmacol., 23, 551-557 (1983).10. Lazareno, S. and Birdsall, N.J.M., Mol. Pharmacol., 48, 362-378 (1995).11. Lazareno, S., et al., Mol. Pharmacol., 53, 573-589 (1998).12. Birdsall, N.J.M., et al., Mol. Pharmacol., 55, 778-786 (1999).13. Gharagozloo, P., et al., J. Med. Chem., 45, 1259-1274 (2002).14. Lazareno, S., et al., Life Sci., 64, 519-526 (1999).15. Lazareno, S., et al., Mol. Pharmacol., 58, 194-207 (2000).16. Lazareno, S., et al., Mol. Pharmacol., 62, 1492-1505 (2002).17. Leppik, R.A., et al., Mol. Pharmacol., 53, 916-925 (1998).18. Leppik, R.A., et al., Mol. Pharmacol., 57, 436-445 (2000).19. Leppik, R.A. and Birdsall, N.J.M., Mol. Pharmacol., 58, 1091-1099 (2000).20. Ehlert, F.J., Mol. Pharmacol., 33, 187-194 (1988).21. Hall, D.A., Mol. Pharmacol., 58, 1412-1423 (2000).22. Urwyler, S., et al., Mol. Pharmacol., 60, 963-971 (2001).23. Parmentier, M-L., et al., Trends Pharmacol. Sci., 23, 268-274 (2002).24. Lazareno, S., et al., Mol. Pharmacol., 65, 257-266 (2004).25. Hoare, S,R.J., et al., Mol. Pharmacol., 63, 751-765 (2003).26. Urwyler, S., et al., J. Pharmacol. Exp. Ther., 307 322-330 (2003).27. Cryan, J.F., et al., J. Pharmacol. Exp. Ther., 310, 952-963 (2004).28. Nemeth, E.F., Cell Calcium, 35, 283-289 (2004).29. Shoback, D.M., et al., J. Clin. Endocrinol. Metab., 88, 5644-5649 (2003).30. Block, G.A., et al., New Engl. J. Med., 350, 1516-1525 (2004).31. Zhang, Z., et al., J. Biol. Chem., 277, 33736-33741 (2002).32. Potter, L.T., et al., Mol. Pharmacol., 35, 652-660 (1989).33. Bruns, R.F. and Fergus, J.H., Mol. Pharmacol., 38, 939-949 (1990).34. Zahn, K., et al., J. Pharmacol. Exp. Ther., 301, 720-728 (2002).35. Pagano, A., et al., J. Biol. Chem., 275, 33750-33758 (2000).36. Birdsall, N.J.M., et al., Br. J. Pharmacol., 78, 257-259 (1983).37. Spalding, T.A., et al., Mol. Pharmacol., 61, 1297-1302 (2001).38. Sur, C., et al., Proc. Natl. Acad. Sci. USA, 100, 13674-13679 (2003).39. Gao, Z., IJzerman, A.P., Biochem. Pharmacol., 60, 669-676 (2000).40. Gao, Z.G., et al., Biochem. Pharmacol., 65, 525-534 (2003).41. Hoare, S.R.J. and Strange, P.G., Mol. Pharmacol., 50, 1295-1308 (1996).42. Hoare, S.R.J., et al., Br. J. Pharmacol., 130, 1045-1059 (2000).43. Fawzi, A.B., et al., Mol. Pharmacol., 59, 30-37 (2001).44. Lanzafame, A. and Christopoulos, A., J. Pharmacol. Exp. Ther., 308, 830-837
(2004).45. Swaminath, G., et al., Mol. Pharmacol., 61, 65-72 (2002).46. Schetz, J.A. and Sibley, D.R., J. Neurochem., 68, 1990-1997 (1997).47. Conigrave, A.D., et al., Proc. Natl. Acad. Sci. USA, 97, 4814-4819 (2000).48. Thomas, E.A., et al., Proc. Natl. Acad. Sci. USA, 94, 14115-14119 (1997).49. Hedlund, P.B., et al., Biochem. Pharmacol., 58, 1807-1813 (1999).50. Fillion, G., et al., Behav. Brain Res., 73, 313-317 (1996).51. Massot, O., et al., Ann. NY Acad. Sci., 861, 174-182 (1998).52. Bhattacharya, S. and Linden, J., Biochim. Biophys. Acta, 1265, 15-21 (1995).53. Kollias-Baker, C.A., et al., J. Pharmacol. Exp. Ther., 281, 761-768 (1997).54. Kourounakis, A., et al., Biochem. Pharmacol., 61, 137-144 (2001).55. Musser, B., et al., J. Pharmacol. Exp. Ther., 288, 446-454 (1999).56. Gao, Z.G., et al., Mol. Pharmacol., 60, 1057-1063 (2001).57. Gao, Z.G., et al., Mol. Pharmacol., 62, 81-89 (2002).58. Waugh, D.J., et al., J. Pharmacol. Exp. Ther., 291, 1164-1171 (1999).59. Molderings, G.J., Br. J. Pharmacol., 130, 1706-1712 (2000).60. Hammerland, L.G., et al., Mol. Pharmacol., 53, 1083-1088 (1998).61. Nemeth, E.F., J. Mol. Endocrinol., 29, 15-21 (2000).62. Petrel, C., et al., J. Biol. Chem., 279, 18990-18997 (2004).63. Sabroe, I., et al., J. Biol. Chem., 275, 25985-25992 (2000).64. Zhao, J., et al., J. Exp. Med., 190, 101-111 (1999).65. Bertini, R., et al., Proc. Natl. Acad. Sci. USA, 101, 11791-11796 (2004).66. Cox, M.A., et al., Mol. Pharmacol., 59, 707-715 (2001).67. Talbodec, A., et al., Mol. Pharmacol., 57, 797-804 (2000).68. Blandin, V., et al., Mol. Pharmacol., 58, 1461-1469 (2000).69. Carroll, F.Y., et al., Mol. Pharmacol., 59, 965-973 (2001).70. Litschig, S., et al., Mol. Pharmacol., 55, 453-461 (1999).71. Lavreysen, H., et al., Mol. Pharmacol., 63, 1082-1093 (2003).72. Malherbe, P., et al., J. Biol. Chem., 278, 8340-8347 (2003).73. Knoflach, F., et al., Proc. Natl. Acad. Sci. USA, 98, 13402-13407 (2001).74. Johnson, M.P., et al., J. Med. Chem., 46, 3189-3192 (2003).
Ligands Interacting with Allosteric Sites... (continued)
®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
5
Allo
ster
ic L
igan
ds
to
G P
rote
in-C
ou
ple
d R
ecep
tors
References (continued on page 8)
®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
6A
llost
eric
Lig
and
s to
G
Pro
tein
-Co
up
led
Rec
epto
rs
Ligands Interacting with Allosteric Sites... (continued)
Receptor Subtype Ligands Allosteric Effect Prod. Code References
Adenosine
A1 PD 81,723 Positive [33,52-55]
Amilorides Negative A 7410 [40]
A2A Amilorides Negative A 7410 [39]
A3 VU5455 Positive [56]
VU8504 Positive [56]
DU124183 Positive [57]
Adrenoceptor
α1A Benzodiazepines Positive [58]
Amilorides Negative A 7410 [18]
α2A Amilorides Negative A 7410 [17,19]
α2D Agmatine Positive A 7127 [59]
β2 Zinc Positive A 6014 [43]
Calcium-Sensing
NPS 467 Positive [60]
NPS 568 Positive [60]
AMG 073 [Cinacalcet] Positive [28-30]
NPS 2143 Negative [61]
Calhex231 Negative [62]
L-Amino acids Positive [31,47]
Chemokine
CCR1/3 UCB35625 Negative [63]
CCR4/5 Trichosanthin Positive [64]
CXCR1/2 Repertaxin Negative [65]
CXCR3 IP-10 Negative C 8490 [66]
CXCR3 I-TAC Negative [66]
CXCR4 Trichosanthin Positive [64]
CorticotrophinReleasing Factor CRF1 Antalarmin Negative A 8727 [25]
NBI 27914 Negative N 3911 [25]
NBI 35965 Negative [25]
DMP-696 Negative [25]
Dopamine
D1/2 Zinc Negative A 6014 [46]
D2/3/4 Amilorides Negative A 7410 [41,42]
Endothelin
ETA Aspirin Negative A 5376 [67]
Salicylic acids Negative [67,68]
GABAB
CGP-7930 Positive C 0862 [22]
CGP-13501 Positive C 0987 [22]
GS39783 Positive G 5919 [26,27]
Table 2. Some allosteric ligands that act as GPCRs.Positive: refers to enhancement of agonist/endogenous ligand binding and/or functionNegative: refers to allosteric inhibition of agonist/endogenous ligand binding and/or functionNeutral: refers to neutral cooperativity with acetylcholine
Ligands Interacting with Allosteric Sites... (continued)
®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
7
Allo
ster
ic L
igan
ds
to
G P
rote
in-C
ou
ple
d R
ecep
tors
Receptor Subtype Ligands Allosteric Effect Prod. Code References
Glutamate
mGlu1 BAY 36-7620 Negative [69,70]
CPCCOEt Negative C 9611 [70,71]
R214127 Negative [71]
NPS 2390 Negative N 4786 [71]
EM-TBPC Negative [72]
Ro 01-6128 Positive [73]
Ro 67-7476 Positive [73]
mGlu2 LY-487379 Positive [74]
LY-181837 Positive [75]
Phenyl-tetrazolyl acetophenones Positive [76]
mGlu4 SIB1893 Positive S 9311 [77]
MPEP Positive M 5435 [77]
(–)-PHCCC Positive [78,79]
mGlu5 MPEP Negative M 5435 [35]
3,3'-Difluorobenzaldazine Positive [80]
CPHHA Positive [81]
Muscarinic
M1-M5 Gallamine Negative G 8134 [9,82]
Alcuronium Negative [83]
Strychnine Negative S 0532, S 8753 [10]
Brucine Positive M1 B 0253 [11,12]
Brucine N-oxide Positive M3/4 R158658 [11,12]
W-84 Negative [84]
Staurosporine Negative S 4400 [15]
KT 5720 Positive M1 K 3761 [15]
WIN 51,708 Negative W-103 [16]
WIN 62,577 Positive M3 W-104 [16]
Thiochrome Positive M4 T 7891 [24]Neutral M1/2/3/5
Tacrine Negative A 3773 [32]
McN-A-343 Negative C 7041 [36]
Neurokinin
NK1 Heparin Positive H 0777, H 3393 [85]
Purine
P2Y1 2,2'-Pyridylisatogen tosylate Negative [86-88]
Serotonin
5-HT1B/D 5-HT Moduline Negative [50,51]
5-HT2A Oleamide Positive O 2136 [48]
5-HT2C PNU-69176E Positive [89]
5-HT7 Oleamide Negative O 2136 [48,49]
Table 2. Some allosteric ligands that act as GPCRs. (continued)Positive: refers to enhancement of agonist/endogenous ligand binding and/or functionNegative: refers to allosteric inhibition of agonist/endogenous ligand binding and/or functionNeutral: refers to neutral cooperativity with acetylcholine

