
CCHHAAPPTTEERR -- IIIIshodhganga.inflibnet.ac.in/bitstream/10603/5385/9/09_chapter2.pdf · flame...
28
C C H H A A P P T T E E R R - - I I I I
Transcript of CCHHAAPPTTEERR -- IIIIshodhganga.inflibnet.ac.in/bitstream/10603/5385/9/09_chapter2.pdf · flame...

CCHHAAPPTTEERR -- IIII

CHAPTER – II
SYNTHESIS OF 1-HEPTA-O-ACETYL--D-MALTOSYL-3-
SUBSTITUTED BENZOTHIAZOLYL CARBAMIDES AND N-HEPTA-O-
ACETYL-β-D-MALTOSYL-O-ALKYL CARBAMATES
ABSTRACT:
1-hepta-O-acetyl-β-D-maltosyl-3-substituted benzothiazolyl
carbamides and N-hepta-O-acetyl-β-D-maltosyl-O-alkyl carbamates have been prepared
by the interaction of Hepta-O-acetyl-β-D-maltosyl isocyanate and 2-amino
benzothiazole/substituted benzothiazoles and various alcohols respectively. These
compounds have been characterized through usual chemical transformations and IR, 1HNMR and Mass Spectral studies. The polarimetric study of the title compounds have
been carried out.
A small and simple benzothiazole nucleus is present in compounds involved
in research aimed at evaluating new products that possess interesting biological activities
such as antitumor, antimicrobial, anti-thelmintic, antileishmanial, anticonvulsant and anti-
inflammatory1. Sugar thioureas containing an N-azolyl substitutent, such as thiazolyl,
thiazoline or benzoxazole ring, have been the subject to attention in connection with the
interest in azole nucleosides analogs as antineoplastic2 and antiviral compounds
3.
Carbamides and their derivatives shows strong antibacterial activity and are
also versatile reagent in organic synthesis4.
Benzothiazoles are bicyclic ring system with multiple applications. In
1950’s a number of 2-amino benzothiazoles were intensively studied as central muscle
relaxants. After that benzothiazole derivatives have been studied extensively and found to
have diverse chemical reactivity and broad spectrum of biological activity.
Although they have been known from long ago to be biologically active5-7
their varied biological features are still of great scientific interest. Benzothiazoles show
antitumor8-10
activity, especially phenyl substituted benzothiazoles. While condensed
pyrimido benzothiazoles and benzothiazolo quinazolines exert antiviral activity11
. Bis-

substituted amidino benzothiazoles acts as potential anti-HIV agents12
. Substituted 6-nitro
and 6-amino-benzothiazoles show antimicrobial activity13
. 2-amino substituted
benzothiazoles also show anti malarial activity14
. Also Schiff base of benzothiazoles
possess antitubercular, anticancer, antitumor, antipyretic, and sterase inhibitory
activities15-16
.
During past few years, in our laboratory N-glucosylated and N-lactosylated
benzothiazolyl thiocarbamides17-18
and carbamides19
have been reported and tested for
their biological activity.
Hence it is quite interesting to synthesized carbamides having
benzothiazolyl substitutent.
Also the addition of alcohols to carbohydrate isocyanate is a general
method for the preparation of linear N-sugar, O-alkyl carbamate. This reaction is
frequently used as a tool for structure determination20
. The exhaustive literature survey
revealed that N-maltosylated benzothiazolyl carbamides and carbamates have not been
prepared earlier. Therefore, synthesis of these two types of compounds, involving the
intereaction of hepta-O-acetyl-β-D-maltosyl isocyanate (I) and 2-amino
benzothiazole/substituted benzothiazoles and alcohols have been carried out. The present
chapter deals with these syntheses.
When the interaction of hepta-O-acetyl-β-D-maltosyl isocyanate (I) and 2-
amino benzothiazole (II) have been carried out in boiling benzene medium for about 4 hr.
and the solvent benzene was distilled off, a semisolid mass was isolated, this when
triturated several times with petroleum ether (60-80°c) afforded a solid. It was
crystallized from ethanol-water, m.p. 190-192°C. The elemental analysis indicated its
molecular formula C34H41O18N3S.
Examination of the Product with m.p. 190-192°C (C34H41O18N3S)

