Caspase-8 promotes NLRP1/NLRP3 inflammasome activation … · some pathway and a...
6
Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1β production in acute glaucoma Wei Chi a,1 , Fei Li a,1 , Hongrui Chen a,1 , Yandong Wang a,1 , Yingting Zhu a , Xuejiao Yang a , Jie Zhu b , Frances Wu b , Hong Ouyang b , Jian Ge a , Robert N. Weinreb b , Kang Zhang b,2 , and Yehong Zhuo a,2 a State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou 510060, China; and b Department of Ophthalmology and Shiley Eye Center, University of California, San Diego, La Jolla, CA 92093 Edited* by Napoleone Ferrara, University of California, San Diego, La Jolla, CA, and approved June 11, 2014 (received for review February 25, 2014) Acute glaucoma is a sight-threatening condition characterized by a sudden and substantial rise in intraocular pressure (IOP) and consequent retinal ganglion cell (RGC) death. Angle closure glau- coma, a common cause of glaucoma in Asia that affects tens of millions of people worldwide, often presents acutely with loss of vision, pain, and high IOP. Even when medical and surgical treatment is available, acute angle closure glaucoma can cause permanent and irreversible loss of vision. Toll-like receptor 4 (TLR4) signaling has been previously implicated in the pathogenesis of IOP-induced RGC death, although the underlying mechanisms are largely unknown. In the present study, we used an acute IOP elevation/glaucoma model to investigate the underlying mechanism of RGC death. We found that TLR4 leads to increased caspase-8 expression; this elevation increases IL-1β expression and RGC death via a caspase-1–dependent pathway involving Nod-like receptor family, pyrin domain contain- ing 1 (NLRP1)/NLRP3 inflammasomes and a caspase-1–independent pathway. We show that inhibition of caspase-8 activation signifi- cantly attenuates RGC death by down-regulating the activation of NLRP1 and NLRP3, thus demonstrating the pivotal role of caspase-8 in the TLR4-mediated activation of inflammasomes. These findings demonstrate collectively a critical role of caspase-8 in transducing TLR4-mediated IL-1β production and RGC death and highlight signal transduction in a caspase-1–dependent NLRP1/NLRP3 inflamma- some pathway and a caspase-1–independent pathway in acute glaucoma. These results provide new insight into the pathogenesis of glaucoma and point to a treatment strategy. retinal ischemia/reperfusion injury | cell apoptosis A cute glaucoma is a significant cause of permanent vision loss and irreversible blindness worldwide (1). It is most common among people of Asian descent, in part due to their having a more crowded anterior chamber (2, 3). With a rapid increase in intraocular pressure (IOP) to levels exceeding retinal perfusion pressure, there is resulting retinal ischemia and retinal ganglion cell (RGC) death. Individuals with angle closure glaucoma are much more likely to lose vision and become blind than those with primary open-angle glaucoma (4). The precise mechanisms by which elevated IOP leads to RGC death are not well understood. Accumulating evidence suggests that overactivated microglia have pivotal roles in triggering neurotoxicity in the CNS, including ret- inal inflammatory responses (5, 6), by producing proinflammatory factors such as IL-1β. IL-1β production is tightly controlled as part of the innate immune response in the CNS (7). Toll-like receptors (TLRs) and Nod-like receptors (NLRs) are two key pattern recognition receptors (PRRs) in the initiation of the innate immune response (8–10). TLR4 has been shown to have a central role in retinal and CNS ischemia/reperfusion (I/R) injuries (11–13). Neuronal death following ischemic injury acti- vates intense inflammation, which triggers TLR4 signaling. It has been demonstrated that TLR4-deficient mice are protected against ischemic brain damage (14, 15). TLRs are membrane-spanning receptors and NLRs are cytoplasmic sensors that oligomerize to form a platform for the inflammasome, a multisubunit complex that activates caspase-1 to process pro–IL-1β into its mature form. IL-1β secretion is a well-characterized outcome of TLR and inflammasome cooperation. TLRs induce transcriptional acti- vation of pro–IL-1β, which is proteolytically processed via inflammasome activation (16, 17). Studies have shown that neu- tralization of inflammasome activity results in less severe tissue pathologies following thromboembolic stroke (18, 19). Currently, the exact function, complex molecular mechanisms, and precise interaction between TLRs and inflammasomes in the development of retinal ischemic injury remain elusive. Caspases are a family of aspartate-specific cysteine proteases with a well-characterized function in apoptotic signaling. Caspase- 8 is implicated as an initiator caspase in death receptor-induced signaling of apoptosis (20, 21). Recent reports have uncovered novel nonapoptotic functions of caspase-8 and caspase-3, showing that they