®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
8A
llost
eric
Lig
and
s to
G
Pro
tein
-Co
up
led
Rec
epto
rs
References (continued from page 5)75. Barda, D.A., et al., Bioorg. Med. Chem. Lett., 14, 3099-3102 (2004).76. Pinkerton, A.B., et al., J. Med. Chem., 47, 4595-4599 (2004).77. Mathiesen, J.M., et al., Br. J. Pharmacol., 138, 1026-1030 (2003).78. Maj, M., et al., Neuropharmacology, 45, 895-906 (2003).79. Marino, M.J., et al., Proc. Natl. Acad. Sci. USA, 100, 13668-13673 (2003).80. O'Brien, J.A., et al., Mol. Pharmacol., 64, 731-740 (2003).81. O'Brien, J.A., et al., J. Pharmacol. Exp. Ther., 309, 568-577 (2004).82. Clark, A.L. and Mitchelson, F., Br. J. Pharmacol., 58, 323-331 (1976).83. Proska, J. and Tucek, S., Mol. Pharmacol., 45, 709-717 (1994).84. Lüllman, H., et al., Eur. J. Pharmacol., 6, 241-247 (1969).85. Knaus, G.A., et al., Eur. J. Pharmacol., 207, 267-270 (1991).86. Spedding, M., et al., Br. J. Pharmacol., 53, 575-583 (1975).87. King, B.F., et al., Br. J. Pharmacol., 117, 1111-1118 (1996).88. Gao, Z.G., et al., Biochem. Pharmacol., 68, 231-237 (2004).89. Im, W.B., et al., Mol. Pharmacol., 64, 78-84 (2003).
Ligands Interacting with Allosteric Sites... (continued)
Antibodies
A-268 Anti-A1 Adenosine Receptor
A-269 Anti-A2A Adenosine Receptor
A-270 Anti-α1 Adrenergic Receptor
A-271 Anti-β1 Adrenergic Receptor
D-187 Monoclonal Anti-D1 Dopamine Receptor, Clone 1-1-F11 s.E6
E 3651 Anti-ETA Endothelin Receptor
G 5915 Anti-GABAB Receptor
G 5293 Anti-phospho-GABAB Receptor [pSer892] R2 Subunit
G 9665 Anti-Glutamate Receptor 1A, Metabotropic (mGluR1A)
G 9790 Anti-Glutamate Receptor 2/3, Metabotropic(mGluR2/3)
G 9915 Anti-Glutamate Receptor 5, Metabotropic (mGluR5)
M 9808 Anti-Muscarinic Acetylcholine Receptor (M1)
M 9558 Anti-Muscarinic Acetylcholine Receptor (M2)
M-272 Monoclonal Anti-Muscarinic Acetylcholine Receptor(M2), Clone 31-1D1
P 6487 Anti-P2Y1 Purinergic Receptor
S 4812 Anti-5-HT2A Serotonin Receptor
S 5062 Anti-5-HT7 Serotonin Receptor
S 8305 Anti-NK1 Tachykinin (Substance P) Receptor
Receptors
E-005 Benzodiazepine Receptor
D-152 D3 Dopamine Receptor, human
D-181 D3 Dopamine Receptor, rat
A-213 α2A Adrenergic Receptor
D-178 D1 Dopamine Receptor
D-180 D2long Dopamine Receptor
D-177 D4.2 Dopamine Receptor, human, CHO cells
D 2439 D4.2 Dopamine Receptor, human, Sf9 cells
M 4560 M2 Muscarinic Acetylcholine Receptor
M-176 M3 Muscarinic Acetylcholine Receptor
M 4810 M4 Muscarinic Acetylcholine Receptor
S -177 5-HT7 Serotonin Receptor, human
S-162 5-HT7 Serotonin Receptor, rat
GPCR Receptors and Antibodies available from Sigma-RBI
About the AuthorNigel Birdsall received his graduate and postgraduate degreesfrom Cambridge University. After postdoctoral research on thechemical mechanisms of carcinogenesis at the Sloan-KetteringInstitute for Cancer Research in New York, he returned toCambridge and the new MRC Molecular Pharmacology Unit.There he studied the structure and dynamics of lipids and lipid-protein interactions in membranes, primarily using NMR tech-niques. He relocated to the National Institute for Medical Researchat Mill Hill in London and began to investigate the detailed bind-ing and functional properties of G protein-coupled receptors,especially muscarinic acetylcholine receptors. He is currentlyProgram Leader in the Division of Physical Biochemistry at NIMRand an Honorary Reader in Pharmacology at University College,London.
Sigma-RBI is especially proud of its association with Dr.Ciechanover who in September 2003 authored an articleentitled “The Ubiquitin-Proteasome System: Death ofProteins is Required for Life of Cells” for the division’squarterly Celltransmissions cell signaling newsletter. Dr.Ciechanover is currently director of the Rappaport FamilyInstitute for Research in Medical Sciences at the Technion(Israel Institute of Technology) in Haifa, Israel.
“We feel honored to be able to include Dr. Ciechanoveramong our Celltransmissions authors,” stated Dr. KeithWatling, Director of Sigma-RBI, “His landmark discoveries,together with Drs. Hershko and Rose, have provided criticalinsight into how proteins are degraded and disposed ofinside the cell.”
Sigma-RBI is the leading supplier of products for ubiquitin-proteasome and cell stress research. Our product lineincludes ubiquitin proteins, inhibitors and antibodies, pro-teosome fractions and a range of enzymes, inhibitors andantibodies for nitric oxide, cell stress and protein degrada-tion research.
For more information, please visit:www.sigma-aldrich.com/ubiquitin-proteasome
2004 Nobel Prize in Chemistryawarded to Celltransmissionsauthor for the discovery ofubiquitin-mediated proteindegradation continued from cover

To request more information, please email Sigma-Aldrich at [email protected] call us at 800-333-7306 (USA)
L E A D E R S H I P I N L I F E S C I E N C E , H I G H T E C H N O L O G Y A N D S E R V I C ESIGMA-ALDRICH CORPORATION • BOX 14508 • ST. LOUIS • MISSOURI • 63178 • USAsigma-aldrich.com
Whether you are looking to acquire a large diver-
sity library or fill chemical space within an
established collection, the Sigma-Aldrich Screening
Collection is a rich source of high-quality compounds.
Consisting of over 90,000 compounds predicted to be
“drug-like” based on Lipinski’s Rule of Five, our collection
is in stock and ready for immediate delivery. By focusing
on structural diversity, purity, and reliability of resupply,
Sigma-Aldrich is one of the preferred screening com-
pound suppliers for biotechnology, pharmaceutical, and
agrochemical companies. When you think about what
you want in your library, see for yourself why drug dis-
covery researchers worldwide trust what we have in ours.
• High level of diversity Maximize your coverage of chemicalspace in a stand alone library or withinan existing collection.
• Drug-like compounds Screen compounds with predicted drug-like properties to increase the potentialfor development of leads with favorableADME profiles.
• High-purity samples Ensure that observed activity isattributable to compound of interestand not to impurities.
• All compounds in stock Order compounds when you need them.Compounds are ready for immediateformatting and shipment.
• Guaranteed re-supply Have confidence knowing that yourwork won't be interrupted by theinconvenience of re-sourcing orresynthesis.
• Flexible formatting Choose your preferred packaging.Receive compounds in the format thatworks best for you with short deliverytimes.
• No reactives Simplify your screening results byreducing the number of false positives.

®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
10N
ew P
rod
uct
Hig
hlig
hts
Antibodies to Potassium Channels
Antibodies to the KIR Family of Potassium Channels
The action of potassium (K+) channels is regulated by voltage,calcium and a variety of neurotransmitters. Each subfamily gener-ally consists of a primary pore-forming α-subunit that is associat-ed with several regulatory subunits [1]. To date, 70 different genesthat encode the α-subunits of K+ channels have been identified.The vast family of K+ channels has been subdivided into the threemain subfamilies: the 2 transmembrane (TM), 4 TM and 6 TM K+
channels [2].
Inwardly rectifying potassium (KIR) channels belong to the 2 TMfamily. They form a diverse family with seven main subtypes foundto date. They have two main physiological roles, to mediate trans-port across the cell membrane and to stabilize the restingmembrane potential near the K+ equilibrium potential. KIR 2 typechannels are partially responsible for controlling excitability of cells
within the heart and brain, while KIR 6 type channels are impor-tant in the regulation of insulin secretion, the response to cardiacischemia and the control of vascular smooth muscle tone [3].
The voltage-gated K+ (Kv) channels belong to the 6 TM family ofK+ channels. The KCNQ family of voltage-gated K+ channelsincludes five known members referred to as KV7.1 - KV7.5(KCNQ1 to KCNQ5) [4]. The KCNQ1 channel is important incardiac function. Mutations on KCNQ1 can be responsible forlong QT syndrome, a cardiac disorder that causes arrhythmias andsudden death [5]. KCNQ2/KCNQ3 heteromultimers are believed tobe the molecular correlates of the so-called M current [6].Mutations in either gene are associated with a form of epilepsyknown as benign familial neonatal convulsions [5].
Anti-Potassium Channel KIR2.2, Prod. Code P 4496Anti-Potassium Channel KIR2.2 is developed in rabbit using apeptide corresponding to amino acid residues 391-409 of ratKIR2.2 as immunogen. The epitope is conserved in mouse andhuman. Anti-Potassium Channel KIR2.2 specifically recognizes thePotassium Channel KIR2.2 protein in rat brain and kidney mem-branes by immunoblotting.
Anti-Potassium Channel KIR6.2, Prod. Code P 4621Anti-Potassium Channel KIR6.2 is developed in rabbit using apeptide corresponding to amino acid residues 372-385 of ratKIR6.2 as immunogen. The epitope is highly conserved in mouseand human. Anti-Potassium Channel KIR6.2 specifically recognizesthe Potassium Channel KIR6.2 protein in rat pancreas membranesby immunoblotting, and does not cross react with KIR6.1.
Antibodies to the KCNQ Family of Voltage-Gated Potassium Channels
Anti-Potassium Channel KV7.1 (KCNQ1), Prod. Code P 5372Anti-Potassium Channel KV7.1 (KCNQ1) is developed in rabbitusing a peptide corresponding to amino acid residues 661-676 ofhuman KCNQ1 as immunogen. This sequence has 14/16 residuesidentical in rat and mouse. Anti-Potassium Channel KV7.1 specifi-cally recognizes the Potassium Channel KV7.1 protein in rat heartmembranes by immunoblotting.
Anti-Potassium Channel KCNQ2 (KV7.2, Voltage-gatedPotassium Channel QKT Subfamily Member 2), Prod. Code P 4371 Anti-Potassium Channel KCNQ2 (KV7.2) is developed inrabbit using a peptide corresponding to amino acid residues 578-593 of rat KCNQ2 as immunogen. The epitope is highly conservedin human. Anti-Potassium Channel KCNQ2 specifically recognizesKCNQ2 protein in KCNQ2 transfected HEK-293 cells byimmunoblotting. It does not cross react with KCNQ3 or other QKTproteins. There are at least nine recognized splice variants of ratKCNQ2. This antibody recognizes all except splice variants B and G.
Anti-Potassium Channel KCNQ3 (KV7.3, Voltage-gatedPotassium Channel QKT Subfamily Member 3), Prod. Code P 5121Anti-Potassium Channel KCNQ3 (KV7.3) is developed in rabbitusing a peptide corresponding to amino acid residues 668-686 ofrat KCNQ3. The epitope is highly conserved in mouse and human.Anti-Potassium Channel KCNQ3 specifically recognizes KCNQ3protein in rat brain and may be used for the detection ofPotassium Channel KCNQ3 by immunoblotting and immunohisto-chemistry.References1. Alexander, S.P., et al., Guide to receptors and channels, 1st edition., Br. J.
Pharmacol., 141, Suppl 1:S1-S126 (2004).2. Gutman, G.A., et al., International Union of Pharmacology. XLI. Compendium
of voltage-gated ion channels: potassium channels., Pharmacol. Rev., 55, 583-586 (2003).
3. Ashcroft, F.M., ed, Ion Channels and Disease, Academic Press, 2000.4. Robbins, J., KCNQ potassium channels: physiology, pathophysiology, and
pharmacology., Pharmacol. Ther., 90, 1-19 (2001).5. Hubner, C.A. and Jentsch, T.J., Ion channel diseases., Hum. Mol. Genet., 11,
2435-2345 (2002).6. Wang, H.S., et al., KCNQ2 and KCNQ3 potassium channel subunits: molecular
correlates of the M-channel., Science, 282, 1890-1893 (1998).
Explore our NEW Antibody Guide!
• Over 250 pages• Over 3,000 quality antibodies all rigorously
tested• Hundreds of NEW antibodies• Organized by area of study for easy reference• World-renowned technical service• Timely delivery anywhere
Fill out and return the enclosed Business ReplyCard to receive your Guide: Antibodies, Toolsfor Life Science Research!