1. Solubility:- The product was found insoluble in water and petroleum ether, while
appreciably soluble in ethanol, acetone, chloroform and benzene.
2. Action of alkaline plumbite solution:- On boiling with alkaline plumbite the
product was found non-desulphurisable.
3. Action of conc. sulphuric acid:- The presence of maltosyl group was confirmed
on the basis of its charring property when warm with conc. sulphuric acid.
4. Optical activity:- The compound was found optically active and its specific
rotation was found to be []D30
= +99.75° (c, 0.948 mol in CHCl3).
5. TLC: The purity of the product was checked by TLC and recorded Rf value 0.78
(Hexane: EtOAc, 1:1).
6. IR, 1HNMR and Mass Spectral Studies:
a) IR spectral analysis21-25
: The IR spectrum of the product distinctly showed the
bands due to υ N-H, υ C=N, υ C=O, υ C-N, and bands due to maltosyl unit.
b) 1HNMR spectral analysis
26-30: The
1HNMR spectrum of the product distinctly
shows signals due to N-H proton aromatic protons and maltosyl protons.
c) Mass spectral analysis31-38
: In its Mass spectrum the molecular ion peaks and
other important fragment peaks are tabulated in table –3(See Page No. 95)
All these facts clearly indicated that the reaction product with m.p.
190-192°C was 1-hepta-O-acetyl-β-D-maltosyl-3-(2)-benzothiazolyl carbamide
(IIIa).
When reaction of hepta-O-acetyl-β-D-maltosyl isocyanate was
extended to several other substituted benzothiazoles the corresponding 1-hepta-O-acetyl-
β-D-maltosyl-3-substituted benzothiazolyl carbamides (IIIb–IIIg) have been isolated
(Table –1).
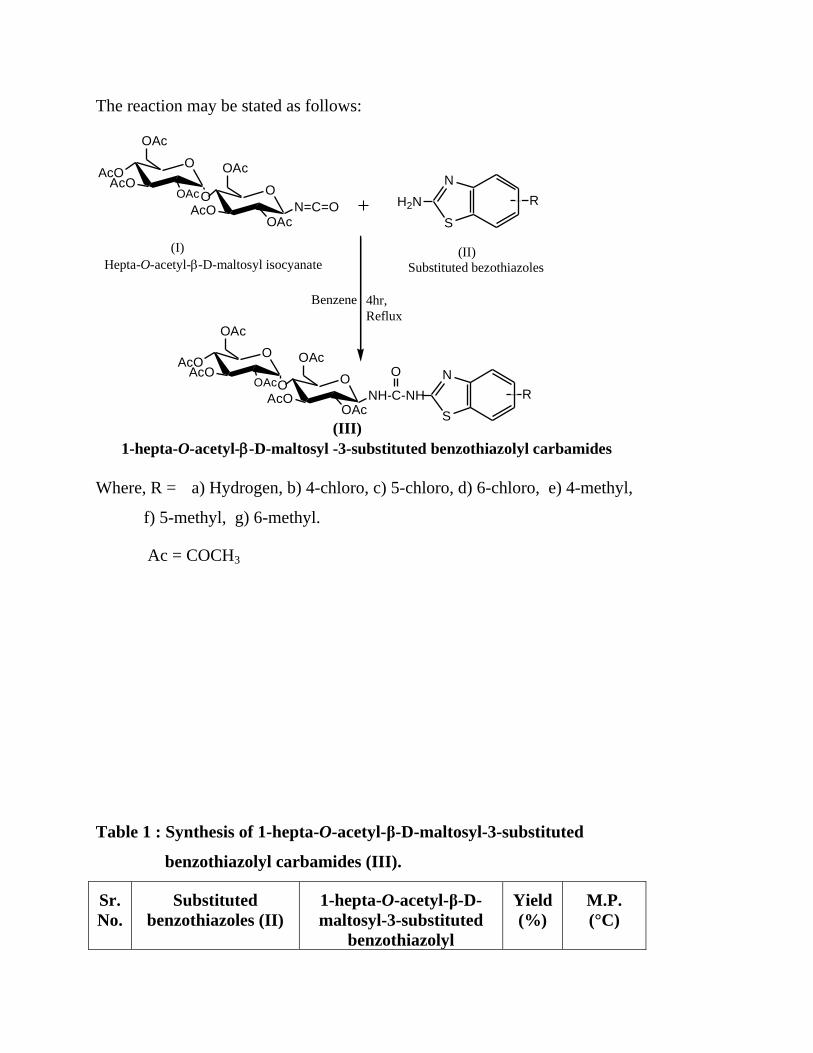
The reaction may be stated as follows:
OAcO
AcOOOAc
OAc
O
AcOOAc
OAc
N=C=OR
N
S
H2N
OAcO
AcOOOAc
OAc
OO
AcOOAc
OAc
NH-C-NH R
N
S
(I)
Hepta-O-acetyl--D-maltosyl isocyanate
Benzene 4hr,
Reflux
(II)
Substituted bezothiazoles
(III)
1-hepta-O-acetyl--D-maltosyl -3-substituted benzothiazolyl carbamides
Where, R = a) Hydrogen, b) 4-chloro, c) 5-chloro, d) 6-chloro, e) 4-methyl,
f) 5-methyl, g) 6-methyl.
Ac = COCH3
Table 1 : Synthesis of 1-hepta-O-acetyl-β-D-maltosyl-3-substituted
benzothiazolyl carbamides (III).
Sr.
No.
Substituted
benzothiazoles (II)
1-hepta-O-acetyl-β-D-
maltosyl-3-substituted
benzothiazolyl
Yield
(%)
M.P.
(°C)
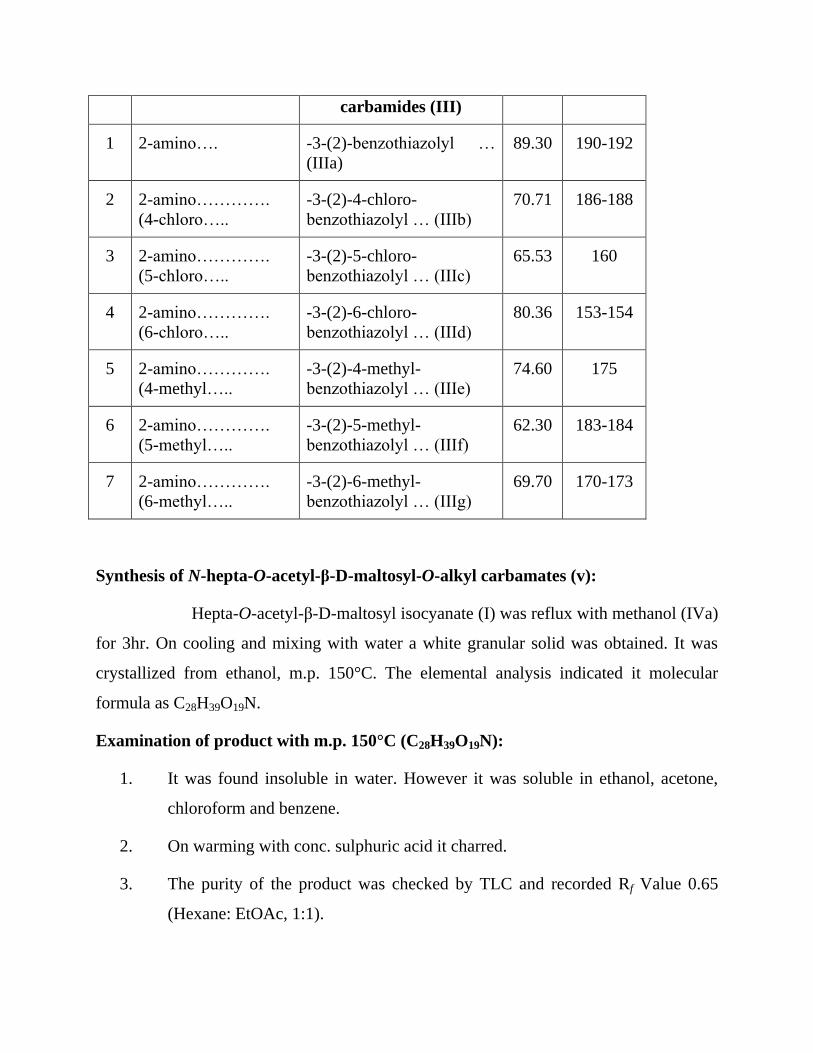
carbamides (III)
1 2-amino…. -3-(2)-benzothiazolyl …
(IIIa)
89.30 190-192
2 2-amino………….
(4-chloro…..
-3-(2)-4-chloro-
benzothiazolyl … (IIIb)
70.71 186-188
3 2-amino………….
(5-chloro…..
-3-(2)-5-chloro-
benzothiazolyl … (IIIc)
65.53 160
4 2-amino………….
(6-chloro…..
-3-(2)-6-chloro-
benzothiazolyl … (IIId)
80.36 153-154
5 2-amino………….
(4-methyl…..
-3-(2)-4-methyl-
benzothiazolyl … (IIIe)
74.60 175
6 2-amino………….
(5-methyl…..
-3-(2)-5-methyl-
benzothiazolyl … (IIIf)
62.30 183-184
7 2-amino………….
(6-methyl…..
-3-(2)-6-methyl-
benzothiazolyl … (IIIg)
69.70 170-173
Synthesis of N-hepta-O-acetyl-β-D-maltosyl-O-alkyl carbamates (v):
Hepta-O-acetyl-β-D-maltosyl isocyanate (I) was reflux with methanol (IVa)
for 3hr. On cooling and mixing with water a white granular solid was obtained. It was
crystallized from ethanol, m.p. 150°C. The elemental analysis indicated it molecular
formula as C28H39O19N.
Examination of product with m.p. 150°C (C28H39O19N):
1. It was found insoluble in water. However it was soluble in ethanol, acetone,
chloroform and benzene.
2. On warming with conc. sulphuric acid it charred.
3. The purity of the product was checked by TLC and recorded Rf Value 0.65
(Hexane: EtOAc, 1:1).