induce microglia neurotoxicity and the production of IL- 1β without causing microglia death (22, 23). However, the un- derlying mechanism of caspase-8 activation in promoting IL-1β secretion is unclear. Thus, the potential roles of TLR4, inflam- masomes, and caspase-8 signaling in the development of IOP- induced retinal damage and RGC death remain to be clarified. In this study, we investigated the signaling pathway mediating RGC death and identified the key players between TLR4 and IL- 1β. It is demonstrated that IOP-induced retinal ischemia directly increases TLR4 expression, triggering caspase-8 signaling to activate Nod-like receptor family, pyrin domain containing 1 (NLRP1) and NLRP3 inflammasomes and IL-1β production in both mouse and rat models. Inhibition of caspase-8 could completely suppress IL-1β Significance Retinal damage and resulting irreversible vision loss are feared complications of rapid and substantially elevated intraocular pressure (IOP) in acute glaucoma. An inflammatory response to retinal ischemia/reperfusion injury involving Toll-like receptor 4 (TLR4) and IL-1β has been implicated in disease pathogenesis; however, the underlying mechanisms remain incompletely understood. This study demonstrates the critical role of caspase- 8 in IOP-induced cell death in rodent models of acute glaucoma. TLR4 signaling, mediated by caspase-8, was crucial for the acti- vation of Nod-like receptor family, pyrin domain containing 1 (NLRP1)/NLRP3 inflammasomes and processing of IL-1β. In- hibition of either TLR4 or caspase-8 signaling significantly blocked production of IL-1β and attenuated retinal ischemic damage. These findings identify a mechanism of retinal retinal ganglion cell death and provide a previously unidentified treatment strategy to preserve vision in acute glaucoma. Author contributions: K.Z. and Y. Zhuo designed research; W.C., F.L., H.C., Y.W., Y. Zhu, X.Y., J.Z., F.W., and H.O. performed research; J.G. contributed new reagents/analytic tools; W.C., F.L., F.W., J.G., R.N.W., K.Z., and Y. Zhuo analyzed data; and W.C., F.L., F.W., K.Z., and Y. Zhuo wrote the paper. The authors declare no conflict of interest. *This Direct Submission article had a prearranged editor. 1 W.C., F.L., H.C., and Y.W. contributed equally to this work. 2 To whom correspondence may be addressed. Email: [email protected] or zhuoyh@ mail.sysu.edu.cn. This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10. 1073/pnas.1402819111/-/DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1402819111 PNAS | July 29, 2014 | vol. 111 | no. 30 | 11181–11186 NEUROSCIENCE
Transcript of Caspase-8 promotes NLRP1/NLRP3 inflammasome activation … · some pathway and a...

Caspase-8 promotes NLRP1/NLRP3 inflammasomeactivation and IL-1β production in acute glaucomaWei Chia,1, Fei Lia,1, Hongrui Chena,1, Yandong Wanga,1, Yingting Zhua, Xuejiao Yanga, Jie Zhub, Frances Wub,Hong Ouyangb, Jian Gea, Robert N. Weinrebb, Kang Zhangb,2, and Yehong Zhuoa,2
aState Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou 510060, China; and bDepartment ofOphthalmology and Shiley Eye Center, University of California, San Diego, La Jolla, CA 92093
Edited* by Napoleone Ferrara, University of California, San Diego, La Jolla, CA, and approved June 11, 2014 (received for review February 25, 2014)
Acute glaucoma is a sight-threatening condition characterized bya sudden and substantial rise in intraocular pressure (IOP) andconsequent retinal ganglion cell (RGC) death. Angle closure glau-coma, a common cause of glaucoma in Asia that affects tens ofmillions of people worldwide, often presents acutely with loss ofvision, pain, and high IOP. Evenwhenmedical and surgical treatmentis available, acute angle closure glaucoma can cause permanent andirreversible loss of vision. Toll-like receptor 4 (TLR4) signaling hasbeen previously implicated in the pathogenesis of IOP-induced RGCdeath, although the underlying mechanisms are largely unknown. Inthe present study, we used an acute IOP elevation/glaucoma modelto investigate the underlying mechanism of RGC death. We foundthat TLR4 leads to increased caspase-8 expression; this elevationincreases IL-1β expression and RGC death via a caspase-1–dependentpathway involving Nod-like receptor family, pyrin domain contain-ing 1 (NLRP1)/NLRP3 inflammasomes and a caspase-1–independentpathway. We show that inhibition of caspase-8 activation signifi-cantly attenuates RGC death by down-regulating the activation ofNLRP1 and NLRP3, thus demonstrating the pivotal role of caspase-8in the TLR4-mediated activation of inflammasomes. These findingsdemonstrate collectively a critical role of caspase-8 in transducingTLR4-mediated IL-1β production and RGC death and highlight signaltransduction in a caspase-1–dependent NLRP1/NLRP3 inflamma-some pathway and a caspase-1–independent pathway in acuteglaucoma. These results provide new insight into the pathogenesisof glaucoma and point to a treatment strategy.