®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
11
New
Pro
du
ct H
igh
ligh
ts
SR 12813: PXR agonist and HMG-CoAreductase inhibitor
The pregnane X receptor (PXR), amember of the nuclear receptor fami-ly of ligand-activated transcriptionfactors, is a transcriptional regulatorof multiple cytochrome P450 andmultidrug resistance-associated pro-teins. SR12813 activates both humanand rabbit PXR [1,2]. SR12813
inhibits cholesterol synthesis by reducing cellular HMG-CoAreductase activity, displaying an IC50 value of 0.85 µM [3].
References1. Jones, S., et al., The pregnane X receptor: a promiscuous xenobiotic receptor
that has diverged during evolution., Mol. Endocrinol., 14, 27-39 (2000).2. Watkins, R.E., et al., The human nuclear xenobiotic receptor PXR: structural
determinants of directed promiscuity., Science, 292, 2329-2333 (2001).3. Berkhout, T.A., et al., The novel cholesterol-lowering drug SR-12813 inhibits
cholesterol synthesis via an increased degradation of 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase., J. Biol. Chem., 271, 14376-14382 (1996). YM-53601: Squalene synthase inhibitor
Squalene synthase is an enzymevital for cholesterol biosynthesis.YM-53601, a squalene synthaseinhibitor, lowered not only plasmacholesterol, but also plasma triglyc-eride levels. YM-53601 equallyinhibited squalene synthase func-
tion in hepatic microsomes prepared from several animal speciesand suppressed cholesterol biosynthesis in rats (ED50 32 mg/kg).In rhesus monkeys, when dosed at 50 mg/kg twice daily for 21 days, YM-53601 decreased plasma non-HDL-C (high densitylipoprotein cholesterol) by 37%, whereas the HMG-CoA reduc-tase inhibitor, pravastatin, when dosed at 25 mg/kg twice dailyfor 28 days, failed to do so.
Reference1. Ugawa, T., et al., YM-53601, a novel squalene synthase inhibitor, reduces
plasma cholesterol and triglyceride levels in several animal species., Br. J.Pharmacol., 131, 63-70 (2000).
sPLA2 inhibitor Available First from Sigma-RBI
Orally active, potentsecretory PhospholipaseA2 (sPLA2; Group IIa)inhibitor - IC50 = 29 nMagainst human recombi-nant nonpancreatic sPLA2.
References1. Arumugam, T.V., et al., Comparative protection against rat intestinal reper-
fusion injury by a new inhibitor of sPLA2, COX-1 and COX-2 selectiveinhibitors, and an LTC4 receptor antagonist., Br. J. Pharmacol., 140, 71-80(2003).
2. Hansford, K.A., et al., D-Tyrosine as a chiral precusor to potent inhibitors ofhuman nonpancreatic secretory phospholipase A2 (IIa) with anti-inflamma-tory activity., Chembiochem., 4, 181-185 (2003).
O
CO2H
HN
O
Prod. Code Y 0128
Prod. Code S 3319
Exo 1: Reversible inhibitor of exocytosis
Exo 1 [2-(4-fluorobenzoylamino)benzoic acidmethyl ester] is a cell-permeable methylan-thranilate analog that reversibly inhibitsvesicular traffic from the ER (endoplasmicreticulum) to Golgi in mammalian cells. It is aselective and potent modifier of Golgi ADP-ribosylation factor (ARF) 1 GTPase activityand has no effect on other endocytic
organelles such as endosomes and trans-Golgi network (TGN).Exo 1 inhibits exocytosis displaying an IC50 value of 20 µM. [1].
Reference1. Feng, Y., et al., Exo1: a new chemical inhibitor of the exocytic pathway.
Proc. Natl. Acad. Sci. USA, 100, 6469-6474 (2003).
NH
O
F
N
HCl
Prod. Code S 4194
Prod. Code E 8280
HO
P(O)(OC2H5)2
P(O)(OC2H5)2H3C
H3CH3C
CH3
H3C CH3
COOCH3
NH
O
F
DAPT (LY-374973): γ-secretase inhibitor
HN
NH
O
CH3
F
F
O
COO-t-Bu
Ph
Alzheimer’s disease (AD)accounts for the majority of thedementia diagnosed over theage of 60 and represents thelargest unmet medical need inneurology with over 12 millionsufferers worldwide. The
pathogenesis of AD is believed to result from the progressiveaccumulation in the brain of β-amyloid (Aβ), a 4 kDa protein. Aβoriginates from the proteolytic cleavage of amyloid precursorprotein (APP) by β-secretase followed by γ-secretase cleavage [1].Because inhibition of γ-secretase blocks the production of Aβ,the identification of compounds that block the activity of thisenzyme has become a major focus of AD research.
Sigma-RBI is pleased to offer DAPT, a γ-secretase inhibitor. DAPTinhibits γ-secretase in cultured cells, thus reducing Aβ levels. Intransgenic mice, which develop amyloid plaques due to the over-expression of mutant βAPP, DAPT dose-dependently (10, 30, 100mg/kg, sc) reduces the Aβ burden within 3 hr post administrationand maintains reduced levels 18 hr after treatment [2]. In addi-tion, DAPT causes Notch phenotyping in zebrafish embryos andDrosophila due to the inhibition of Notch intracellular domaingeneration, which is involved in gene transcription regulation,processing, translocation and signaling [3,4].
References1. Lammich, S., et al., Presenilin-dependent intramembrane proteolysis of
CD44 leads to the liberation of its intracellular domain and the secretion ofan Abeta-like peptide., J. Biol. Chem., 277, 44754-44759 (2002).
2. Dovey, H.F., et al., Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain., J. Neurochem., 76, 173-181 (2001).
3. Micchelli, C.A., et al., Gamma-secretase/presenilin inhibitors for Alzheimer'sdisease phenocopy Notch mutations in Drosophila., FASEB J. 17, 79-81 (2003).
4. Sastre, M., et al., Presenilin-dependent gamma-secretase processing ofbeta-amyloid precursor protein at a site corresponding to the S3 cleavageof Notch., EMBO Rep., 2, 835-841 (2001).
Related Product
Name Description Prod. Code
L-685,458 Potent, selective, structurally novel γ-secretase L 1790inhibitor; equipotent in inhibition of both Aβ1-42 and Aβ1-40 peptide fragment production
Prod. Code D 5942
HN
NH
O
CH3
F
F
O
COO-t-Bu
Ph

®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
12N
ew P
rod
uct
s
APOPTOSIS AND CELL CYCLE
D 5817 DMXAAApoptosis inducer; anti-vascular.
E 8405 EB-47Potent inhibitor of PARP.
I 1159 trans-4-Iodo-4'-boranyl-chalconeMDM2 inhibitor; immunomodulator.
M 5318 MC2p
M 5443 NLS-MC2p
R 3528 Anti-phospho-Rb [pThr356] (rabbit)
R 3403 Anti-phospho-Rb [pSer249/pThr252] (rabbit)
R 3278 Anti-phospho-Rb [pSer612] (rabbit)
R 4028 Anti-phospho-Rb [pSer780] (rabbit)
R 3778 Anti-phospho-Rb [pSer807] (rabbit)
R 3903 Anti-phospho-Rb [Ser807/pSer811] (rabbit)
R 4278 Anti-phospho-Rb [pSer811] (rabbit)
R 4153 Anti-phospho-Rb [pThr821] (rabbit)
R 4403 Anti-phospho-Rb [pThr826] (rabbit)
T 2825 TIQ-APotent inhibitor of PARP; anti-apoptotic.
ASSAY KITS
CS0400 Casein kinase assay kit
CS0340 Dihydrofolate reductase (DHFR) assay kit
CS0260 Glutathione assay kit
CS0410 Glutathione S-transferase (GST) assay kit
CS0380 JNK activity assay kit
CS0250 p38 MAPK activity assay kit
CS0390 Mitochondria staining kit
CS0430 Tau Elisa, mouse
CS0440 Phospho-Tau [pSer199] Elisa, mouse
CS0170 Thioredoxin reductase assay kit
CYTOKINES, GROWTH FACTORS AND HORMONES
A 9853 Anti-Androgen Receptor (rabbit)
C 3866 Anti-Calcitonin Receptor-Like Receptor (rabbit)
M 7068 MPP dihyrochlorideSpecific ERα estrogen receptor antagonist.
P 9121 Prolactin Receptor Fc Chimerahuman, recombinant, expressed in NSO cells
V 5014 Anti-phospho-VEGFR2 [pTyr1054/pTyr1059] (rabbit)
V 5239 Anti-phospho-VEGFR2 [pTyr951] (rabbit)
V 5264 Anti-phospho-VEGFR2 [pTyr1214] (rabbit)
V 5389 Anti-phospho-VEGFR2 [pTyr1054] (rabbit)
CYTOSKELETON AND EXTRACELLULAR MATRIX
A 3854 Monoclonal Anti-β-Actin-PeroxidaseClone AC-15 (mouse)
C 5615 Monoclonal Anti-phospho β-Catenin [pSer45]Clone BCS-45 (mouse)
C 0864 Anti-phospho-Cortactin [pTyr466], mouse (rabbit)
C 0739 Anti-phospho-Cortactin [pTyr421], mouse (rabbit)
G 8419 GW311616APotent, intracellular human neutrophil elastase inhibitor.Sold for research purposes under agreement fromGlaxoSmithKline.
H 4663 HMBAInhibitor of microtubule polymerization; apoptosis inducer.
P 8996 Monoclonal Anti-Pinch-1, Clone PINCH-C58 (mouse)
P 0372 Anti-Podocin (rabbit)
P 7496 Anti-Protein Disulfide Isomerase (PDI) (rabbit)
R 3653 Anti-Radixin (rabbit)
T 1450 Anti-γ-Tubulin (LL-17) (rabbit)
T 0950 Anti-γ-Tubulin (QG-17) (rabbit)
T 2200 Anti-β III Tubulin (rabbit)
V 4139 Anti-Vinculin (rabbit)
V 4889 Anti-phospho-Vinculin [pTyr822] (rabbit)
G PROTEINS AND CYCLIC NUCLEOTIDES
B 4560 6-Bnz-cAMPMembrane permeable and selective cAMP-dependentprotein kinase (PKA) activator.
A 2104 8-AHA-cAMPSelective cAMP-dependent protein kinase (PKA) activator.
C 8990 Sp-8-pCPT-cAMPSelective cAMP-dependent protein kinase (PKA) activator.
F 9553 FTI-276Highly potent Ras CAAX peptidomimetic, antagonizes Hand K-Ras oncogenic signaling; inhibitor of farnesyl-transferase (FTase) in vitro.
F 9803 FTI-277Highly potent Ras CAAX peptidomimetic; inhibitor offarnesyltransferase.
G 5169 GGTI-298CAAX peptidomimetic; inhibitor of geranylgeranyl-transferase I (GGTase).
G 5294 GGTI-2133CAAX peptidomimetic; inhibitor of geranylgeranyl-transferase I (GGTase I).
G 2420 Anti-G Protein-Coupled Receptor BG37/TGR5 (rabbit)
G 2670 Anti-G Protein-Coupled Receptor AGR9 (rabbit)
I 8283 ICI 63197Phosphodiesterase IV (PDE IV) inhibitor.
P 0747 8-MA-cAMPSelective cAMP-dependent protein kinase (PKA) activator.
P 0622 PET-cGMPcGMP-dependent protein kinase (PKG I and PKG II) activator.
P 0872 8-PIP-cAMPSelective cAMP-dependent protein kinase (PKA) activator.
S 6069 Sp-5,6-DCI-cBIMPSSelective cAMP-dependent protein kinase (PKA) activator.
New Products for Cell Signaling & Neuroscience