4. Its [α]D30
was found to be +60.32°C (c, 0.667 mol in CHCl3).
5. IR, 1HNMR and Mass Spectral Studies :
a) IR spectral analysis: The IR spectrum of the product clearly indicated the
presence of υ N-H, υ C=O, υ C-O and bands due to maltosyl unit.
b) 1HNMR spectral analysis: The
1HNMR spectrum of the products shows
signals due to N-H proton aliphatic protons and protons due to maltosyl
ring.
c) Mass spectral analysis: The mass spectrum of the product was also
recorded, the molecular ion peaks was also present in mass spectrum. Some
important fragment peaks were distinctly noticed.
All above facts clearly demonstrated the structure of the product with m.p.
150°C as N-hepta-O-acetyl-β-D-maltosyl-O-methyl carbamate (Va).
This reaction of hepta-O-acetyl- β -D-maltosyl isocyanate was also
extended to several other alcohols and the corresponding N-hepta-O-acetyl- β -D-
maltosyl-O-alkyl carbamates (Vb – Ve) have been isolated. (Table No. 2).
The reaction of (I) and alcohols (IV) leading to (V) may be stated as
follows.
OAcO
AcOOOAc
OAc
O
AcOOAc
OAc
N=C=O R-OH
OAcO
AcOOOAc
OAc
O
AcOOAc
OAc
NH-C-O-R
O
(I)
Hepta-O-acetyl--D-maltosyl
isocyanate
(IV)
Alcohols
(V)
1-hepa-O-acetyl--D-maltosyl
-O-alkyl carbamates
Where, R = a) methyl, b) ethyl, c) iso-propyl, d) iso-amyl, e) n-butyl.
Table 2 : N-hepta-O-acetyl- β -D-maltosyl-O-alkyl carbamates (V).
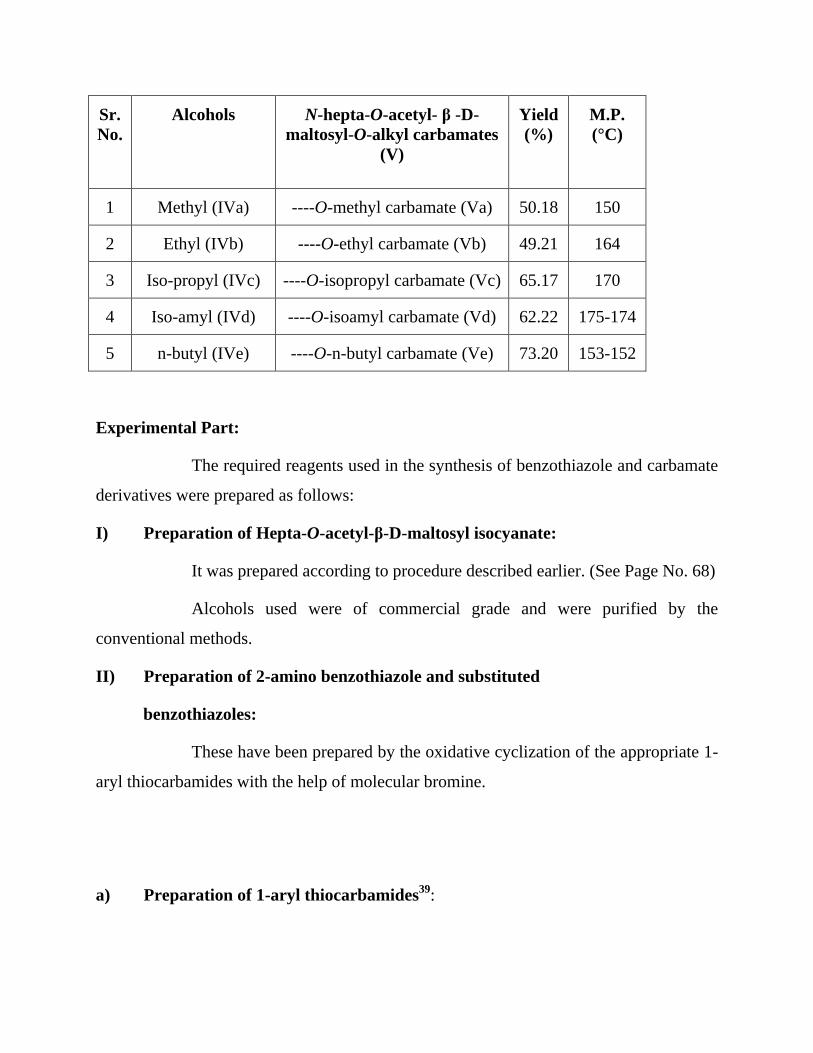
Sr.
No.
Alcohols N-hepta-O-acetyl- β -D-
maltosyl-O-alkyl carbamates
(V)
Yield
(%)
M.P.
(°C)
1 Methyl (IVa) ----O-methyl carbamate (Va) 50.18 150
2 Ethyl (IVb) ----O-ethyl carbamate (Vb) 49.21 164
3 Iso-propyl (IVc) ----O-isopropyl carbamate (Vc) 65.17 170
4 Iso-amyl (IVd) ----O-isoamyl carbamate (Vd) 62.22 175-174
5 n-butyl (IVe) ----O-n-butyl carbamate (Ve) 73.20 153-152
Experimental Part:
The required reagents used in the synthesis of benzothiazole and carbamate
derivatives were prepared as follows:
I) Preparation of Hepta-O-acetyl-β-D-maltosyl isocyanate:
It was prepared according to procedure described earlier. (See Page No. 68)
Alcohols used were of commercial grade and were purified by the
conventional methods.
II) Preparation of 2-amino benzothiazole and substituted
benzothiazoles:
These have been prepared by the oxidative cyclization of the appropriate 1-
aryl thiocarbamides with the help of molecular bromine.
a) Preparation of 1-aryl thiocarbamides39
:

These have been prepared by the interaction of ammonium thiocyanate and
appropriate aryl amine hydrochlorides. Details of typical preparation (where,
aryl=phenyl) are as follows:
Aniline (52ml) and conc. hydrochloric acid (52ml) were warmed in a
500ml of round bottom flask till its hydrochloride was formed. The hydrochloride was
dissolved in water and to this solution was added a solution of ammonium thiocyanate
(40g in 150ml water). After mixing properly the mixture was boiled over gentle bunsen
flame until the boiling solution become turbid due to sepration of phenyl thiocarbamide.
This hot solution was poured with stirring in 250ml of cold water. When 1-phenyl
thiocarbamide was obtained (40g) as a white solid. It was crystallized from boiling water,
m.p. 154°C. Melting points of other aryl thiocarbamides prepared are tabulated below.
Sr. No. 1-aryl thiocarbamides M.P. (°C) Lit. M.P. (°C)
1 1-phenyl thiocarbamide 154 154
2 1-o-tolyl thiocarbamide 160 161
3 1-m-tolyl thiocarbamide 110 111
4 1-p-tolyl thiocarbamide 189 189
5 1-o-Cl-phenyl thiocarbamide 143 143
6 1-m-Cl-phenyl thiocarbamide 140 141
7 1-p-Cl-phenyl thiocarbamide 182 183
b) Preparation of substituted benzothiazoles40-41
:
These have been prepared by oxidative cyclisation of aryl thiocarbamides
with the help of molecular bromine.
To a chloroformic paste of phenyl thiocarbamide (5g in 10ml) was added
gradually a chloroformic solution of bromine (20%) with constant stirring until a slight
excess of bromine was added as evident from an orange red colour. It was then allowed
to stand for 5-6 hrs. The resultant acidic solid (a hydrobromide) was treated with cold
ethanol, the solid, went into solution. On basification with dilute ice cold ammonium
hydroxide, 2-amino benzothiazole was separated out as a white solid (3g) m.p. 128°C.
The other substituted 2-amino benzothiazioles were prepared by the
analogous procedure. Their melting points are listed below:
Sr. No. Benzothiazole M.P. (°C)
1 2-amino benzothiazole 128
2 2-amino-(4-methyl) benzothiazole 185
3 2-amino-(5-methyl) benzothiazole 119
4 2-amino-(6-methyl) benzothiazole 140
5 2-amino-(4-chloro) benzothiazole 194
6 2-amino-(5-chloro) benzothiazole 180
7 2-amino-(6-chloro) benzothiazole 164
Interaction of 2-amino benzothiazole and hepta-O-acetyl-β-D-maltosyl isocyanate:
Formation of 1-hepta-O-acetyl-β-D-maltosyl-3-substituted benzothiazolyl
carbamides (III):
Experiment No. 1 : Synthesis of 1-hepta-O-acetyl-β-D-maltosyl-3-(2)-
benzothiazolyl carbamide (IIIa).
A benzene solution of hepta-O-acetyl- β -D-maltosyl isocyanate (0.005M,
3.3g, in 15ml) was mixed with the suspension of 2-amino benzothiazole (0.005M, 0.8g in
10ml) and mixture was reflux over boiling water bath 4 hr. After heating, benzene was
distilled off and sticky mass obtained as a residue was triturated several time with
petroleum ether to afford white solid. It was crystallized from ethanol-water, m.p. 190-
192°C. [Found: C, 50.29; H, 4.98; N, 5.14; S, 3.63, C34H41O18N3S; requires: C, 50.30, H,
5.05; N, 5.17; S, 3.94%]
The product was found soluble in ethanol, acetone, chloroform and benzene
while insoluble in water and petroleum ether. It charred when heated with conc. sulphuric
acid. It was non-desulphurisable when boiled with alkaline plumbite solution. It was
found to be optically active and its specific rotation [α]D30
= +99.75° (c, 0.978 mol in
CHCl3). The purity was checked by TLC and recorded Rf value 0.78 (Hexane: EtOAc,
1:1).
IR, 1HNMR and Mass Spectral Studies:
IR spectral analysis:
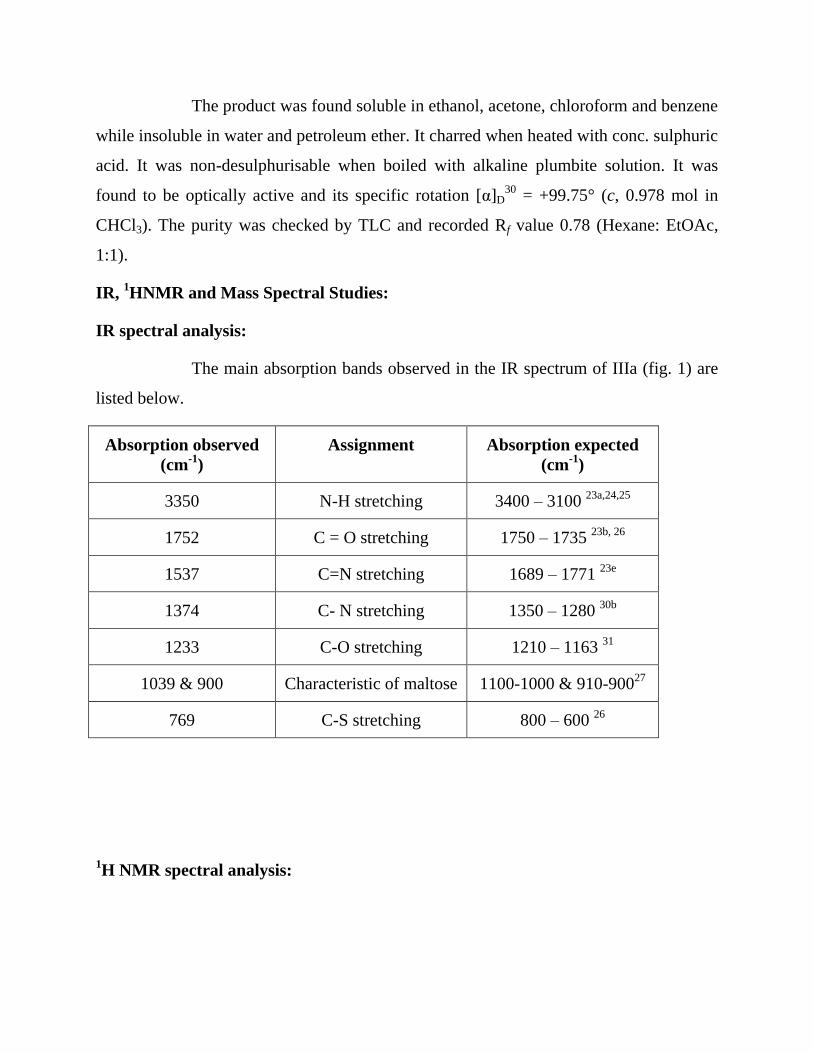
The main absorption bands observed in the IR spectrum of IIIa (fig. 1) are
listed below.
Absorption observed
(cm-1
)
Assignment Absorption expected
(cm-1
)
3350 N-H stretching 3400 – 3100 23a,24,25
1752 C = O stretching 1750 – 1735 23b, 26
1537 C=N stretching 1689 – 1771 23e
1374 C- N stretching 1350 – 1280 30b
1233 C-O stretching 1210 – 1163 31
1039 & 900 Characteristic of maltose 1100-1000 & 910-90027
769 C-S stretching 800 – 600 26
1H NMR spectral analysis:
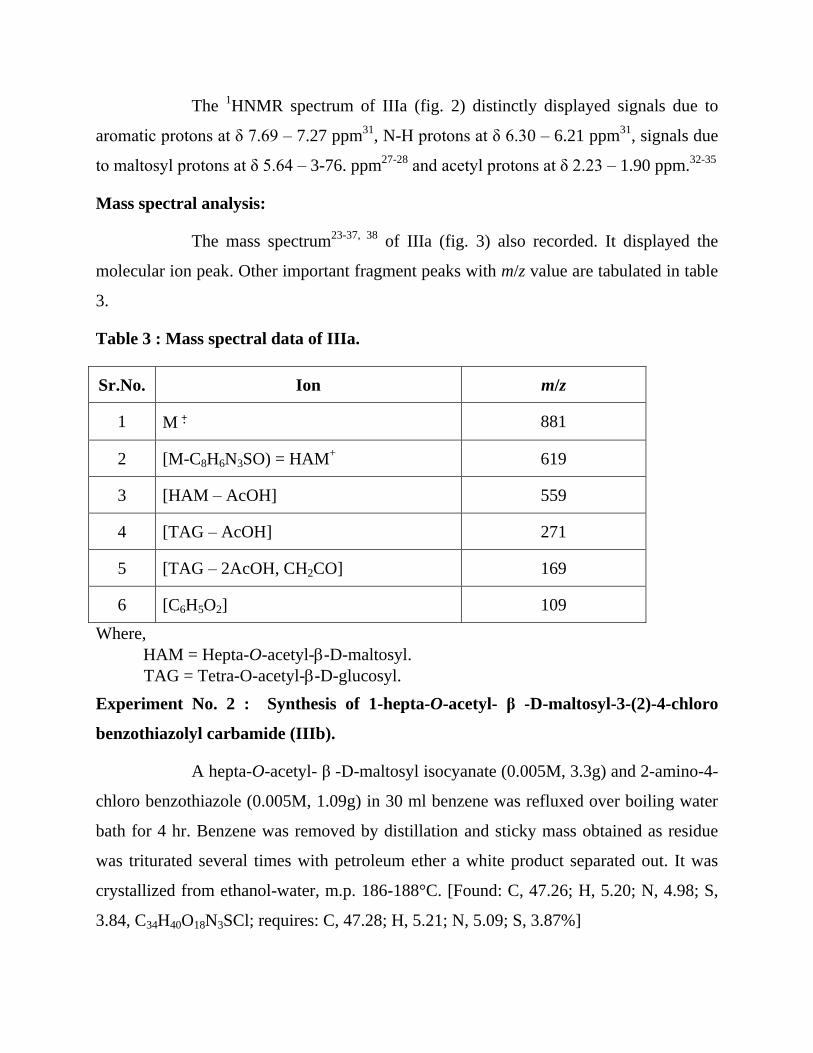
The 1HNMR spectrum of IIIa (fig. 2) distinctly displayed signals due to
aromatic protons at δ 7.69 – 7.27 ppm31
, N-H protons at δ 6.30 – 6.21 ppm31
, signals due
to maltosyl protons at δ 5.64 – 3-76. ppm27-28
and acetyl protons at δ 2.23 – 1.90 ppm.32-35
Mass spectral analysis:
The mass spectrum23-37, 38
of IIIa (fig. 3) also recorded. It displayed the
molecular ion peak. Other important fragment peaks with m/z value are tabulated in table
3.
Table 3 : Mass spectral data of IIIa.
Sr.No. Ion m/z
1 M 881
2 [M-C8H6N3SO) = HAM+ 619
3 [HAM – AcOH] 559
4 [TAG – AcOH] 271
5 [TAG – 2AcOH, CH2CO] 169
6 [C6H5O2] 109
Where,
HAM = Hepta-O-acetyl--D-maltosyl.
TAG = Tetra-O-acetyl--D-glucosyl.
Experiment No. 2 : Synthesis of 1-hepta-O-acetyl- β -D-maltosyl-3-(2)-4-chloro
benzothiazolyl carbamide (IIIb).
A hepta-O-acetyl- β -D-maltosyl isocyanate (0.005M, 3.3g) and 2-amino-4-
chloro benzothiazole (0.005M, 1.09g) in 30 ml benzene was refluxed over boiling water
bath for 4 hr. Benzene was removed by distillation and sticky mass obtained as residue
was triturated several times with petroleum ether a white product separated out. It was
crystallized from ethanol-water, m.p. 186-188°C. [Found: C, 47.26; H, 5.20; N, 4.98; S,
3.84, C34H40O18N3SCl; requires: C, 47.28; H, 5.21; N, 5.09; S, 3.87%]
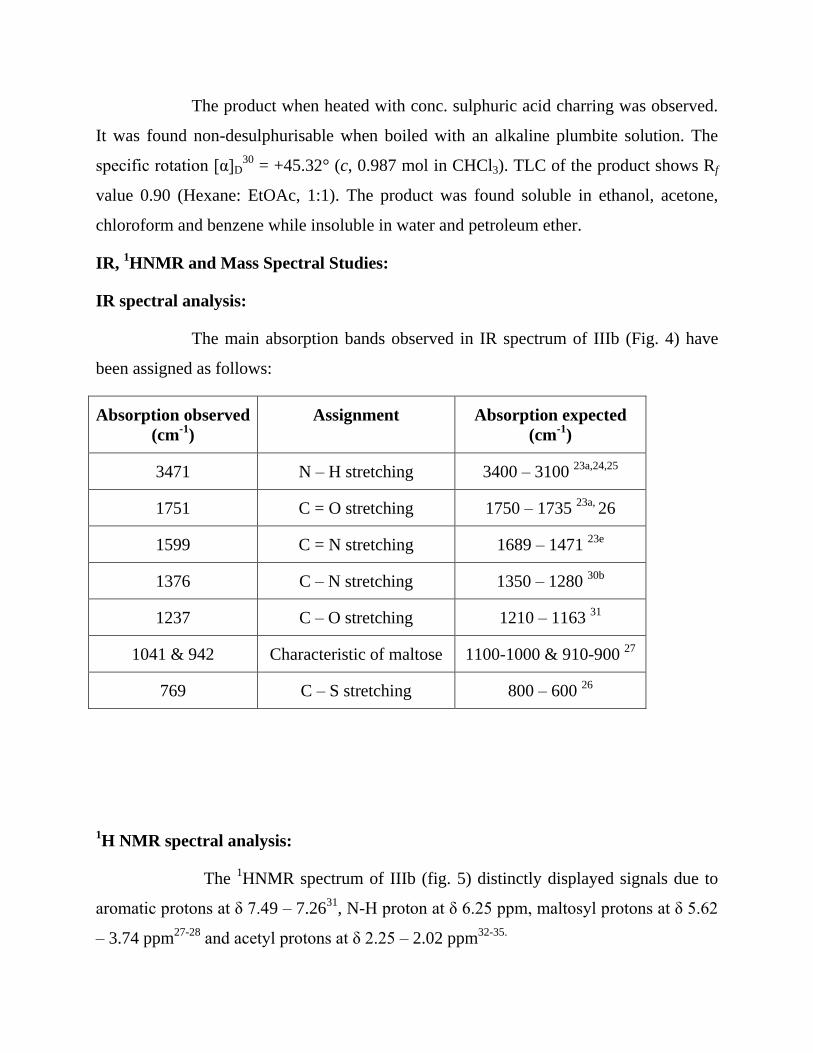
The product when heated with conc. sulphuric acid charring was observed.
It was found non-desulphurisable when boiled with an alkaline plumbite solution. The
specific rotation [α]D30
= +45.32° (c, 0.987 mol in CHCl3). TLC of the product shows Rf
value 0.90 (Hexane: EtOAc, 1:1). The product was found soluble in ethanol, acetone,
chloroform and benzene while insoluble in water and petroleum ether.
IR, 1HNMR and Mass Spectral Studies:
IR spectral analysis:
The main absorption bands observed in IR spectrum of IIIb (Fig. 4) have
been assigned as follows:
Absorption observed
(cm-1
)
Assignment Absorption expected
(cm-1
)
3471 N – H stretching 3400 – 3100 23a,24,25
1751 C = O stretching 1750 – 1735 23a,
26
1599 C = N stretching 1689 – 1471 23e
1376 C – N stretching 1350 – 1280 30b
1237 C – O stretching 1210 – 1163 31
1041 & 942 Characteristic of maltose 1100-1000 & 910-900 27
769 C – S stretching 800 – 600 26
1H NMR spectral analysis:
The 1HNMR spectrum of IIIb (fig. 5) distinctly displayed signals due to
aromatic protons at δ 7.49 – 7.2631
, N-H proton at δ 6.25 ppm, maltosyl protons at δ 5.62
– 3.74 ppm27-28
and acetyl protons at δ 2.25 – 2.02 ppm32-35.
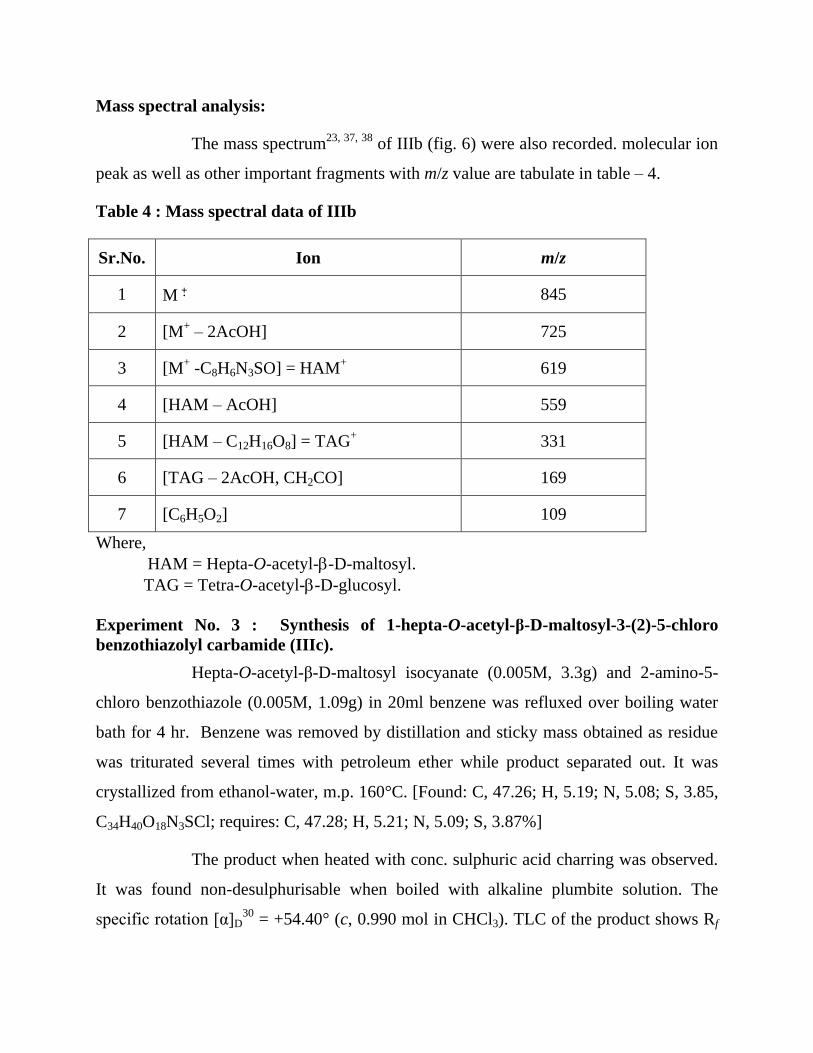
Mass spectral analysis:
The mass spectrum23, 37, 38
of IIIb (fig. 6) were also recorded. molecular ion
peak as well as other important fragments with m/z value are tabulate in table – 4.
Table 4 : Mass spectral data of IIIb
Sr.No. Ion m/z
1 M 845
2 [M+ – 2AcOH] 725
3 [M+ -C8H6N3SO] = HAM
+ 619
4 [HAM – AcOH] 559
5 [HAM – C12H16O8] = TAG+ 331
6 [TAG – 2AcOH, CH2CO] 169
7 [C6H5O2] 109
Where,
HAM = Hepta-O-acetyl--D-maltosyl.
TAG = Tetra-O-acetyl--D-glucosyl.
Experiment No. 3 : Synthesis of 1-hepta-O-acetyl-β-D-maltosyl-3-(2)-5-chloro
benzothiazolyl carbamide (IIIc).
Hepta-O-acetyl-β-D-maltosyl isocyanate (0.005M, 3.3g) and 2-amino-5-
chloro benzothiazole (0.005M, 1.09g) in 20ml benzene was refluxed over boiling water
bath for 4 hr. Benzene was removed by distillation and sticky mass obtained as residue
was triturated several times with petroleum ether while product separated out. It was
crystallized from ethanol-water, m.p. 160°C. [Found: C, 47.26; H, 5.19; N, 5.08; S, 3.85,
C34H40O18N3SCl; requires: C, 47.28; H, 5.21; N, 5.09; S, 3.87%]
The product when heated with conc. sulphuric acid charring was observed.
It was found non-desulphurisable when boiled with alkaline plumbite solution. The
specific rotation [α]D30
= +54.40° (c, 0.990 mol in CHCl3). TLC of the product shows Rf