retinal ischemia/reperfusion injury | cell apoptosis
Acute glaucoma is a significant cause of permanent vision lossand irreversible blindness worldwide (1). It is most common
among people of Asian descent, in part due to their having amore crowded anterior chamber (2, 3). With a rapid increase inintraocular pressure (IOP) to levels exceeding retinal perfusionpressure, there is resulting retinal ischemia and retinal ganglioncell (RGC) death. Individuals with angle closure glaucoma aremuch more likely to lose vision and become blind than those withprimary open-angle glaucoma (4). The precise mechanisms bywhich elevated IOP leads to RGC death are not well understood.Accumulating evidence suggests that overactivated microglia havepivotal roles in triggering neurotoxicity in the CNS, including ret-inal inflammatory responses (5, 6), by producing proinflammatoryfactors such as IL-1β. IL-1β production is tightly controlled as partof the innate immune response in the CNS (7).Toll-like receptors (TLRs) and Nod-like receptors (NLRs) are
two key pattern recognition receptors (PRRs) in the initiation ofthe innate immune response (8–10). TLR4 has been shown tohave a central role in retinal and CNS ischemia/reperfusion (I/R)injuries (11–13). Neuronal death following ischemic injury acti-vates intense inflammation, which triggers TLR4 signaling. It hasbeen demonstrated that TLR4-deficient mice are protected againstischemic brain damage (14, 15). TLRs are membrane-spanningreceptors and NLRs are cytoplasmic sensors that oligomerize toform a platform for the inflammasome, a multisubunit complexthat activates caspase-1 to process pro–IL-1β into its mature form.IL-1β secretion is a well-characterized outcome of TLR and
inflammasome cooperation. TLRs induce transcriptional acti-vation of pro–IL-1β, which is proteolytically processed viainflammasome activation (16, 17). Studies have shown that neu-tralization of inflammasome activity results in less severe tissuepathologies following thromboembolic stroke (18, 19). Currently,the exact function, complex molecular mechanisms, and preciseinteraction between TLRs and inflammasomes in the developmentof retinal ischemic injury remain elusive.Caspases are a family of aspartate-specific cysteine proteases
with a well-characterized function in apoptotic signaling. Caspase-8 is implicated as an initiator caspase in death receptor-inducedsignaling of apoptosis (20, 21). Recent reports have uncoverednovel nonapoptotic functions of caspase-8 and caspase-3, showingthat they induce microglia neurotoxicity and the production of IL-1β without causing microglia death (22, 23). However, the un-derlying mechanism of caspase-8 activation in promoting IL-1βsecretion is unclear. Thus, the potential roles of TLR4, inflam-masomes, and caspase-8 signaling in the development of IOP-induced retinal damage and RGC death remain to be clarified.In this study, we investigated the signaling pathway mediating
RGC death and identified the key players between TLR4 and IL-1β. It is demonstrated that IOP-induced retinal ischemia directlyincreases TLR4 expression, triggering caspase-8 signaling to activateNod-like receptor family, pyrin domain containing 1 (NLRP1) andNLRP3 inflammasomes and IL-1β production in both mouse andrat models. Inhibition of caspase-8 could completely suppress IL-1β
Significance
Retinal damage and resulting irreversible vision loss are fearedcomplications of rapid and substantially elevated intraocularpressure (IOP) in acute glaucoma. An inflammatory response toretinal ischemia/reperfusion injury involving Toll-like receptor4 (TLR4) and IL-1β has been implicated in disease pathogenesis;however, the underlying mechanisms remain incompletelyunderstood. This study demonstrates the critical role of caspase-8 in IOP-induced cell death in rodent models of acute glaucoma.TLR4 signaling, mediated by caspase-8, was crucial for the acti-vation of Nod-like receptor family, pyrin domain containing 1(NLRP1)/NLRP3 inflammasomes and processing of IL-1β. In-hibition of either TLR4 or caspase-8 signaling significantlyblocked production of IL-1β and attenuated retinal ischemicdamage. These findings identify a mechanism of retinal retinalganglion cell death and provide a previously unidentifiedtreatment strategy to preserve vision in acute glaucoma.
Author contributions: K.Z. and Y. Zhuo designed research; W.C., F.L., H.C., Y.W., Y. Zhu,X.Y., J.Z., F.W., and H.O. performed research; J.G. contributed new reagents/analytic tools;W.C., F.L., F.W., J.G., R.N.W., K.Z., and Y. Zhuo analyzed data; and W.C., F.L., F.W., K.Z.,and Y. Zhuo wrote the paper.
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.1W.C., F.L., H.C., and Y.W. contributed equally to this work.2To whom correspondence may be addressed. Email: [email protected] or [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1402819111/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1402819111 PNAS | July 29, 2014 | vol. 111 | no. 30 | 11181–11186
NEU
ROSC
IENCE
processing in IOP-induced retinal damage, whereas inhibition ofcaspase-1 only partially reduced levels of IL-1β, implying that cas-pase-8 processed IL-1β via a caspase-1–dependent, NLRP1/NLPR3pathway and a caspase-1-independent pathway. Unlike in vitro LPSstimulating caspase-3 activation in microglia (22), IOP-inducedischemia did not significantly drive caspase-3 activation in theearly stage of acute glaucoma, suggesting that caspase-8– ratherthan caspase-3–induced inflammatory responses may be re-sponsible for RGC death with elevated IOP in the early retinaldamage of acute glaucoma. These results demonstrate that cas-pase-8 is a key link between TLR4 and inflammasomes in IL-1βprocessing and that it contributes to IOP-induced RGC death.Targeted inhibition of TLR4 and caspase-8 signaling could sig-nificantly attenuate retinal ischemic damage and RGC death byregulating the activation of inflammasomes and the productionof IL-1β.