New
Pro
du
cts
GENE REGULATION AND EXPRESSION
D 5817 DMXAAApoptosis inducer; anti-vascular.
A 8103 Monoclonal Anti-hABH1, Clone HABH1-151 (mouse)
A 8228 Monoclonal Anti-hABH2, Clone HABH2-7 (mouse)
A 8353 Monoclonal Anti-hABH3, Clone HABH3-99 (mouse)
A 8853 Anti-phospho-ATF2 [pThr71] (rabbit)
B 3810 Anti-BRCA1-Associated Protein BRAP2 (rabbit)
C 6240 CITCOConstitutive adrostane receptor (CAR) agonist; nuclearreceptor NR113 agonist.
D 4567 Anti-DNMT1 (rabbit)
F 1054 Anti-FOXC2 (rabbit)
G 8794 Anti-GATA4 (rabbit)
G 8669 Anti-GATA5 (rabbit)
G 6669 Monoclonal Anti-Gemin 2, Clone 2E17 (mouse)
G 6544 Monoclonal Anti-Gemin 3, Clone 12H12 (mouse)
H 4788 Anti-HES-2 (rabbit)
H 5038 Anti-HES-3 (rabbit)
H 4913 Anti-HEX (rabbit)
I 8158 Anti-Irx2a (rabbit)
H 4538 Monoclonal Anti-Histone Deacetylase 5 (HDAC5)Clone HDAC5-35 (mouse)
I 9283 ITEEndogenous ligand identified for aryl hydrocarbon receptor; ligand inducible transcription factor.
L 5042 Anti-LHX4 (rabbit)
L 5167 Anti-LIG4 (rabbit)
M 6818 Monoclonal Anti-MeCP2, Clone Mec-168 (mouse)
G 6919 Anti-G9a Methyltransferase (rabbit)
M 6193 Anti-MRE11A (rabbit)
M 5943 Anti-Mtsh1/teashirt (rabbit)
N 8411 Anti-NeuroD2 (rabbit)
N 8286 Anti-NSCL 2 (HEN2) (rabbit)
P 6246 Monoclonal Anti-PABP, Clone 10E10 (mouse)
P 8872 Anti-PACAP Receptor Type 1 (rabbit)
P 6746 Monoclonal Anti-PML, Clone PML-97 (mouse)
P 6871 Anti-PRMT1 (NQ-15) (rabbit)
P 6996 Anti-PRMT1 (TK-16) (rabbit)
P 7497 Anti-Prostate-Specific G Protein-Coupled Receptor(PSGR) (rabbit)
R 6153 Monoclonal Anti-RhoE, Clone 4 (mouse)
R 4528 Monoclonal Anti-hnRNP-A1, Clone 9H10 ( mouse)
R 4653 Monoclonal Anti-hnRNP-A2/B1, Clone DP3B3 (mouse)
R 5028 Monoclonal Anti-hnRNP-C1/C2, Clone 4F4 (mouse)
R 4903 Monoclonal Anti-hnRNP-L, Clone 4D11 (mouse)
R 5653 Monoclonal Anti-hnRNP-Q, Clone 18E4 (mouse)
S 4319 Anti-Scratch (rabbit)
S 1444 Anti-Sprouty 2 (N-terminal) (rabbit)
S 1819 Anti-Sprouty 2 (C-terminal) (rabbit)
GENE REGULATION AND EXPRESSION (continued)
S 4194 SR 12813PXR (NR 112) agonist; cholesterol lowering drug; HMGCoA reductase inhibitor.
U 4758 Monoclonal Anti-U2AF65, Clone MC3 (mouse)
Z 4775 ZebularineCytidine analog; cytidine deaminase inhibitor; DNA demethylating agent.
IMMUNE CELL SIGNALING AND BLOOD
A 1854 (–)-ArctigeninDibenzylbutyrolactone ligand; natural product; viralintegrase and topoisomerase II inhibitor; antioxidant;antiviral; anti-inflammatory and immunomodulator.
B 3685 Monoclonal Anti-BOB78, Clone BOB78 (mouse)
P 8747 Anti-Proteinase-Activated Receptor 2 (rabbit)
T 5700 Anti-Thrombin Receptor (rabbit)
INTRACELLULAR CALCIUM SIGNALING
C 4491 Anti-Calcium-Sensing Receptor CASR (rabbit)
ION CHANNELS
A 6603 Aa1, recombinant, expressed in E. coliBlocks Shaker B channels expressed in Xenopus oocytes;blocks fast (A-type) K+ currents in cerebellar granular cells.
C 2240 Anti-Calcium Channel CaV3.1 (α1G) (rabbit)
C 2615 Chromanol 293BSlow delayed rectifier K+ current blocker acting viaKCNQ1 channels.
H 3038 Anti-HCN1 (rabbit)
I 6783 Isopimaric acidPotent opener of large conductance Ca2+-activated K+
(BK) channels.
K 0514 Monoclonal Anti-KCNK9 (TASK3), Clone KCN-75 (mouse)
M 3318 Anti-MiRP1 (rabbit)
N 7286 Anti-NHERF-1 (rabbit)
N 8161 NS309Ca2+-activated IK/SK K+ channel activator.
P 4996 Anti-Potassium Channel EAG-2 (rabbit)
P 4371 Anti-Potassium Channel KCNQ2 (rabbit)
P 5121 Anti-Potassium Channel KCNQ3 (rabbit)
P 4496 Anti-Potassium Channel Kir2.2 (rabbit)
P 4621 Anti-Potassium Channel Kir6.2 (rabbit)
S 6444 N-SalicyloyltryptamineCa2+-activated K+ channel activator; anticonvulsant;neuroprotectant.
New Products for Cell Signaling & Neuroscience
®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
13
New
Pro
du
cts

®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
14N
ew P
rod
uct
s
ION CHANNELS (continued)
S 0944 Slotoxin, recombinant, expressed in E. coliBlocks Maxi K+ channels.
S 0819 Anti-Sodium Channel NaV1.5 (rabbit)
T 0325 Anti-TRPC5 (rabbit)
LIPID SIGNALING
A 9978 AICARAMP-activated protein kinase (AMPK) activator.
C 4615 CloricromeneAnti-thrombotic coumarin derivative; produces coronarydilation; stimulates prostacyclins and lowers TXB2synthesis in platelets.
C 9115 CP-24879Delta5/Delta6 (∆5/∆6) desaturase inhibitor.
F 1179 FPL-55712LTD4 Leukotriene receptor antagonist.
H 4413 HydroxyfasudilRho-Kinase Inhibitor.
K 0639 Ki16425Subtype-selective, potent and reversible LPA1 and LPA3receptor antagonist.
M 5693 MEDICA 16ATP-citrate lyase inhibitor; potent triacylglycerol-loweringagent.
M 8192 15R-Methyl-prostaglandin D2Potent and selective DP2 prostanoid receptor agonist.
M 5818 Monoclonal Anti-hMps1, Clone N1 (mouse)
S 3319 sPLA2 inhibitorOrally active, potent secretory phospholipase A2 (sPLA2;Group IIa) inhibitor.
N 0912 NAEPASelective lysophosphatidic acid-1 (LPA1) receptor agonist;LPA mimetic.
O 4139 OrlistatPancreatic lipase inhibitor.
P 7622 Anti-Prostacyclin Receptor (rabbit)
P 7872 Anti-DP Prostanoid Receptor (rabbit)
P 7747 Anti-EP2 Prostanoid Receptor (rabbit)
P 8372 Anti-EP3 Prostanoid Receptor (rabbit)
P 8497 Anti-EP4 Prostanoid Receptor (rabbit)
P 8622 Anti-Prostaglandin F2α Receptor (rabbit)
S 4194 SR 12813PXR (NR 112) agonist; cholesterol lowering drug; HMGCoA reductase inhibitor.
S 3944 SW2871Novel and selective human sphingosine-1-phosphate sub-type 1 (S1P1) receptor agonist.
T 5200 Triparanol Desmosterol Delta24 (∆24) reductase inhibitor.
NEUROBIOLOGY
A 0479 AdrafinilWake-promoting agent; modafinil analog.
A 9603 Anti-APH-1aL (rabbit)
B 1935 S(+)-Butorphan mandelateκ/µ opioid receptor agonist.
D 5942 DAPT (LY-374973)γ-secretase inhibitor. Sold under license to US PatentNumbers 6,211,235 and 6,191,166.
N 9161 Notch-2/Fc Chimerarat, recombinant, expressed in NSO cells
N 9036 Notch-3/Fc Chimerahuman, recombinant, expressed in Sf 21 cells
N 8911 Notch-3/Fc Chimeramouse, recombinant, expressed in insect cells
P 9246 PB28 dihydrochlorideSelective σ2 sigma receptor ligand; putative agonist.
R 5403 Monoclonal Anti-Rhodopsin, Clone 1D4 (mouse)
S 5067 β-Secretase (BACE1), human, recombinant
S 2944 Monoclonal Anti-SMN, Clone 2B1 (mouse)
S 2069 Anti-SNAP29 (rabbit)
S 2319 Anti-SNAP23 (DS-19) (rabbit)
S 2194 Anti-SNAP23 (TS-19) (rabbit)
S 4819 Anti-Syntaxin 7 (rabbit)
V 4514 Anti-Vesicle Associated Membrane Protein 4,(VAMP4) (rabbit)
NEUROTRANSMISSION
A 3104 A-331440 L-tartaric acid saltNon-imidazole H3 histamine receptor antagonist. Subjectto US Patent Nos. 6,515,013 and 6,620,839. Sold underlicense from Abbott Laboratories.
A 6853 α2A-Adrenergic Receptor Preparation, human
A 6728 β1-Adrenergic Receptor Preparation, human
C 4366 Anti-Cholecystokinin B Receptor (rabbit)
C 6848 Ciproxifan maleatePotent, selective H3 histamine receptor antagonist.
C 4740 Cisapride5-HT4 serotonin receptor agonist.
C 4241 Anti-Corticotropin-Releasing Factor Receptor2(CRFR2) (rabbit)
D 6317 DMeOBNegative allosteric modulator of mGluR5 metabotropicglutamate receptors.
D 4067 δ Sleep-Inducing PeptideTrp-Ala-Gly-Gly-Asp-Ala-Ser-Gly-Glu
D 2442 Anti-Dopamine Transporter (rabbit)
E 7155 ETA Endothelin Receptor Preparation, human
G 5544 Anti-GABAA Receptor (α6 subunit) (rabbit)
G 5669 Anti-GABAA Receptor (α2 subunit) (rabbit)
G 0170 Anti-GABAA Receptor (α4 subunit), N-terminus (rabbit)
New Products for Cell Signaling & Neuroscience