value 0.93 (Hexane: EtOAc, 1:1). The product was found soluble in ethanol, acetone,
chloroform and benzene while insoluble in water and petroleum ether.
Experiment No. 4 : Synthesis of 1-hepta-O-acetyl- β -D-maltosyl-3-(2)-6-chloro
benzothiazolyl carbamide (IIId).
A benzene solution of hepta-O-acetyl-β-D-maltosyl isocyanate (0.005M,
3.3g in 25ml) and benzene solution of 2-amino-6-chloro benzothiazole (0.005M, 1.09g in
5ml) was mixed and reaction mixture was refluxed over boiling water bath for 4 hr.
Benzene was distilled off and sticky mass obtained as residue was triturated 3 to 4 times
with petroleum ether to afford, a white product. It was crystallized from ethanol-water,
m.p. 153-154°C. [Found: C, 47.26; H, 5.20; N, 5.10; S, 3.85, C34H40O18N3SCl; requires:
C, 47.28; H, 5.21; N, 5.09; S, 3.87%]
The product was found non-desulphurisable when boiled with alkaline
plumbite solution. It charred when warm with conc. sulphuric acid, its specific rotation
[α]D30
= +65.70° (c, 0.993 mol in CHCl3). TLC of the product shows Rf value 0.87
(Hexane: EtOAc, 1:2). The product was found soluble in ethanol, acetone, chloroform
and benzene while insoluble in water and petroleum ether.
Experiment No. 5 : Synthesis of 1-hepta-O-acetyl- β -D-maltosyl-3-(2)-4-methyl
benzothiazolyl carbamide (IIIe).
A benzene suspension of 2-amino-4-methyl benzothiazole (0.005M, 0.82g)
was added to benzene solution of hepta-O-acetyl-β-D-maltosyl isocyanate (0.005M, 3.3g)
and the reaction mixture was refluxed over boiling water bath for 4 hr. Benzene was
removed by distillation and sticky mass obtained as a residue was triturated with
petroleum ether 3 to 4 times. Finally a white product separated out. It was crystallized
from ethanol-water, m.p. 175°C. [Found: C, 50.86; H, 5.20; N, 5.11; S, 3.84,
C35H43O18N3S; requires: C, 50.90; H, 5.21; N, 5.09; S, 3.87%]