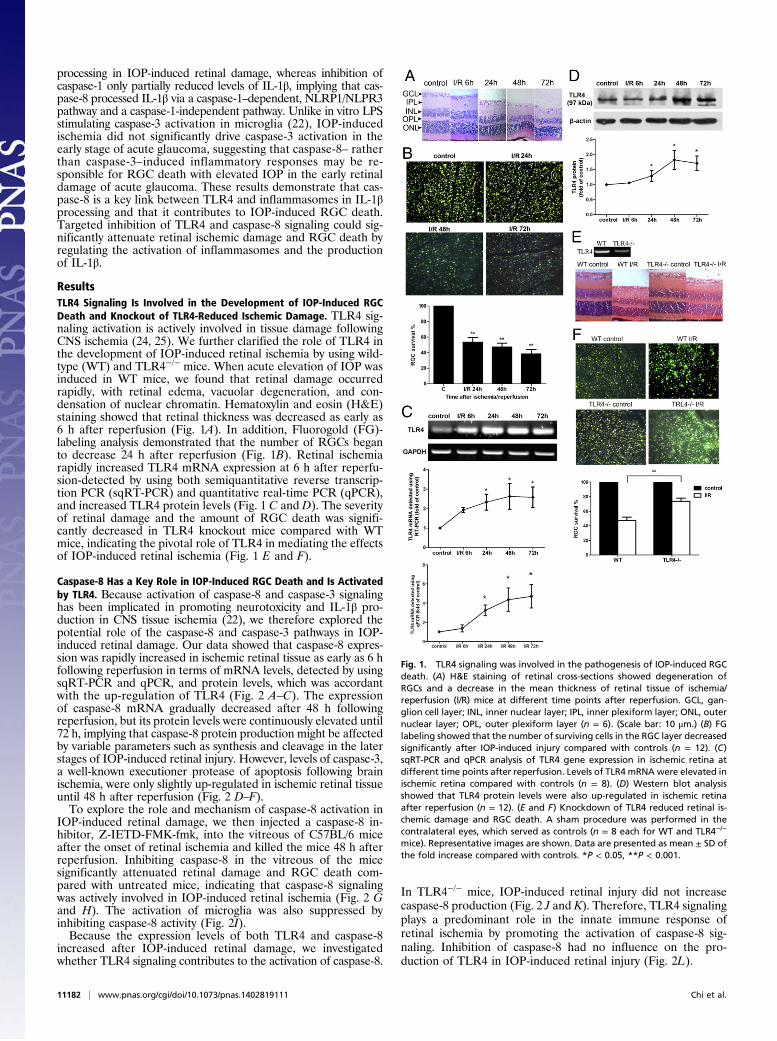
ResultsTLR4 Signaling Is Involved in the Development of IOP-Induced RGCDeath and Knockout of TLR4-Reduced Ischemic Damage. TLR4 sig-naling activation is actively involved in tissue damage followingCNS ischemia (24, 25). We further clarified the role of TLR4 inthe development of IOP-induced retinal ischemia by using wild-type (WT) and TLR4−/− mice. When acute elevation of IOP wasinduced in WT mice, we found that retinal damage occurredrapidly, with retinal edema, vacuolar degeneration, and con-densation of nuclear chromatin. Hematoxylin and eosin (H&E)staining showed that retinal thickness was decreased as early as6 h after reperfusion (Fig. 1A). In addition, Fluorogold (FG)-labeling analysis demonstrated that the number of RGCs beganto decrease 24 h after reperfusion (Fig. 1B). Retinal ischemiarapidly increased TLR4 mRNA expression at 6 h after reperfu-sion-detected by using both semiquantitative reverse transcrip-tion PCR (sqRT-PCR) and quantitative real-time PCR (qPCR),and increased TLR4 protein levels (Fig. 1 C and D). The severityof retinal damage and the amount of RGC death was signifi-cantly decreased in TLR4 knockout mice compared with WTmice, indicating the pivotal role of TLR4 in mediating the effectsof IOP-induced retinal ischemia (Fig. 1 E and F).
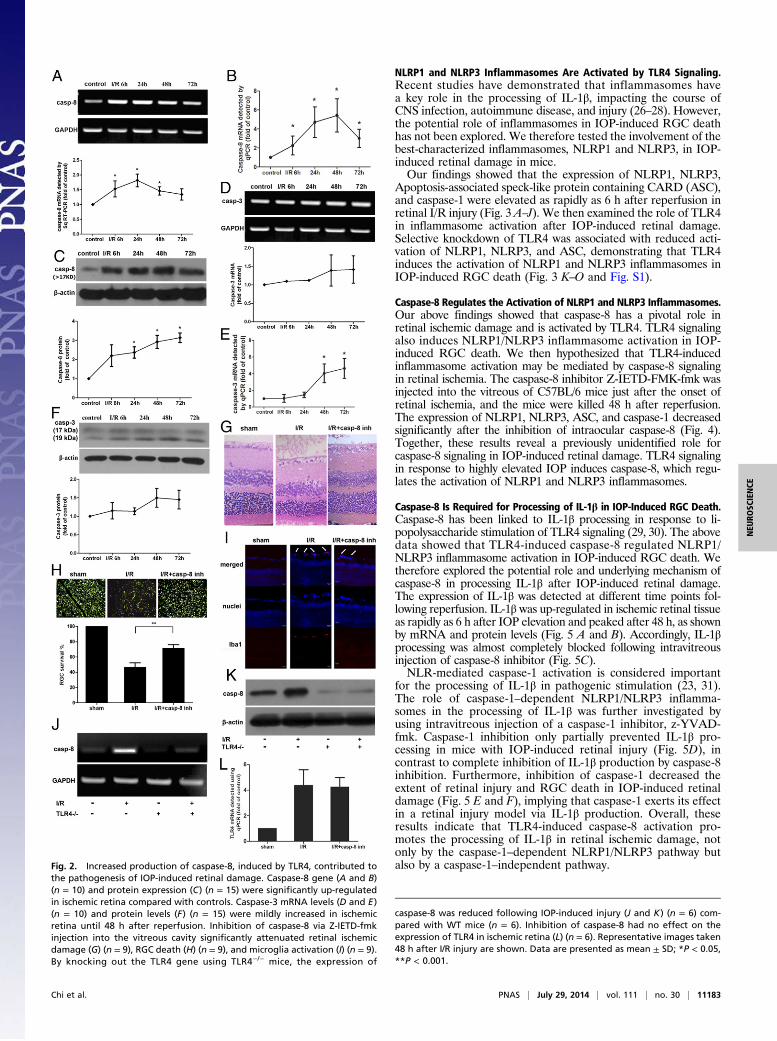
Caspase-8 Has a Key Role in IOP-Induced RGC Death and Is Activatedby TLR4. Because activation of caspase-8 and caspase-3 signalinghas been implicated in promoting neurotoxicity and IL-1β pro-duction in CNS tissue ischemia (22), we therefore explored thepotential role of the caspase-8 and caspase-3 pathways in IOP-induced retinal damage. Our data showed that caspase-8 expres-sion was rapidly increased in ischemic retinal tissue as early as 6 hfollowing reperfusion in terms of mRNA levels, detected by usingsqRT-PCR and qPCR, and protein levels, which was accordantwith the up-regulation of TLR4 (Fig. 2 A–C). The expressionof caspase-8 mRNA gradually decreased after 48 h followingreperfusion, but its protein levels were continuously elevated until72 h, implying that caspase-8 protein production might be affectedby variable parameters such as synthesis and cleavage in the laterstages of IOP-induced retinal injury. However, levels of caspase-3,a well-known executioner protease of apoptosis following brainischemia, were only slightly up-regulated in ischemic retinal tissueuntil 48 h after reperfusion (Fig. 2 D–F).To explore the role and mechanism of caspase-8 activation in