®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
15
New
Pro
du
cts
New Products for Cell Signaling & Neuroscience
NEUROTRANSMISSION (continued)
G 0295 Anti-GABAA Receptor (α6 subunit), cytosolic loop(rabbit)
G 9169 Anti-GABAA Receptor (α4 subunit), cytosolic loop(rabbit)
G 9294 Anti-GABAA Receptor (α5 subunit), cytosolic loop(rabbit)
G 9419 Anti-GABAA Receptor (β1 subunit), cytosolic loop(rabbit)
G 9544 Anti-GABAA Receptor (β2 subunit), cytosolic loop(rabbit)
G 9669 Anti-GABAA Receptor (β3 subunit), cytosolic loop(rabbit)
G 9794 Anti-GABAA Receptor (δ subunit), N-terminus (rabbit)
G 9919 Anti-GABAA Receptor (γ2 subunit), cytosolic loop(rabbit)
G 4419 GalnonNon-peptide galanin receptor agonist.
G 7919 Anti-Glutamate Receptor 4, Metabotropic (mGluR4)(rabbit)
G 5419 Monoclonal Anti-Glutamic acid decarboxylase 67(GAD67), Clone K-87 (mouse)
I 0909 6-IodonordihydrocapsaicinTRPV1 (V1) vanilloid receptor antagonist.
I 7033 6'-Iodoresiniferatoxin (6-I-RTX)VR1 vanilloid receptor partial agonist.
J 3770 JNJ7777120First potent, selective non-imidazole H4 histaminereceptor antagonist.
K 0389 K41498CRF2 corticotropin releasing factor receptor antagonist(peptide); antisauvagine-30 analog.
M 4068 Mesoridazine benzenesulfonateD2 dopamine receptor antagonist; antipsychotic.
M 1818 Molindone hydrochlorideD2 dopamine receptor antagonist; MAO inhibitor.
M 9693 Anti-M2 Muscarinic Acetylcholine Receptor (rabbit)
M 9568 Anti-M3 Muscarinic Acetylcholine Receptor (rabbit)
N 7161 Nogo-66(1-40) antagonist peptidePromotes axonal regeneration by blocking effects ofmyelin-derived axon outgrowth inhibitors such asNogo66 or CNS myelin. Sold under a non-exclusivelicense. For research purposes only, and not fordiagnostic or therapeutic use or for use in humans.
O 5014 Anti-δ1 Opioid Receptor (OPRD1) (rabbit)
O 5139 Anti-µ1 Opioid Receptor (OPRM1) (rabbit)
P 1747 Anti-Phospho-Tyrosine Hydroxylase [Ser31] (rabbit)
P 2247 Anti-Phospho-Tyrosine Hydroxylase [Ser19] (rabbit)
P 1997 Anti-Phospho-Dynamin [Ser774] (rabbit)
P 2122 Anti-Phospho-Dynamin [Ser778] (rabbit)
P 4746 Anti-Purinergic Receptor P2X6 (rabbit)
P 4871 Anti-Purinergic Receptor P2Y12 (rabbit)
S 0694 Anti-Somatostatin Receptor Type 2 (rabbit)(lyophilized powder)
NEUROTRANSMISSION (continued)
S 2694 SB-408124OX1 orexin receptor antagonist. Sold for researchpurposes under agreement from GlaxoSmithKline.
S 1069 (S)-SNAP 5114GAT-2 and GAT-3 GABA transporter inhibitor.
S 1445 Anti-Somatostatin Receptor Type 1 (rabbit)
S 0695 Anti-Somatostatin Receptor Type 2 (rabbit)
S 0820 Anti-Somatostatin Receptor Type 3 (rabbit)
S 0945 Anti-Somatostatin Receptor Type 4 (rabbit)
S 1070 Anti-Somatostatin Receptor Type 5 (rabbit)
T 0575 TopiramateKainate GluR5 glutamate receptor antagonist; anti-convulsant.
T 5950 Anti-NK1 Tachykinin Receptor (rabbit)
T 6075 Anti-NK2 Tachykinin Receptor (rabbit)
T 5825 Anti-NK3 Tachykinin Receptor (rabbit)
V 4264 VK-28Neuroprotectant; brain permeable iron chelator.
NITRIC OXIDE AND CELL STRESS
E 8655 Monoclonal Anti-E6AP, Clone E6AP-330 (mouse)
H 4163 Anti-Heat Shock Factor 1 (HSF1) (rabbit)
H 4038 Anti-Heat Shock Protein 40 (rabbit)
S 5069 Anti-Superoxide Dismutase (MnSOD) (DD-17) (rabbit)
S 5569 Anti-Superoxide Dismutase (MnSOD) (KC-19) (rabbit)
PROTEIN PHOSPHORYLATION
P 5997 Anti-Phospho-CAM Kinase II [pThr305] (rabbit)
P 2372 Protein Kinase A, rat brain, purified
R 2903 Anti-RALT/MIG-6 (SG-16) (rabbit)
S 1819 Anti Sprouty-2 (C-terminal) (rabbit)
S 1444 Anti Sprouty-2 (N-terminal) (rabbit)
T 5575 TG003Potent, specific, reversible and ATP-competitive inhibitorof Cdc2-like kinase (Clk).
T 2949 Anti-mTOR (FRAP) (rabbit)
T 1325 Anti-phospho Tyrosine (rabbit)
T 1575 Anti-phospho Tyrosine (pTyr256) (rabbit)
V 4764 Anti-phospho-Vav1 [pTyr160] (rabbit)
V 4639 Anti-phospho-Vav3 [pTyr173] (rabbit)
Z 0151 Anti-phospho-ZAP-70 [pTyr292] (rabbit)
Z 0276 Anti-phospho-ZAP-70 [pTyr315/pTyr319] (rabbit)

®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
16Monoclonal Anti-PML
Prod. Code P 6746Clone PML-97, developed in mouse Purified mouse immunoglobulin Immunogen: recombinant full-length human PML (promyelocytic leukemia) proteinIsotype: IgG1Species Cross Reactivity: human, mouse, hamster
In acute promyelocytic leukemia (APL) patients, the PML(promyelocytic leukemia) protein is fused with the RARα(Retinoic Acid Receptor α) protein. In normal cells, the PMLprotein is found in nuclear bodies (PML NBs) while in APLpatients, the PML NBs are disrupted and form micro particu-late patterns in the nucleus and cytoplasm. In normal cells, thePML protein and the PML NB play a role in different processessuch as growth control, transcription, DNA repair, DNA replica-tion and RNA transport.
Applications: Western Blot (~90 kDa), ELISA, immunoprecipita-tion and immunocytochemistry
Cyclofenil: ERβ estrogen receptorantagonist
Hydroxyfasudil: Selective Rho kinaseinhibitor
Estrogen receptors are ligand-regulatedtranscription factors. There are twoestrogen receptor subtypes that differin tissue distribution, but exhibit over-lapping physiological functions. ERαexpression is primarily found in thebreast and uterus. In contrast, ERβ isfound mainly in brain, bone andvascular epithelium. Genetic knockout
experiments implicate ERα in reproductive physiology while ERβis implicated in cognitive function and bone metabolism.However, ERα and ERβ can form functional heterodimers andone subtype can partially compensate for the absence of theother. Cyclofenil is a nonsteroidal selective estrogen receptor β(ERβ) modulator (SERM). Cyclofenil displays 4.9-fold affinity forERβ versus ERα in a competitive radiometric binding assay using[3H]-estradiol and full length purified recombinant human ERαand ERβ receptors[1].
Given this complex functional relationship, selective antagonists,such as cyclofenil, are important tools for dissecting the functionsof these receptor subtypes and are potential clinical candidatesthat may selectively modulate bone, connective tissue and central(LH, FSH and prolactin secretion) versus uterine effects of estrogen[2].
References1. Muthyala, R.S., et al., Bridged bicyclic cores containing a 1,1-diarylethylene
motif are high-affinity subtype-selective ligands for the estrogen receptor.,J. Med. Chem., 46, 1589-1602 (2003).
2. Mueller, S.O. and Korach, K.S., Estrogen receptors and endocrine diseases:lessons from estrogen receptor knockout mice., Curr. Opin. Pharmacol., 1,613-619 (2001).
Related Products
Product Name Activities Prod. No.
(R,R)-cis-Diethyltetrahydro-2,8-chrysenediol ERβ antagonist D 8690
1,3,5-tris(4-Hydroxyphenyl)-4-propyl-1H-pyrazole ERα agonist H 6036
Raloxifene hydrochloride ER modulator R 1402
Prod. Code H 4413
The small GTPase Rho (Prod. Code R 2900),and its downstream effectors, the serine/-threonine Rho kinases family, have beenshown to be involved in various cellularfunctions including actin cytoskeletonorganization, cell adhesion, cell motility, aswell as cytokinesis [1-2]. Rho kinase has alsobeen shown to play a role in vascular and
smooth muscle contraction [2] and it has been suggested thatRho/Rho kinase inhibition may be a novel therapeutic target inthe treatment of various diseases such as hypertension [2],arteriosclerosis [2], bronchial asthma [3], cancer [4] andAlzheimer’s disease [5].
The Rho kinase inhibitor hydroxyfasudil (Prod. Code H 4413) isthe latest introduction to the Sigma-RBI portfolio of Rho kinaseinhibitors. Hydroxyfasudil is an active metabolite of HA-1077(fasudil; Prod. Code H-139) that is produced in the liver and isequally potent against Rho kinase (Ki = 560 nM), and moreselective for Rho kinase vs PKC (Ki = 18 µM) or myosin lightchain (MLC) kinase (Ki = 140 µM) than HA-1077 (Ki = 350 nM,3.3 µM and 36 µM, respectively) [6]. In rabbit aortic smoothmuscle, hydroxyfasudil and HA-1077 have been shown toequally inhibit receptor activation dependent PGF2-inducedcontraction (IC50 values of 4.0 µM and 3.7 µM, respectively),MLC kinase dependent KCl-induced contraction (IC50 values of18.8 µM and 22 µM, respectively) and PKC dependent 12-deoxyphorbol 13-isobutyrate (DPB)-induced contraction (IC50 values of 13.0 µM and 4.6 µM, respectively) [6]. Foradditional comparison, Rho kinase inhibitor Y-27632 (Prod. Code Y 0503) displayed IC50 values of 0.85, 2.8 and 1.23 µM,respectively [6].
These results suggest that hydroxyfasudil should serve as a usefultool for studying the Rho/Rho kinase pathway’s role in smoothmuscle contraction.
References1. Uehata, M., et al., Calcium sensitization of smooth muscle mediated by a
Rho-associated protein kinase in hypertension., Nature, 389, 990-994(1997).
2. Takahara, A., et al., Cardiovascular effects of Y-27632, a selective Rho-asso-ciated kinase inhibitor, assessed in the halothane-anesthetized caninemodel., Eur. J. Pharmacol., 460, 51-57 (2003).
3. Nakahara, T., et al., Y-27632 potentiates relaxant effects of beta 2-adreno-ceptor agonists in bovine tracheal smooth muscle., Eur. J. Pharmacol., 389,103-106 (2000).
4. Somlyo, A.V., et al., Rho kinase and matrix metalloproteinase inhibitorscooperate to inhibit angiogenesis and growth of human prostate cancerxenotransplants., FASEB J., 17, 223-234 (2003).
5. Zhou, Y., et al., Nonsteroidal anti-inflammatory drugs can lower amyloido-genic Abeta42 by inhibiting Rho., Science, 302, 1215-1217 (2003).
6. Ito, K., et al., Essential role of Rho kinase in the Ca2+ sensitization ofprostaglandin F(2alpha)-induced contraction of rabbit aortae., J. Physiol.546, 823-836 (2003).
New
Pro
du
ct H
igh
ligh
ts
OCOCH3
CH3COO
Prod. Code C 3490
N
HN
S OO
HN
O
HCl