The product charred when heated with conc. sulphuric acid. On boiling
with alkaline plumbite solution, it was found non-desulphurisable. Its specific rotation
[α]D30
= +123.20° (c, 0.991 mol in CHCl3). TLC of the product carried out and recorded
of Rf value 0.79 (Hexane: EtOAc, 1:1). The product was found soluble in ethanol,
acetone, chloroform and benzene while insoluble in water and petroleum ether.
Experiment No. 6 : Synthesis of 1-hepta-O-acetyl- β -D-maltosyl-3-(2)-5-methyl
benzothiazolyl carbamide (IIIf).
2-amino-5-methyl benzothiazole (0.005M, 0.82g) was added to benzene
solution of hepta-O-acetyl-β-D-maltosyl isocyanate (0.005M, 3.3g) in 20ml benzene and
reaction was carried out over boiling water bath for 4 hr. Benzene was distilled off and
sticky residue obtained. This on trituration with petroleum ether, a white product was
separated out. It was crystallized from ethanol-water, m.p. 183.184°C. [Found: C, 50.89;
H, 5.20; N, 5.08; S, 3.86, C35H43O18N3S; requires: C, 50.90; H, 5.21; N, 5.09; S, 3.87%]
The product was found non-disulphurisable when boiled with alkaline
plumbite solution. It charred when warm with conc. sulphuric acid. Its [α]D30
was found
to be +141.51° (c, 0.982 mol in CHCl3). The purity was checked by TLC and recorded
Rf value 0.78 (Hexane: EtOAc, 1:1). The product was found soluble in ethanol, acetone,
chloroform and benzene while insoluble in water and petroleum ether.
Experiment No. 7 : Synthesis of 1-hepta-O-acetyl- β -D-glucosyl-3-(2)-6-methyl
benzothiazolyl carbamide (IIIg).
The reaction of hepta-O-acetyl-β-D-maltosyl isocyanate (0.005M, 3.3g in
15ml benzene) and 2-amino-6-methyl benzothiazole (0.005M, 0.82g in 5ml benzene) was
carried out over boiling water bath for 4 hr. Benzene was removed by distillation and the
sticky mass obtain as a residue was triturated with petroleum ether 3 to 4 times. Finally a
white product separated out. It was crystallized from ethanol- water, m.p. 171-173°C.
[Found: C, 50.89; H, 5.20; N, 5.10; S, 3.86, C35H43O18N3S; requires: C, 50.90; H, 5.21;
N, 5.09; S, 3.87%]

The product found non-desulphurisable, when boiled with an alkaline
plumbite solution. It charred on heating with conc. sulphuric acid. Its [α]D30
was found to
be -296.43° (c, 0.933 mol in CHCl3). The purity was checked by TLC and recorded Rf
value 0.82 (Hexane: EtOAc, 1:1). The product was found soluble in ethanol, acetone,
chloroform and benzene while insoluble in water and petroleum ether.
IR, 1HNMR and Mass Spectral Studies:
IR spectral analysis:
IR spectrum of IIIg (Fig. 7) exhibited the presence of following bands.
Absorption observed
(cm-1
)
Assignment Absorption expected
(cm-1
)
3487 N-H stretching 3400 – 3100 23a, 24, 25
1749 C = O stretching 1750 – 1735 23b – 26
1634 C = N stretching 1689 – 1471 23e
1375 C – N stretching 1350 – 1280 30b
1238 C – O stretching 1210 – 1163 31
1042 & 941 Characteristic of maltose 1100-1000 & 910-900 27
774 C – S stretching 800 – 600 26
1H NMR spectral analysis:
The 1HNMR spectrum of IIIg (fig. 8) displayed the signals due to N-H
proton δ 5.62 ppm, aromatic proton at δ 7.36–7.26 ppm31
. Methyl protons at δ 2.37
ppm32
, maltosyl protons at δ 5.59–3.97 ppm27-28
and acetyl protons at 2.23–2.03 ppm32-36
.
Mass spectral analysis:
The Mass spectrum of IIIg (fig. 9) was also recorded. The molecular ion
peak as well as other important fragment peaks with m/z value listed in table – 5.
Table 5 : Mass spectral data of IIIg

Sr.No. Ion m/z
1 M 825
2 [M+ – 2AcOH] 705
3 [M+ -C8H6N3SO] = HAM
+ 619
4 [HAM – AcOH] 559
5 [HAM – C12H16O8] = TAG+ 331
6 [TAG – 2AcOH, CH2CO] 169
Where,
HAM = Hepta-O-acetyl--D-maltosyl
TAG = Tetra-O-acetyl--D-glucosyl
Interaction of Hepta-O-acetyl-β-D-maltosyl isocyanate and various Alcohols:
Formation of N-hepta-O-acetyl-β-D-maltosyl-O-alkyl carbamates (V):
Experiment No. 1 : Synthesis of N-hepta-O-acetyl-β-D-maltosyl-O-methyl
carbamate (Va).
Hepta-O-acetyl-β-D-maltosyl isocyanate (0.005M, 3.3g) was added to
methyl alcohol (20ml) and the reaction mixture was refluxed over boiling water bath for
3 hr. It was then allowed to cool and pour it in water with vigorous stirring, a white
granular solid was separated out. It was crystallized from aqueous ethnaol, m.p. 150°C.
[Found: C, 48.37; H, 5.59; N, 1.98; C28H39O19N; requires: C, 48.48; H, 5.62; N, 2.02%]
It was found soluble in alcohols, acetone, chloroform and benzene while
insoluble in water and petroleum ether. It charred when warmed with conc. sulphuric
acid. The specific rotation was found to be [α]D30
= +60.32° (c, 0.667 mol in CHCl3). The
purity was checked by TLC, and recorded Rf value 0.65 (Hexane: EtOAc, 1:2).
IR, 1HNMR and Mass Spectral Studies:
IR spectral analysis:
The main absorption bands observed in the IR spectrum of Va (Fig. 10) are listed below:
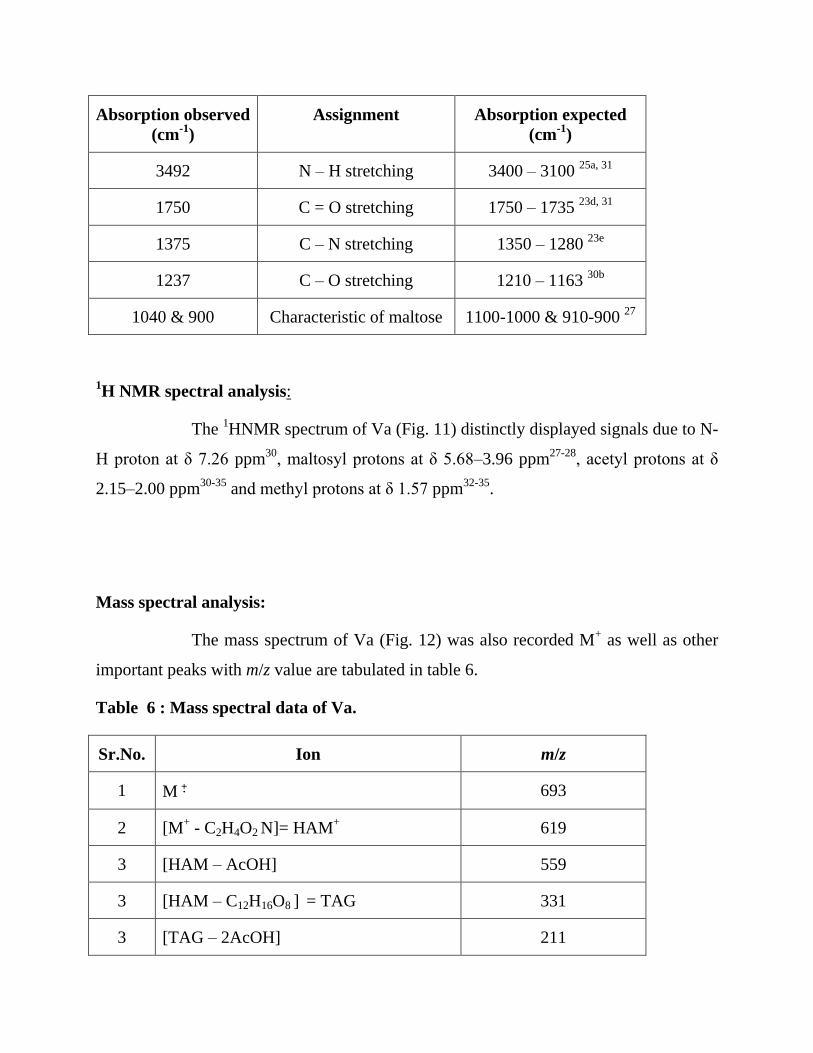
Absorption observed
(cm-1
)
Assignment Absorption expected
(cm-1
)
3492 N – H stretching 3400 – 3100 25a, 31
1750 C = O stretching 1750 – 1735 23d, 31
1375 C – N stretching 1350 – 1280 23e
1237 C – O stretching 1210 – 1163 30b
1040 & 900 Characteristic of maltose 1100-1000 & 910-900 27
1H NMR spectral analysis:
The 1HNMR spectrum of Va (Fig. 11) distinctly displayed signals due to N-
H proton at δ 7.26 ppm30
, maltosyl protons at δ 5.68–3.96 ppm27-28
, acetyl protons at δ
2.15–2.00 ppm30-35
and methyl protons at δ 1.57 ppm32-35
.
Mass spectral analysis:
The mass spectrum of Va (Fig. 12) was also recorded M+ as well as other
important peaks with m/z value are tabulated in table 6.
Table 6 : Mass spectral data of Va.
Sr.No. Ion m/z
1 M 693
2 [M+ - C2H4O2 N]= HAM
+ 619
3 [HAM – AcOH] 559
3 [HAM – C12H16O8 ] = TAG 331
3 [TAG – 2AcOH] 211