IOP-induced retinal damage, we then injected a caspase-8 in-hibitor, Z-IETD-FMK-fmk, into the vitreous of C57BL/6 miceafter the onset of retinal ischemia and killed the mice 48 h afterreperfusion. Inhibiting caspase-8 in the vitreous of the micesignificantly attenuated retinal damage and RGC death com-pared with untreated mice, indicating that caspase-8 signalingwas actively involved in IOP-induced retinal ischemia (Fig. 2 Gand H). The activation of microglia was also suppressed byinhibiting caspase-8 activity (Fig. 2I).Because the expression levels of both TLR4 and caspase-8
increased after IOP-induced retinal damage, we investigatedwhether TLR4 signaling contributes to the activation of caspase-8.
In TLR4−/− mice, IOP-induced retinal injury did not increasecaspase-8 production (Fig. 2 J and K). Therefore, TLR4 signalingplays a predominant role in the innate immune response ofretinal ischemia by promoting the activation of caspase-8 sig-naling. Inhibition of caspase-8 had no influence on the pro-duction of TLR4 in IOP-induced retinal injury (Fig. 2L).
Fig. 1. TLR4 signaling was involved in the pathogenesis of IOP-induced RGCdeath. (A) H&E staining of retinal cross-sections showed degeneration ofRGCs and a decrease in the mean thickness of retinal tissue of ischemia/reperfusion (I/R) mice at different time points after reperfusion. GCL, gan-glion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outernuclear layer; OPL, outer plexiform layer (n = 6). (Scale bar: 10 μm.) (B) FGlabeling showed that the number of surviving cells in the RGC layer decreasedsignificantly after IOP-induced injury compared with controls (n = 12). (C)sqRT-PCR and qPCR analysis of TLR4 gene expression in ischemic retina atdifferent time points after reperfusion. Levels of TLR4 mRNA were elevated inischemic retina compared with controls (n = 8). (D) Western blot analysisshowed that TLR4 protein levels were also up-regulated in ischemic retinaafter reperfusion (n = 12). (E and F) Knockdown of TLR4 reduced retinal is-chemic damage and RGC death. A sham procedure was performed in thecontralateral eyes, which served as controls (n = 8 each for WT and TLR4−/−
mice). Representative images are shown. Data are presented as mean ± SD ofthe fold increase compared with controls. *P < 0.05, **P < 0.001.
11182 | www.pnas.org/cgi/doi/10.1073/pnas.1402819111 Chi et al.