®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
17Monoclonal Anti-hnRNP-Q
Prod. Code R 5653Clone Name 18E4, developed in mousePurified mouse immunoglobulinImmunogen: recombinant human hnRNP-Q Isotype: IgG1Species Cross Reactivity: human, bovine, canine, rat, mouse,chicken, and Xenopus
In the nucleus, heterogeneous nuclear ribonucleoproteins(hnRNPs) form complexes with RNA polymerase II transcripts.hnRNPs perform several biological functions such as transcrip-tion, pre-mRNA processing, cytoplasmic mRNA translation andturnover.
The hnRNP-Q family of proteins consist of three proteins,referred to as Q1, Q2 and Q3, that are derived by alternativesplicing from the same gene [1]. These proteins interact with thesurvival of motor neuron proteins (SMN) that are mutated inpatients with spinal muscular atrophy (SMA).
Applications: Western blot (55-70 kDa), ELISA, immunoprecipita-tion, and immunohistochemistry
References1. Mourelatos., Z., et al., SMN interacts with a novel family of hnRNP and
spliceosomal proteins. EMBO J., 20, 5443-5452 (2001).
Related Antibodies
Name Host Clone Prod. Code
Monoclonal Anti-hnRNP-A1 Mouse 9H10 R 4528
Monoclonal Anti-hnRNP-A2/B1 Mouse DP3B3 R 4653
Monoclonal Anti-hnRNP-C1/C2 Mouse 4F4 R 5028
Monoclonal Anti-hnRNP-L Mouse 4D11 R 4903
Monoclonal Anti-hnRNP-M1-M4 Mouse HL374 (1D8-2C5) R 3902
Monoclonal Anti-hnRNP-M3-M4 Mouse HL372 (2A6-2H3) R 3777
Monoclonal Anti-hnRNP-U Mouse 3G6 R 6278
Monoclonal Anti-Gemin 2 Mouse 2E17 G 6669
Monoclonal Anti-Gemin 3 Mouse 12H12 G 6544
Anti-Radixin
Prod. Code R 3653Developed in RabbitAffinity isolated antibodyImmunogen: peptide corresponding to amino acid residues 400-409 of human radixinSpecies Cross Reactivity: human, mouse, rat
The ERM (ezrin-radixin-moesin) proteins are closely related mem-bers of the talin-protein 4.1 merlin/schwannomin superfamily.They are general cross-linkers between the plasma membraneand actin filaments. ERM proteins are involved not only incytoskeletal organization, but also in signal transduction andapoptosis.
Applications: Western blot (~80 kDa), immunoprecipitation andimmunofluorescence
New
Pro
du
ct H
igh
ligh
ts
GGTI-298 and FTI-276: Geranylgeranyltrans-ferase and farnesyltransferase inhibitors
Farnesyltransferase (FTase) and geranylgeranyltransferase(GGTase) are involved in critical steps in the post-translationalprocessing of Ras and related guanosine triphosphate-dependentproteins. The GGTase-I inhibitor GGTI-297 arrests cells in G0/G1and induces accumulation of p21WAF in a p53-independent man-ner. The FTase inhibitor FTI-276 interferes with cell cycle events by amechanism that involves neither p21WAF nor p27KIP. FT-276 inhib-ited FTase in Ras-induced malignant transformation in vitro andwas highly selective for FTase over GGTase I, displaying IC50 val-ues of 0.5 nM vs. 50 nM, whereas GGTI-297 was selective forGGTase I over FTase, displaying IC50 values of 55 nM vs. 203 nM.References1. Miquel, K. et al., GGTI-298 induces G0-G1 block and apoptosis whereas FTI-
277 causes G2-M enrichment in A549 cells., Cancer Res., 57, 1846-1850 (1997).2. Cohen-Jonathan, E., et al., The farnesyltransferase inhibitor FTI-277
suppresses the 24-kDa FGF2-induced radioresistance in HeLa cells expressingwild-type RAS., Radiat. Res., 152, 404-411 (1999).
3. Chakrabarti, D., et al., Protein farnesyltransferase and protein prenylationin Plasmodium falciparum., J. Biol. Chem., 277, 42066-42073 (2002).
4. Lantry, L.E., et al., Effect of farnesyltransferase inhibitor FTI-276 on estab-lished lung adenomas from A/J mice induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone., Carcinogenesis, 21, 113-116 (2000).
Related Products
Name Description Prod. Code
GGTI-2133 Geranylgeranyltransferase inhibitor G 5294
L-744,832 Farnesyltransferase inhibitor L 7287
B581 Farnesyltransferase inhibitor B 2559
HN
O
OCH3
O
CH3
CH3
HN
H2N
HS
CF3COOH
NH
NH
O
COOH
NH2
HS
CF3COOH
SCH3
Prod. Codes G 5169 and F 9553
ICI 63197: Phosphodiesterase IV (PDE IV)inhibitor
Prod. Code I 8283
The phosphodiesterase type IV (PDE IV) familyof enzymes, encoded by four genes referred toas PDE4A-D, is characterized by its low Kmvalue for cAMP and its sensitivity to inhibitionby rolipram. Disturbances in cAMP-mediated
signaling have been implicated in several neuropsychiatric dis-orders, and PDE4 inhibitors may possess therapeutic potential in avariety of diseases, particularly asthma and depression. ICI 63197inhibits [3H]-rolipram binding to rat brain displaying an IC50 valueof 35 nM [1-4].
References1. Zhao, Y., et al., Inhibitor binding to type 4 phosphodiesterase (PDE4) assessed
using [3H]piclamilast and [3H]rolipram., J. Pharmacol. Exp. Ther., 305, 565-572(2003).
2. O’Donnell, J.M., et al., Antidepressant-like effects of rolipram and otherinhibitors of cyclic adenosine monophosphate phosphodiesterase onbehavior maintained by differential reinforcement of low response rate., J.Pharmacol. Exp. Ther., 264, 1168-1178 (1993).
3. Lourenco, C.M. et al., Imaging of cAMP-specific phosphodiesterase-IV: com-parison of [11C]rolipram and [11C]Ro 20-1724 in rats., Synapse, 31, 41-50 (1999).
4. Wachtel, H., Potential antidepressant activity of rolipram and other selectivecyclic adenosine 3',5'-monophosphate phosphodiesterase inhibitors.,Neuropharmacology, 22, 267-272 (1983).
Related Products
Name Description Prod. Code
Rolipram Selective cAMP-selective PDE IV inhibitor. R 6520
Ro 20-1724 cAMP phosphodiesterase inhibitor. B 8279
Etazolate Selective cAMP-specific phosphodiesterase inhibitor. E 1896
N
NO
Me N
N
NH2

Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
18
Glutathione Assay Kit (Prod. Code CS0260) for the measurement of total glutathione inbiological samples
Reduced glutathione (GSH), a tripeptide (γ-glutamyl-cysteinyl-glycine), is the major free thiol in most living cells and is involved inmany biological processes such as detoxification of xenobiotics,removal of hydroperoxides, and maintenance of the oxidation stateof protein sulfhydryls. It is the key antioxidant present in animaltissues.
The kit provides all reagents for the simple and quick assay of thelevel of total glutathione (oxidized glutathione and GSH) in a cell,tissue extracts, red blood cells or plasma.
The biological samples are first deproteinized using 5% sulfosalicylicacid solution. The total glutathione content of the sample is thenassayed using a kinetic assay in which catalytic amounts ofglutathione cause a continuous reduction of 5,5’-dithiobis-(2-nitro-benzoic acid (DTNB) to TNB. The oxidized glutathione formed isrecycled by glutathione reductase and NADPH. The product, TNB, isassayed colorimetrically at 412 nm.
Assaying in a 96 well plate maximizes the number of samples thatcan be tested.
Features and Benefits• A quick and simple method• The kit provides all the reagents required for a colorimetric assay
of total glutathione (oxidized + GSH)• The kit includes a deproteination reagent for biological samples• Sufficient reagents are supplied for 700 x 0.2 ml tests of total
glutathione
Standard curve of reduced glutathione as measured in the kineticcolorimetric assay.
Glutathione S-Transferase (GST) Assay Kit (Prod. Code CS0410) for the measurement of GSTin biological samples
Glutathione S-transferases (GSTs) are a group of enzymes impor-tant in the detoxication of many xenobiotics in mammals. Theenzymes protect cells against toxicants by conjugating the thiolgroup of the glutathione to electrophilic xenobiotics, and therebydefend cells against the mutagenic, carcinogenic, and toxic effectsof the compounds. GST activity is present in plants, insects, yeast,bacteria, and in most mammalian tissues, especially in the liver,which plays a key tole in detoxification.
Glutathione S-Transferase (GST) Assay Kit measures total GSTactivity by measuring the conjugation of 1-chloro-2,4-dinitro-benzene (CDNB) with reduced glutathione. Product formation(GS-DNB conjugate) is detected at 340 nm, which makes possiblea spectrophotometric assay for GST activity. The rate of increaseof absorption is directly proportional to the GST activity in thesample.
Features and Benefits• The kit provides all the reagents required for a fast and easy
measurement of total GST activity • The kit includes a GST positive control• The kit can be used to measure GST activity in cell and bacterial
lysates, tissue homogenates and in plasma and erythrocytelysates
• The kit is sufficient for 100 assays in 1 ml cuvette or 500 reactions in 96 well plates
Measurement of Glutathione-S-Transferase (GST) activity
The activity of GST enzyme (Prod. Code G 6794) was measuredat different concentrations in a 1 ml cuvette using the GST AssayKit (Prod. Code CS0410). The graph demonstrates a linear ratiobetween the amount of enzyme (over the range ~0.05-0.5 µg)and the enzyme activity, as measured by the increase in OD340
units per min.
®
®
New
Pro
du
ct H
igh
ligh
ts
0.005
0.01
0.015
0.02
0.025
0.03
0.035
0
.
0 0.2 0.4 0.6
(
0.005
0.01
0.015
0.02
0.025
0.03
0.035
0
.0.005
0.01
0.015
0.02
0.025
0.03
0.035
0
.
0 0.2 0.4 0.6
Reduced Glutathione nmole) O
D/m
in
0
0.02
04
06
0.08
0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4 0.45 0.5
0.1
0
0.02
00..04
00..06
0.08
0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4 0.45 0.5
µ
0.1
OD
340/m
in
g GST
Assay Kits for Cell Signaling & Neuroscience