4 [TAG – 2AcOH, CH2CO] 169
5 [C6H5O2] +
109
Where,
HAM = Hepta-O-acetyl--D-maltosyl.
TAG = Tetra-O-acetyl--D-glucosyl.
Experiment No. 2 : Synthesis of N-hepta-O-acetyl-β-D-maltosyl-O-ethyl carbamate
(Vb).
Hepta-O-acetyl-β-D-maltosyl isocyanate (0.005M, 3.3g) was mixed with
ethyl alcohol (20ml) and the reaction mixture was reflux over boiling water bath for 3 hr.
After heating the reaction mixture was allowed to cool and pour it in ice cold water, a
granular white solid was isolated. It was crystallized from aqueous ethanol, m.p. 164°C.
[Found: C, 49.20; H, 5.75; N, 1.97, C29H41O19N; requires: C, 49.22; H, 5.79; N, 1.98%]
The product charred when heated with conc. sulphuric acid. Its [α]D30
= -
40.50° (c, 0.667 mol in CHCl3). The purity of product was checked by TLC and recorded
Rf value 0.63 (Hexane: EtOAc, 1:1).
Experiment No. 3 : Synthesis of N-hepta-O-acetyl-β-D-maltosyl-O-isoporpyl
carbamate (Vc).
Hepta-O-acetyl-β-D-maltosyl isocyanate (0.005M, 3.3g) was mixed with
isopropyl alcohol (20ml) and the reaction was carried out over boiling water bath for 3 hr.
After heating the reaction mixture was allowed to cool. It was then pour into ice cold
water with stirring a granular white solid was separated out. It was crystallized from
aqueous ethanol, m.p. 170° C. [Found: C, 49.89; H, 5.93; N, 1.95, C30H43O19N; requires:
C, 49.93; H, 5.96; N, 1.94%]
It was found soluble in ethanol, acetone, chloroform and benzene while
insoluble in water and petroleum ether. It charred when warmed with conc. sulphric acid.
The product was optically active and its specific rotation was found to be [α]D30
= -70.35°
(c, 0.668 mol in CHCl3). The purity of product checked by TLC and recorded Rf value
0.71 (Hexane: EtOAc, 1:1).

Experiment No. 4 : Synthesis of N-hepta-O-acetyl-β-D-maltosyl-O-isoamyl
carbamate (Vd).
Hepta-O-acetyl-β-D-maltosyl isocyanate (0.005M, 3.3g) was added to
isoamyl alcohol and the reaction was carried out at 130-150°C. Afterwards, reaction
mixture was allowed to cool and pour in ice cold water, a granular solid separated out. It
was crystallized from ethanol-water, m.p. 174-175°C. [Found: C, 50.96; H, 6.21; N, 1.90;
for C32H47O19N; requires: C, 51.26; H, 6.27; N, 1.86%]
The product was soluble in ethanol, acetone, chloroform and benzene while
insoluble in water and petroleum ether. It charred when heated with conc. sulphuric acid.
The product was optically active and its specific rotation was found to be [α]D30
=
+104.13° (c, 0.674 mol in CHCl3). The purity of the product checked by TLC and
recorded Rf value 0.77 (Hexane: EtOAc, 1:1).
Experiment No. 5 : Synthesis of N-hepta-O-acetyl-β-D-maltosyl-O-n-butyl
carbamate (Ve).
n-Butyl alcohol (20ml) was mixed with hepta-O-acetyl-β-D-maltosyl
isocyanate (0.005M, 3.3g) and reaction was carried out at 120-150°C. Afterwards, the
reaction mixture was allowed to cool and pour into ice cold water, a granular solid
separated out. It was crystallized from aqueous ethanol, m.p. 152-153°C. [Found: C,
50.59; H, 6.09; N, 1.89; C31H45O19N; requires: C, 50.61; H, 6.12; N, 1.90%]
The product was soluble in ethanol, acetone, chloroform and benzene while
insoluble in water and petroleum ether. It charred when heated with sulphuric acid. The
product was optically active and its [α]D30
= +64.62° (c, 0.667 mol in CHCl3). The purity
of the product checked by TLC and recorded Rf value 0.80 (Hexane: EtOAc, 1:1)
IR, 1HNMR and Mass Spectral Studies:
IR spectral analysis:
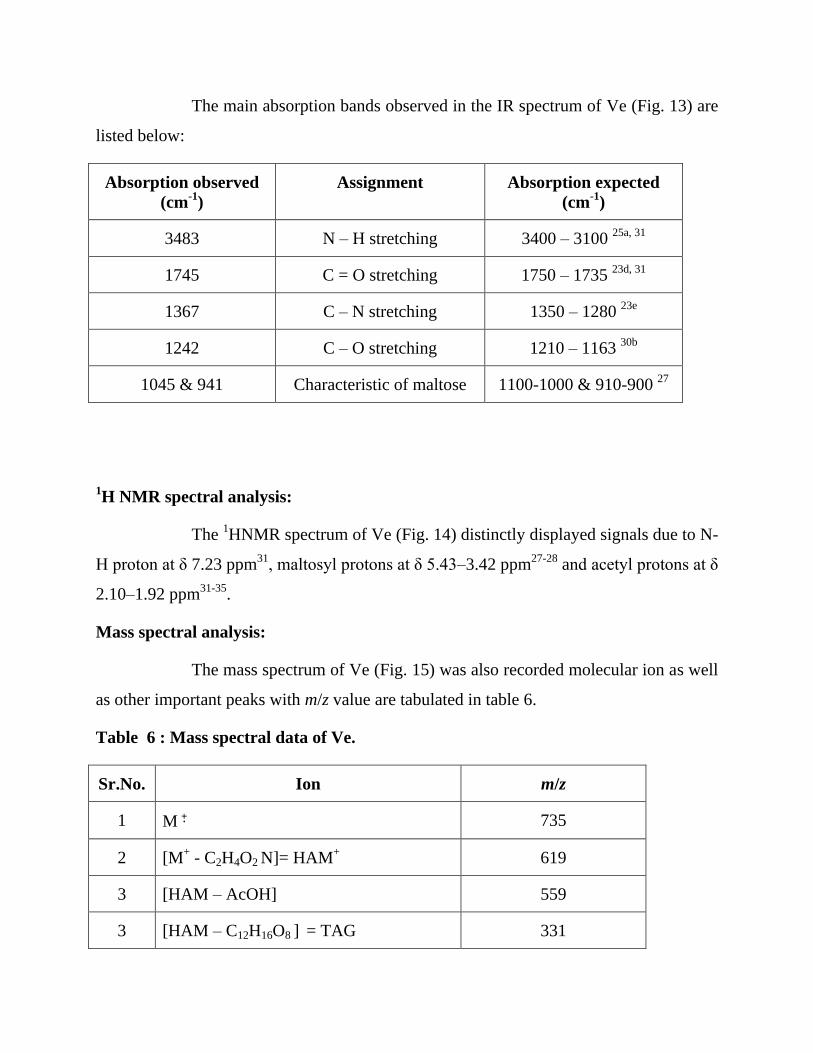
The main absorption bands observed in the IR spectrum of Ve (Fig. 13) are
listed below:
Absorption observed
(cm-1
)
Assignment Absorption expected
(cm-1
)
3483 N – H stretching 3400 – 3100 25a, 31
1745 C = O stretching 1750 – 1735 23d, 31
1367 C – N stretching 1350 – 1280 23e
1242 C – O stretching 1210 – 1163 30b
1045 & 941 Characteristic of maltose 1100-1000 & 910-900 27
1H NMR spectral analysis:
The 1HNMR spectrum of Ve (Fig. 14) distinctly displayed signals due to N-
H proton at δ 7.23 ppm31
, maltosyl protons at δ 5.43–3.42 ppm27-28
and acetyl protons at δ
2.10–1.92 ppm31-35
.
Mass spectral analysis:
The mass spectrum of Ve (Fig. 15) was also recorded molecular ion as well
as other important peaks with m/z value are tabulated in table 6.
Table 6 : Mass spectral data of Ve.
Sr.No. Ion m/z
1 M 735
2 [M+ - C2H4O2 N]= HAM
+ 619
3 [HAM – AcOH] 559
3 [HAM – C12H16O8 ] = TAG 331