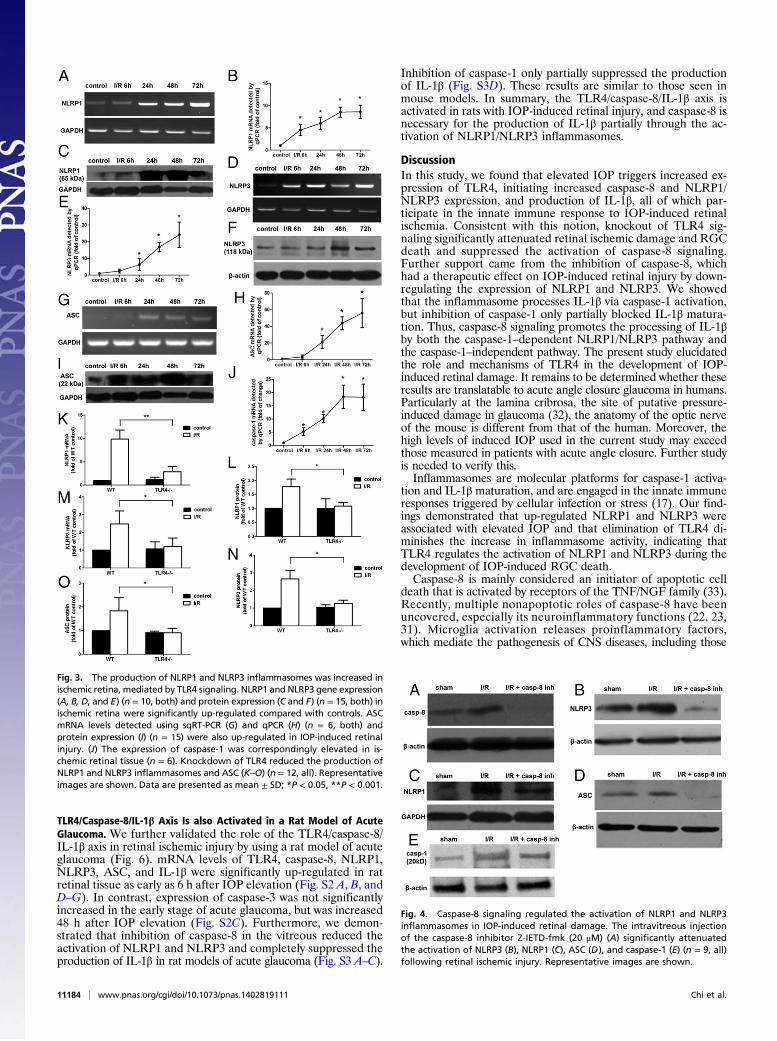
NLRP1 and NLRP3 Inflammasomes Are Activated by TLR4 Signaling.Recent studies have demonstrated that inflammasomes havea key role in the processing of IL-1β, impacting the course ofCNS infection, autoimmune disease, and injury (26–28). However,the potential role of inflammasomes in IOP-induced RGC deathhas not been explored. We therefore tested the involvement of thebest-characterized inflammasomes, NLRP1 and NLRP3, in IOP-induced retinal damage in mice.Our findings showed that the expression of NLRP1, NLRP3,
Apoptosis-associated speck-like protein containing CARD (ASC),and caspase-1 were elevated as rapidly as 6 h after reperfusion inretinal I/R injury (Fig. 3 A–J). We then examined the role of TLR4in inflammasome activation after IOP-induced retinal damage.Selective knockdown of TLR4 was associated with reduced acti-vation of NLRP1, NLRP3, and ASC, demonstrating that TLR4induces the activation of NLRP1 and NLRP3 inflammasomes inIOP-induced RGC death (Fig. 3 K–O and Fig. S1).
Caspase-8 Regulates the Activation of NLRP1 and NLRP3 Inflammasomes.Our above findings showed that caspase-8 has a pivotal role inretinal ischemic damage and is activated by TLR4. TLR4 signalingalso induces NLRP1/NLRP3 inflammasome activation in IOP-induced RGC death. We then hypothesized that TLR4-inducedinflammasome activation may be mediated by caspase-8 signalingin retinal ischemia. The caspase-8 inhibitor Z-IETD-FMK-fmk wasinjected into the vitreous of C57BL/6 mice just after the onset ofretinal ischemia, and the mice were killed 48 h after reperfusion.The expression of NLRP1, NLRP3, ASC, and caspase-1 decreasedsignificantly after the inhibition of intraocular caspase-8 (Fig. 4).Together, these results reveal a previously unidentified role forcaspase-8 signaling in IOP-induced retinal damage. TLR4 signalingin response to highly elevated IOP induces caspase-8, which regu-lates the activation of NLRP1 and NLRP3 inflammasomes.