®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
19Assay Kits for Cell Signaling & Neuroscience
Casein kinases (CK) are highly conserved serine/threoninekinases, ubiquitous in eukaryotic organisms, and defined by theirsubstrate preference for casein. They are insensitive to cyclicnucleotides, calcium, and phospholipids. Casein kinases aregrouped into two main families, type I and type II, according totheir structure, specificity and response to effectors.
Features and Benefits
• The kit provides an easy method for measuring the activity ofthe different casein kinases by in vitro phosphorylation of α-casein
• The kit includes CK I and CK II specific inhibitors (IC261 andheparin, respectively) to allow the distinction between the CKactivities
• The kit can be used for CK activity measurement in celllysates, tissue homogenates, column fractions or a purifiedenzyme
• Sufficient for 100 tests
Casein Kinase activity in rat liver extract was measured in thepresence or absence of CK I or/and CK II inhibitors using caseinkinase assay kit, general. The CK phosphorylation activity wasdetermined by measuring the incorporation of radioactive 32Pinto the CK substrate, α-casein.
1. Control CK activity2. Inhibition of CK activity by CK I inhibitor, IC261 (Prod. Code
I 0658)3. Inhibition of CK activity by CK II inhibitor, heparin (Prod. Code
H 0777 and H 3393)4. Inhibition of CK activity by the combination of CK I and CK II
inhibitors.
The use of CK I and CK II inhibitors enable the discriminationbetween CK I and CK II specific activity.
DHFR Assay Kit (Prod. Code CS0340)For the detection of Dihydrofolate Reductase (DHFR) activity and for screening of DHFRinhibitors
DHFR is an enzyme catalyzing a reaction essential for the biosyn-thesis of nucleotidic bases of DNA. Blockade of the DHFRenzyme causes cell death as a result of inhibition of DNAsynthesis. For this reason, DHFR is considered a target for thedevelopment of anti-tumor drugs.
The assay is based on the ability of DHFR to catalyze thereversible NADPH-dependent reduction of dihydrofolic acid totetrahydrofolic acid. The reaction progress is followed by moni-toring the decrease in absorbance at 340 nm.
DHFR
Dihydrofolic acid + NADPH + H+ Tetrahydrofolic acid + NADP+
Features and Benefits• A quick and simple method• The kit contains all the reagents required for a colorimetric
assay of DHFR activity in cell lysates, tissue homogenates, orcolumn fractions of purified enzyme
• The kit includes a purified enzyme to be used as a positivecontrol and for screening of DHFR inhibitors
• The kit includes methotrexate (MTX), a prokaryotic andeukaryotic DHFR specific inhibitor, which exhibits anti-tumoractivities
• The kit was tested using A431, NIH 3T3 and CHO cell lines,rat liver, kidney, brain, and skeletal muscle tissue extracts andrecombinant DHFR
• Sufficient reagents are supplied for 50-100 1ml tests
Activity of recombinant Dihydrofolate Reductase (DHFR) in thepresence of the inhibitor methotrexate (MTX; Prod. Code A 7019). Specific activity (Unit/mg protein) of recombinant DHFRwas measured in the absence or in the presence of differentconcentrations of MTX. 50% inhibition of the enzyme isobserved at a concentration of 0.025 µM MTX.
Casein Kinase Assay Kit (Prod. Code CS0400)
New
Pro
du
ct H
igh
ligh
ts
IC261 - - ++Heparin - - ++
3
4
0
5000
10000
15000
20000
25000
1 2 3 4
CK
act
ivit
y (C
PM)
0
0.5
1
1.5
2
2.5
3
3.5
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 1.1
M of MTX0
0.5
1
1.5
2
2.5
3
3.5
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 1.1
µM of MTX
Spec
ific
act
ivit
y(u
nit
s/m
g P
rote
in)

®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
20N
ew P
rod
uct
Hig
hlig
hts
YIC-C8-434: Acyl-CoA: Cholesterol O-Acyltransferase (ACAT) inhibitor
Acyl CoA cholesterolO-acyltransferase(ACAT, EC 2.3.1.26)is the enzymeresponsible for the
intracellular esterification of cholesterol. In the liver, ACAT-derivedcholesteryl esters are secreted as a component of very low-densi-ty lipoprotein (VLDL). In the gastrointestinal tract, ACAT-mediatedcholesterol esterification is the rate-limiting step in the absorptionof food- and bile-derived cholesterol. YIC-C8-434, an ACATinhibitor, inhibits the formation of cholesteryl esters andcholesterol both in human colon adenocarcinoma Caco2 cellsand in human hepatoma HepG2 cells, displaying IC50 values of0.38 and 0.49 µM, respectively [1]. Oral administration of YIC-C8-434 to rats at a dose of 8.3 mg/kg/d inhibited [14C] cholesterol absorption by 17% (p<0.01) and reducedintestinal cholesterol absorption and hepatic VLDL cholesterolsecretion by direct inhibition of ACAT [1].
References1. Ohishi, K., et al., Inhibitory effects of N-(3,5-dimethoxy-4-n-octyloxycin-
namoyl)-N'-(3,4-dimethylphenyl)piperazine (YIC-C8-434), an acyl-CoA:choles-terol O-acyltransferase inhibitor, on cholesterol esterification in the intes-tine and liver., Biol. Pharm. Bull., 26, 1125-1128 (2003).
2. Kaneko, K., et al., Sex-dependent toxicity of a novel acyl-CoA:cholesterolacyltransferase inhibitor, YIC-C8-434, in relation to sex-specific forms ofcytochrome P450 in rats., Toxicol. Sci., 64, 259-268 (2001).
Related Product
Name Description Prod. Code
Sandoz 58-035 ACAT inhibitor S 9318
SB-408124: Selective non-peptide OX1orexin receptor antagonist
Orexin-A and Orexin-B, also known asHypocretin-1 andHypocretin-2, areneuropeptides thathave been isolated
from the rat hypothalamus [1]. They have been implicated in thecontrol of feeding, drinking, regulation of arousal and the sleep-wake cycle [1-4].
SB-408124 is a selective, non-peptide OX1 orexin receptor antag-onist that displays Ki values of 27 nM and 99 nM in the mem-brane scintillation proximity assay (SPA) format and whole-cellassay, respectively [5]. This compound acts as a functional antag-onist, as determined using a calcium mobilization assay and dis-plays a Kb value of 21.7 nM at the human OX1 orexin receptorexpressed in Chinese hamster ovary cells [5]. It also dose-dependently blocks orexin-A induced grooming following oraladministration in rats [6].
SB-408124 will serve as a unique tool with which to elucidatethe function of the OX1 orexin receptor.
Sold for research purposes under agreement with GlaxoSmithKline
References1. Sakurai, T., et al., Orexins and orexin receptors: a family of hypothalamic
neuropeptides and G protein-coupled receptors that regulate feedingbehavior. Cell, 92, 573-585 (1998).
2. Smart, D. and Jerman, J., The physiology and pharmacology of the orexins.Pharmacol. Ther., 94, 51-61 (2002).
3. Hagan, J.J., et al., Orexin A activates locus coeruleus cell firing and increas-es arousal in the rat. Proc. Natl. Acad. Sci., USA, 96, 10911-10916 (1999).
4. Piper, D.C., et al., The novel brain neuropeptide, orexin-A, modulates thesleep-wake cycle of rats. Eur. J. Neurosci., 12, 726-730 (2000).
5. Langmead, C.J., et al., Characterization of the binding of [3H]-SB-674042, anovel non-peptide antagonist to the human orexin-1 receptor. Br. J.Pharmacol., 141, 340-346 (2004).
6. Upton, N., et al., Evidence that orexin-A evoked grooming, but not waterintake, in the rat is mediated by orexin-1 (OX1) receptor., Society forNeuroscience abstract 320.1 (2001).
Topiramate: Novel anticonvulsant andkainate GluR5 receptor antagonistAvailable First from Sigma-RBI
Topiramate is a structurally novel anti-convulsant [1] that may also be effica-cious in the treatment of chronic pain,obesity [2], post-traumatic stress disor-der and migraine [3]. Topiramateappears to act as an antagonist at theGluR5 subtype of the kainate receptor[4,5] and displays an IC50 value of
0.5 µM in whole cell recording of the principal neurons of therat basolateral amygdala [5].
References1. LaRoche, S.M., et al., The new antiepileptic drugs: scientific review. J. Am.
Med. Assoc., 291, 605-614 (2004).2. Appolinario, J.C., et al., Psychotropic drugs in the treatment of obesity:
what promise? CNS Drugs, 18, 629-651 (2004).3. Diener, H.C., et al., Topiramate in migraine prophylaxis: Results from a
placebo-controlled trial with propranolol as an active control. J. Neurol.,251, 943-950 (2004).
4. Kaminski, R.M., et al., Topiramate selectively protects against seizuresinduced by ATPA, a GluR5 kainate receptor agonist. Neuropharmacology,46, 1097-1104 (2004).
5. Gryder, D.S. and Rogawski, M.A., Selective antagonism of GluR5 kainate-receptor-mediated synaptic currents by topiramate in rat basolateralamygdala neurons. J. Neurosci., 23, 7069-7074 (2003).
H3C O
H3CO
OCH3
N
O
N CH3
CH3
Prod. Code Y 0628
Prod. Code T 0575
O
H3CO
OCH3
N
O
N CH3
CH3
Prod. Code S 2694
O
OO
S
O O
NH2
O
H
O
O
Me
Me
H
H
Me
Me
GW311616A: Human neutrophil elastase(HNE) inhibitor
GW311616A is a potent,cell permeable, orallybioavailable and selectiveinhibitor of human neu-trophil elastase (HNE) [1].HNE is serine protease ofthe trypsin class. Normallung maintains a balancebetween trypsin class
enzymes and endogenous anti-trypsin (α1-proteinase inhibitor,α1-PI). In smokers’ lungs, the recruitment of inflammatory leuko-cytes such as neutrophils with the release of HNE and the inacti-vation of α1-PI by tobacco products lead to the disruption of thisprotective mechanism and pathologic destruction of the lungextracellular matrix. Small molecule HNE inhibitors are part of ageneral anti-proteinase strategy used in treatments for emphyse-ma and COPD (Chronic Obstructive Pulmonary Disease) [2].
The Ki value of GW311616A for purified HNE is 0.31 nM [3]. Incontrast, its IC50 values for chymotrypsin (Prod. No. C 6423),trypsin and cathepsin G are 4.2 µM, >100 µM and >100 µM,respectively [3]. In addition, GW311616A inhibited HNE inhuman whole blood, possessing an IC50 value of 0.67 mM, inaddition to inhibiting HNE in vivo in hamster and canine follow-ing oral administration [3].
GW311616A is sold for research purposes under agreementfrom GlaxoSmithKline
References1. Macdonald, S.J., et al., The discovery of a potent, intracellular, orally
bioavailable, long duration inhibitor of human neutrophil elastase-GW311616A a development candidate., Bioorg. Med. Chem. Lett., 11, 895-898 (2001).
2. Ohbayashi, H., Neutrophil elastase inhibitors as treatment for COPD. ExpertOpin. Investig. Drugs., 11, 965-980 (2002).
3. Macdonald, S.J., et al., Discovery of further pyrrolidine trans-lactams asinhibitors of human neutrophil elastase (HNE) with potential as develop-ment candidates and the crystal structure of HNE complexed with aninhibitor (GW475151)., J. Med. Chem. 45, 3878-3890 (2002).
N
O
N
NSO2Me
O
HCL
Prod. Code G 8419

Accelerate your drug discovery process by validating your biochemical and cell-basedassays with the Sigma-RBI Library of Pharmacologically Active Compounds. Have confi-
dence taking your critical discovery projects to screen with assays validated by well-character-ized, high-purity compounds representing all major target classes. See for yourself whyLOPAC has become the gold standard for assay validation:
• 1,280 pharmacologically active compounds• Marketed drugs and pharmaceutically relevant
structures• Guaranteed Sigma-Aldrich quality and easy re-supply
• Searchable structure/activity database*• Pre-solubilized and normalized compounds• ANSI/SBS-compliant storage• Robust performance
The Library of Pharmacologically Active CompoundsProduct Number LO1280 (LO3300 outside the U.S.†)
Pharmacological ActivitiesSDFile provided to construct a database containing:• Structure• Pharmacological Activity• Name• Product Number• Rack Position
Convenient FormatCompounds are arranged in solution (250 µl at 10 mM in DMSO) in 96-well format in 16 racks of 80, one compound per well.
For more information, please visit the Drug Discovery link on the Sigma-Aldrich web site: sigma-aldrich.com
*Technical information for each compound is provided in standard SDFile format for use with ISIS/Base or other compatible software (software not provided).†Due to import/export restrictions on a small number of LOPAC1280 compounds, the International Version (LO3300) will vary slightly from the U.S. Version (LO1280).