3 [TAG – 2AcOH] 211
4 [TAG – 2AcOH, CH2CO] 169
5 [C6H5O2] +
109
Where,
HAM = Hepta-O-acetyl--D-maltosyl
TAG = Tetra-O-acetyl--D-glucosyl
REFERENCES:
1 A. Rana, M. Diddiqui and S. A.
Khan
: Indian J. Pharma. Sci., 69(1), 10-
17 (2007).
2 Ya. V. Rashkes and Vu. M.
Milgon
: Khim. Geterotsikil.soedin.; 1539-
1543 (1989); Chem.Abst.; 122,
235728 (1990).
3 J. Fuentes Mota, J. M. Garcia
Fernandez, C. O. Mellet, M.A.
Pradera Adrian, and T. Cueves
Lorite
: J. Carbohydr. Chem.; 9, 837
(1990).
4 L. H. Cao, C. J. Zhou, H. Y.
Goa, and Y. T. Liu
: J. Clin. Chem. Soc., 48, 207-210
(2001).
5 M. Lacova, J. Chavancova, O.
Hybloova and S. Varkonda
: Chem. Pap., 45, 411 (1991).
6 I. Chaluk, V. Sutorius, and S.
Sekarka
: Chem. Pap., 44, 131 (1990).

7 T. Papenfuns : Ger. Offen, De., 3, 528 (1987).
8 T. D. Bradshow, M. C. Bibby, J.
A. Double, I. Fichtner, P. A.
Cooper, M. C. Alley, S.
Donohue, S.F. Stinson, J.E.
Tomaszewjski, E.A.Sausville,
and M. F. G. Stevens
: Mol. Cancer. Therapeautics., 1,
239 (2002).
9 T. D. Bradshow, M. S. Chua, H.
L. Browne, V. Trapani, E. A.
Sausville, and M.F.G. Stevens
: Brit. J. Cancer., 86, 1348 (2002).
10 I. Hutchinson, S. A. Jennings, B.
R. Vishnuvajjala, A. D.
Westwell, and M.F.G. Stevens
: J. Med. Chem., 45, 744 (2002).
11 M. A. El-Sherbeny : Arzencim-Forsch, 50, 843
(2002).
12 L. Racane, V. Tralickulenovic,
L.Fiseo-Jakie, D. W. Boykin,
and G. Karmiski-Zamola
: Heterocycles, 55, 2085 (2001).
13 Mahmood-ul-Hasan, Z. H.
Chohan and C. T. Supuran
: Main Group Met. Chem., 25, 291
(2002).
14 S. Hout, M. Azas, A. Darque,
M. Robin, C. Di, Giorgio, M.
Gasquet, J. Galy and P. Trimon-
David
: Paractiology, 129, 525-542
(2004).

15 M. S. Shingare and D. B. Ingle : J. Ind. Chem. Soc., 53, 1036
(1976).
16 B. Dash and M. Patra : Indian J. Chem., 19B, 894
(1980).
17 G. V. Korpe, S. P. Deshmukh
and A. K. Fokmare
: Indian J. Heterocyclic Chem., 10,
287-290 (2001).
18 P. V. Tale and S. P. Deshmukh : Heteroatom chemistry., 17(4),
304-309 (2006).
19 Y. I. Chikawa, Y. Matukawa, T.
Mishiyama, and M. Isobe
: Eur. J. Org. Chem., 586-591
(2004).
20 A. G. Sarap and S. P. Deshmukh : Int. J. Chem. Sci., 7(4), 2389-
2397 (2009).
21 Arnold Weissbeger : “Physical method of organic
chemistry”, Part-II, 2nd
Ed.
Interscience Pub. INC New York
(1949).
22 B. Fried and S. H. Sherama : Thin-layer chromatography, 2nd
Ed., Chromotography Science
series, New York Decker 35
(1986).
23 R. M. Silverstein, G. C. Bassler
and T. C. Morrill
: "Spectroscopic Identification of
Organic Compounds", 5th
Ed.,
John Wiley and Sons, Inc., New
York (1991) p. a) 108, b) 119, C,

120, d) 123, e) 127.
24 Margareta Avram and G. H.
Mateesau
: “Infra-Red spectroscopy.
Applications in organic
chemistry”, John Wielly and
Son, INC, New York (1970).
25
D. H. Williams and I. Flemming
:
"Spectroscopic Methods in
Organic Chemistry", Vol. IV,
Tata McGraw Hill (1991) p. a)
42, b) 47, c) 55.
26 R. Varma, S. Y. Kulkarni, C. I.
Jose and V. S. Pansare
: Carbohydr. Res., 133, 25 - 32
(1984).
27 B. H. koeppen : Carbohydr. Res., 13, 193-198
(1970).
28 S. A. Barker, E. J. Bourne, R.
Stephens and D. H. Whiffen
: J. Chem. Soc., 3468 (1954).
29 K. Biemann, D. C. Dejongh, and
H. K. Schones
: J. Am. Chem. Soc., 85, 1763
(1963).
30 J. R. Dyer : "Applications of Absorption
Spectroscopy of Organic
Compounds", 8th
Ed., Prentice
Hall (1991) p. a) 36, b) 37, c) 38,
d) 88.
31 C. Suitz-Barriu, A-Torres : Synlett, 12, 1891-1894 (1999).

Pinedo and F. Santoya-Gonzaler
32 R. Kassab, C. Felix, H. Parrot-
Lopez, and R. Bonaly
: Tetrahedron Lett., 38(43), 7555
(1997).
33 C. Prata, N. Mora, J-M.
Lacombe, J-C.Maurizis and B.
Pucci
: Carbohydr. Res., 321, 4 (1999).
34 J. J. Garcia-Lopez, F.
Hernandez Mateo, J. Isca-
Garcia, J.M.Kim, R. Roy, F.
Santoyo-Gonzaler and A.
Vargas-Berenguel
: J. Org. Chem., 64, 522 (1999).
35 J. Isac-Garcia, F. G. Calvo-
Flores, F. Hernandez-Mateo and
F. Santoyo-Gonzalez
: Eur. J. Org. Chem., 282 (2001).
36 M. A. Saleh : Sulfur. Lett., 25(6), 235 (2002).
37 M. A. Maier, C. G.
Yannopoulos, N. Mohamed, A.
Roland, H. Fritz, V. Mohan, G.
Just and M. Manoharan
: Bioconjugate Chem., 14, 18
(2003).
38 H. Budzikie, Wicz, C. Djerassi
and D. H. Williams
: “Structural elucidation of natural
product by mass spectroscopy”,
Vol.II, Holden-Day (1964).
39 H. Krall, and R. D. Gupta : J. Indian Chem. Soc., 12, 629
(1935).

40 R. F. Hunter : J. Chem. Soc. 127, 2003-2007
(1928).
41 H. P. Kaufman : Arch, Pharma., 266-197 (1925).














![Case Report Initial Biological Evaluations of [18F]KS-7-51 to … · 2020. 9. 22. · and initial biological evaluations of [18F]KS-7-51, a p-fluoroethoxy phenyl derivative in a murine](https://static.fdocument.org/doc/165x107/601e58f23cdaba46814221b9/case-report-initial-biological-evaluations-of-18fks-7-51-to-2020-9-22-and.jpg)