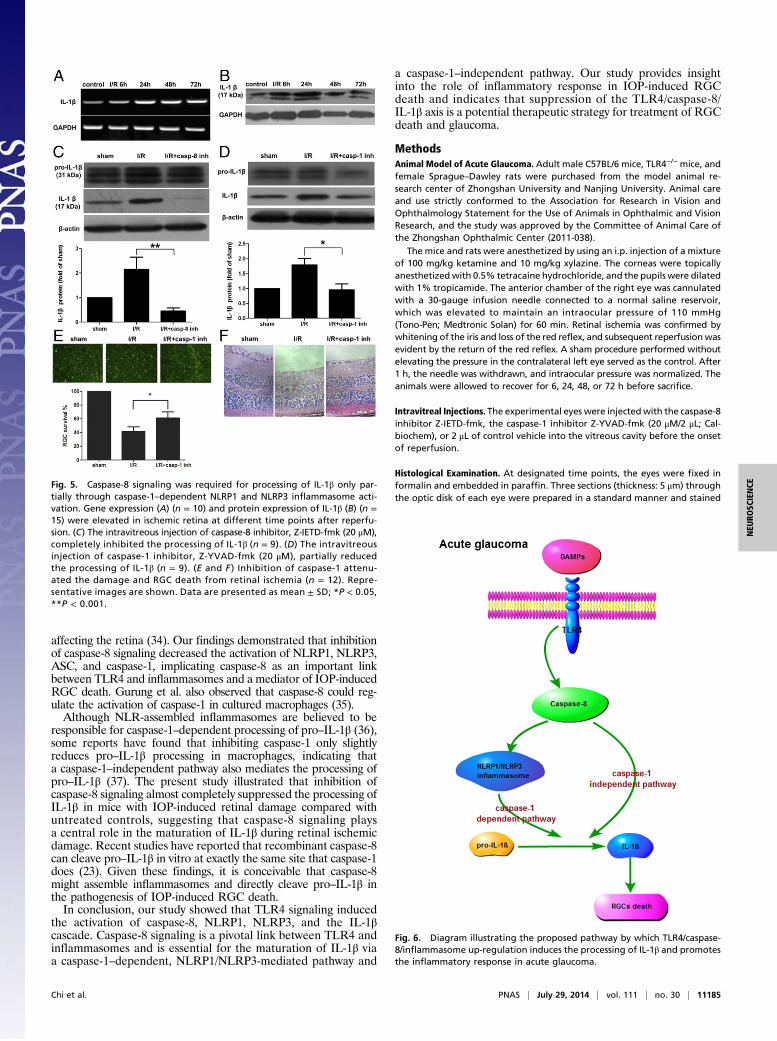
Caspase-8 Is Required for Processing of IL-1β in IOP-Induced RGC Death.Caspase-8 has been linked to IL-1β processing in response to li-popolysaccharide stimulation of TLR4 signaling (29, 30). The abovedata showed that TLR4-induced caspase-8 regulated NLRP1/NLRP3 inflammasome activation in IOP-induced RGC death. Wetherefore explored the potential role and underlying mechanism ofcaspase-8 in processing IL-1β after IOP-induced retinal damage.The expression of IL-1β was detected at different time points fol-lowing reperfusion. IL-1β was up-regulated in ischemic retinal tissueas rapidly as 6 h after IOP elevation and peaked after 48 h, as shownby mRNA and protein levels (Fig. 5 A and B). Accordingly, IL-1βprocessing was almost completely blocked following intravitreousinjection of caspase-8 inhibitor (Fig. 5C).NLR-mediated caspase-1 activation is considered important
for the processing of IL-1β in pathogenic stimulation (23, 31).The role of caspase-1–dependent NLRP1/NLRP3 inflamma-somes in the processing of IL-1β was further investigated byusing intravitreous injection of a caspase-1 inhibitor, z-YVAD-fmk. Caspase-1 inhibition only partially prevented IL-1β pro-cessing in mice with IOP-induced retinal injury (Fig. 5D), incontrast to complete inhibition of IL-1β production by caspase-8inhibition. Furthermore, inhibition of caspase-1 decreased theextent of retinal injury and RGC death in IOP-induced retinaldamage (Fig. 5 E and F), implying that caspase-1 exerts its effectin a retinal injury model via IL-1β production. Overall, theseresults indicate that TLR4-induced caspase-8 activation pro-motes the processing of IL-1β in retinal ischemic damage, notonly by the caspase-1–dependent NLRP1/NLRP3 pathway butalso by a caspase-1–independent pathway.Fig. 2. Increased production of caspase-8, induced by TLR4, contributed to
the pathogenesis of IOP-induced retinal damage. Caspase-8 gene (A and B)(n = 10) and protein expression (C) (n = 15) were significantly up-regulatedin ischemic retina compared with controls. Caspase-3 mRNA levels (D and E)(n = 10) and protein levels (F) (n = 15) were mildly increased in ischemicretina until 48 h after reperfusion. Inhibition of caspase-8 via Z-IETD-fmkinjection into the vitreous cavity significantly attenuated retinal ischemicdamage (G) (n = 9), RGC death (H) (n = 9), and microglia activation (I) (n = 9).By knocking out the TLR4 gene using TLR4−/− mice, the expression of
caspase-8 was reduced following IOP-induced injury (J and K) (n = 6) com-pared with WT mice (n = 6). Inhibition of caspase-8 had no effect on theexpression of TLR4 in ischemic retina (L) (n = 6). Representative images taken48 h after I/R injury are shown. Data are presented as mean ± SD; *P < 0.05,**P < 0.001.
Chi et al. PNAS | July 29, 2014 | vol. 111 | no. 30 | 11183
NEU
ROSC
IENCE
TLR4/Caspase-8/IL-1β Axis Is also Activated in a Rat Model of AcuteGlaucoma. We further validated the role of the TLR4/caspase-8/IL-1β axis in retinal ischemic injury by using a rat model of acuteglaucoma (Fig. 6). mRNA levels of TLR4, caspase-8, NLRP1,NLRP3, ASC, and IL-1β were significantly up-regulated in ratretinal tissue as early as 6 h after IOP elevation (Fig. S2 A, B, andD–G). In contrast, expression of caspase-3 was not significantlyincreased in the early stage of acute glaucoma, but was increased48 h after IOP elevation (Fig. S2C). Furthermore, we demon-strated that inhibition of caspase-8 in the vitreous reduced theactivation of NLRP1 and NLRP3 and completely suppressed theproduction of IL-1β in rat models of acute glaucoma (Fig. S3 A–C).