22
PEPscreen®: Custom Peptide LibrariesHigh-throughput peptide solution for
generating fast leads in screening applications
Sigma-Genosys has developed a proprietary peptide synthesis platform using state-of-the-arttechnology. This technology allows the fastest and most efficient high-throughput parallelsynthesis of milligram quantities of peptide libraries. The PEPscreen® service providesresearchers with the ability to order large numbers of extremely affordable custom peptidesdelivered in less than 7 working days.
Ordering Information
For more information refer to www.sigma-genosys.com/CTSCREEN
For technical inquiries please contact [email protected]
Place your order by emailing your sequence in an 8-channel orientation (peptide #1 = A1, peptide #2 = B1, peptide #8 = H1, peptide #9 = A2, etc.)
Prices are subject to change without notification.
L E A D E R S H I P I N L I F E S C I E N C E , H I G H T E C H N O L O G Y A N D S E R V I C ESIGMA-ALDRICH CORPORATION • BOX 14508 • ST. LOUIS • MISSOURI 63178 • USAsigma-genosys.com
Features and Benefits• Compatible with robotic automation for resuspension and dispensing
in screening applications
• Ideal for performing solution-phase assays
• Suitable for robotic production of peptide microarrays
• N-Terminal modifications and non-standard residue availabilityallows for maximum flexibility in project design
• Delivery of product in less than 7 working days allows for fastertarget identification
Product Specifications• Quantity: 0.5-2 mg
• Length: 6-20 residues
• Purity: ~70% (based on 15 residues)
• Analysis: MALDI-TOF mass spectroscopy on 100% of peptides
• Format: Lyophilized in 96-well format (2-D bar-coded tubes)
• Shipped with electronic PDF files of QC data and peptidesequence map
• Minimum order size is 48 peptides
Modifications Available• N-Terminal acetylation
• N-Terminal biotin
• N-Terminal fluorescein
• D-Amino acids
• Non-standard residues
• Phosphorylated residues
• C-Terminal acid or amide
PEPscreen® is a registered trademark of Sigma-Genosys, L.P.

Editorial BoardScientific Editor: Keith J. Watling, Ph.D.Managing Editor: Julie L. CoughlanLayout/Design/Production: Diane Ward
Three Strathmore Road, Natick, MA 01760-2447 USANet: sigma-aldrich.com© 2004 by Sigma-RBI, A Member of the Sigma-Aldrich® Family. All rights reserved. No part of this publication may bereproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, photocopying, record-ing, or otherwise, without the prior written permission of the copyright holder. Sigma brand products are sold throughSigma-Aldrich, Inc. Sigma-Aldrich, Inc. warrants that its products conform to the information contained in this andother Sigma-Aldrich publications. Purchaser must determine the suitability of the product(s) for their particular use.Additional terms and conditions may apply. Please see reverse side of the invoice or packing slip.
®
®
Celltr
ansm
issi
ons
Vol 2
0, N
o. 3
, 20
04
Ord
er:
1-80
0-32
5-30
10
Tech
nica
l Ser
vice
: 1-
800-
325-
5832
s
igm
a-al
dric
h.co
m/c
ells
igna
ling
23
New
Lit
erat
ure
Poster detailing:• α-Secretase pathway• β-Secretase pathway• Immune response• Over 200 products listed
• Brochure covering 17 ion channel-related topics:AquaporinsCyclic Nucleotide-Gated ChannelsIon Exchangers and Co-TransportersLigand-Gated ChannelsVoltage-Gated Channels
• Over 600 products, including over 100 NEW products
• Over 3,000 quality antibodies all rigorously tested• Hundreds of NEW antibodies• Organized by area of study for easy reference• World-renowned technical service• Timely delivery anywhere
New Cell Signaling & Neuroscience Literature
Fill out & return the Business Reply Card to receive your new Sigma-RBI Literature!

The SIGMA-ALDRICH Family
World Headquarters • 3050 Spruce St., St. Louis, MO 63103 • (314) 771-5765
Order/Customer Service 1-800-325-3010 • Fax 1-800-325-5052Technical Service 1-800-325-5832 • sigma-aldrich.com/techservice
Development/Bulk Manufacturing Inquiries 1-800-336-9719
HSM01796-502193
0124
We are committed to the success of our Customers, Employees and Shareholders through leadership in Life Science, High Technology and Service.
©2004 Sigma-Aldrich Co. Printed in USA Sigma brand products are sold through Sigma-Aldrich, Inc.Sigma-Aldrich, Inc. warrants that its products conform to the information contained in this and other Sigma-Aldrich publications. Purchaser must determine the suitability of the product(s) for their particular use. Additional terms and conditions may apply. Please see reverse side of the invoice or packing slip. SIGMA and - are registered trademarks of Sigma-Aldrich Co. and its division Sigma-Aldrich Biotechnology LP. Riedel-de Haën®: trademark under license from Riedel-de Haën GmbH.
ArgentinaSIGMA-ALDRICH DE ARGENTINA, S.A.Tel: 54 11 4556 1472Fax: 54 11 4552 1698
AustraliaSIGMA-ALDRICH PTY., LIMITEDFree Tel: 1800 800 097 Free Fax: 1800 800 096Tel: 612 9841 0555Fax: 612 9841 0500
AustriaSIGMA-ALDRICH HANDELS GmbHTel: 43 1 605 81 10Fax: 43 1 605 81 20
BelgiumSIGMA-ALDRICH NV/SA.Free Tel: 0800-14747Free Fax: 0800-14745Tel: 03 899 13 01Fax: 03 899 13 11
BrazilSIGMA-ALDRICH BRASIL LTDA.Tel: 55 11 3732-3100Fax: 55 11 3733-5151
CanadaSIGMA-ALDRICH CANADA LTD.Free Tel: 800-565-1400Free Fax: 800-265-3858Tel: 905-829-9500Fax: 905-829-9292
ChinaSIGMA-ALDRICH CHINA INC.Tel: 86-21-6386 2766Fax: 86-21-6386 3966
Czech RepublicSIGMA-ALDRICH s.r.o.Tel: 246 003 200Fax: 246 003 291
DenmarkSIGMA-ALDRICH DENMARK A/STel: 43 56 59 10Fax: 43 56 59 05
FinlandSIGMA-ALDRICH FINLANDTel: 09-3509250Fax: 09-350-92555
FranceSIGMA-ALDRICH CHIMIE S.à.r.l.Tel appel gratuit: 0800 211 408Fax appel gratuit: 0800 031 052
GermanySIGMA-ALDRICH CHEMIE GmbHFree Tel: 0800-51 55 000Free Fax: 0800-649 00 00
GreeceSIGMA-ALDRICH (O.M.) LTDTel: 30 210 9948010Fax: 30 210 9943831
HungarySIGMA-ALDRICH KftTel: 06-1-235-9054Fax: 06-1-269-6470Ingyenes zöld telefon: 06-80-355-355Ingyenes zöld fax: 06-80-344-344
IndiaSIGMA-ALDRICH CHEMICALS PRIVATE LIMITEDTelephoneBangalore: 91-80-5112-7272New Delhi: 91-11-2616 5477Mumbai: 91-22-2570 2364Hyderabad: 91-40-5531 5488FaxBangalore: 91-80-5112-7473New Delhi: 91-11-2616 5611Mumbai: 91-22-2579 7589Hyderabad: 91-40-5531 5466
IrelandSIGMA-ALDRICH IRELAND LTD.Free Tel: 1800 200 888Free Fax: 1800 600 222Tel: 353 1 4041900Fax: 353 1 4041910
IsraelSIGMA-ALDRICH ISRAEL LTD.Free Tel: 1-800-70-2222Tel: 08-948-4100Fax: 08-948-4200
ItalySIGMA-ALDRICH S.r.l.Telefono: 02 33417310Fax: 02 38010737Numero Verde: 800-827018
JapanSIGMA-ALDRICH JAPAN K.K.Tokyo Tel: 03 5796 7300Tokyo Fax: 03 5796 7315
KoreaSIGMA-ALDRICH KOREATel: 031-329-9000Fax: 031-329-9090
MalaysiaSIGMA-ALDRICH (M) SDN. BHDTel: 603-56353321Fax: 603-56354116
MexicoSIGMA-ALDRICH QUÍMICA, S.A. de C.V.Free Tel: 01-800-007-5300Free Fax: 01-800-712-9920
The NetherlandsSIGMA-ALDRICH CHEMIE BVTel Gratis: 0800-0229088Fax Gratis: 0800-0229089Tel: 078-6205411Fax: 078-6205421
New ZealandSIGMA-ALDRICH PTY., LIMITEDFree Tel: 0800 936 666Free Fax: 0800 937 777
NorwaySIGMA-ALDRICH NORWAY ASTel: 23 17 60 60Fax: 23 17 60 50
PolandSIGMA-ALDRICH Sp. z o.o.Tel: (+61) 829 01 00Fax: (+61) 829 01 20
PortugalSIGMA-ALDRICH QUÍMICA, S.A.Free Tel: 800 202180Free Fax: 800 202178Tel: 21 9242555Fax: 21 9242610
RussiaSIGMA-ALDRICH RUSSIAOOO SAF-LABTel: 095 975-1917/3321Fax: 095 975-4792
SingaporeSIGMA-ALDRICH PTE. LTD.Tel: 65-67791200Fax: 65-67791822
South AfricaSIGMA-ALDRICH SOUTH AFRICA (PTY) LTD.Free Tel: 0800 1100 75Free Fax: 0800 1100 79Tel: 27 11 979 1188Fax: 27 11 979 1119
SpainSIGMA-ALDRICH QUÍMICA S.A.Free Tel: 900 101376Free Fax: 900 102028Tel: 91 661 99 77Fax: 91 661 96 42
SwedenSIGMA-ALDRICH SWEDEN ABTel: 020-350510Fax: 020-352522Outside Sweden Tel: +46 8 7424200Outside Sweden Fax: +46 8 7424243
SwitzerlandFLUKA CHEMIE GmbHSwiss Free Call: 0800 80 00 80Tel: +41 81 755 2828Fax: +41 81 755 2815
United KingdomSIGMA-ALDRICH COMPANY LTD.Free Tel: 0800 717181Free Fax: 0800 378785Tel: 01747 833000Fax: 01747 833313
United StatesSIGMA-ALDRICHP.O. Box 14508St. Louis, Missouri 63178Toll-free: 800-325-3010Call Collect: 314-771-5750Toll-Free Fax: 800-325-5052Tel: 314-771-5765Fax: 314-771-5757Internet:sigma-aldrich.com
sigma-aldrich.com


