Inhibition of caspase-1 only partially suppressed the productionof IL-1β (Fig. S3D). These results are similar to those seen inmouse models. In summary, the TLR4/caspase-8/IL-1β axis isactivated in rats with IOP-induced retinal injury, and caspase-8 isnecessary for the production of IL-1β partially through the ac-tivation of NLRP1/NLRP3 inflammasomes.
DiscussionIn this study, we found that elevated IOP triggers increased ex-pression of TLR4, initiating increased caspase-8 and NLRP1/NLRP3 expression, and production of IL-1β, all of which par-ticipate in the innate immune response to IOP-induced retinalischemia. Consistent with this notion, knockout of TLR4 sig-naling significantly attenuated retinal ischemic damage and RGCdeath and suppressed the activation of caspase-8 signaling.Further support came from the inhibition of caspase-8, whichhad a therapeutic effect on IOP-induced retinal injury by down-regulating the expression of NLRP1 and NLRP3. We showedthat the inflammasome processes IL-1β via caspase-1 activation,but inhibition of caspase-1 only partially blocked IL-1β matura-tion. Thus, caspase-8 signaling promotes the processing of IL-1βby both the caspase-1–dependent NLRP1/NLRP3 pathway andthe caspase-1–independent pathway. The present study elucidatedthe role and mechanisms of TLR4 in the development of IOP-induced retinal damage. It remains to be determined whether theseresults are translatable to acute angle closure glaucoma in humans.Particularly at the lamina cribrosa, the site of putative pressure-induced damage in glaucoma (32), the anatomy of the optic nerveof the mouse is different from that of the human. Moreover, thehigh levels of induced IOP used in the current study may exceedthose measured in patients with acute angle closure. Further studyis needed to verify this.Inflammasomes are molecular platforms for caspase-1 activa-
tion and IL-1β maturation, and are engaged in the innate immuneresponses triggered by cellular infection or stress (17). Our find-ings demonstrated that up-regulated NLRP1 and NLRP3 wereassociated with elevated IOP and that elimination of TLR4 di-minishes the increase in inflammasome activity, indicating thatTLR4 regulates the activation of NLRP1 and NLRP3 during thedevelopment of IOP-induced RGC death.Caspase-8 is mainly considered an initiator of apoptotic cell
death that is activated by receptors of the TNF/NGF family (33).Recently, multiple nonapoptotic roles of caspase-8 have beenuncovered, especially its neuroinflammatory functions (22, 23,31). Microglia activation releases proinflammatory factors,which mediate the pathogenesis of CNS diseases, including those
Fig. 3. The production of NLRP1 and NLRP3 inflammasomes was increased inischemic retina, mediated by TLR4 signaling. NLRP1 and NLRP3 gene expression(A, B, D, and E) (n = 10, both) and protein expression (C and F) (n = 15, both) inischemic retina were significantly up-regulated compared with controls. ASCmRNA levels detected using sqRT-PCR (G) and qPCR (H) (n = 6, both) andprotein expression (I) (n = 15) were also up-regulated in IOP-induced retinalinjury. (J) The expression of caspase-1 was correspondingly elevated in is-chemic retinal tissue (n = 6). Knockdown of TLR4 reduced the production ofNLRP1 and NLRP3 inflammasomes and ASC (K–O) (n = 12, all). Representativeimages are shown. Data are presented as mean ± SD; *P < 0.05, **P < 0.001.
Fig. 4. Caspase-8 signaling regulated the activation of NLRP1 and NLRP3inflammasomes in IOP-induced retinal damage. The intravitreous injectionof the caspase-8 inhibitor Z-IETD-fmk (20 μM) (A) significantly attenuatedthe activation of NLRP3 (B), NLRP1 (C), ASC (D), and caspase-1 (E) (n = 9, all)following retinal ischemic injury. Representative images are shown.
11184 | www.pnas.org/cgi/doi/10.1073/pnas.1402819111 Chi et al.
affecting the retina (34). Our findings demonstrated that inhibitionof caspase-8 signaling decreased the activation of NLRP1, NLRP3,ASC, and caspase-1, implicating caspase-8 as an important linkbetween TLR4 and inflammasomes and a mediator of IOP-inducedRGC death. Gurung et al. also observed that caspase-8 could reg-ulate the activation of caspase-1 in cultured macrophages (35).Although NLR-assembled inflammasomes are believed to be
responsible for caspase-1–dependent processing of pro–IL-1β (36),some reports have found that inhibiting caspase-1 only slightlyreduces pro–IL-1β processing in macrophages, indicating thata caspase-1–independent pathway also mediates the processing ofpro–IL-1β (37). The present study illustrated that inhibition ofcaspase-8 signaling almost completely suppressed the processing ofIL-1β in mice with IOP-induced retinal damage compared withuntreated controls, suggesting that caspase-8 signaling playsa central role in the maturation of IL-1β during retinal ischemicdamage. Recent studies have reported that recombinant caspase-8can cleave pro–IL-1β in vitro at exactly the same site that caspase-1does (23). Given these findings, it is conceivable that caspase-8might assemble inflammasomes and directly cleave pro–IL-1β inthe pathogenesis of IOP-induced RGC death.In conclusion, our study showed that TLR4 signaling induced
the activation of caspase-8, NLRP1, NLRP3, and the IL-1βcascade. Caspase-8 signaling is a pivotal link between TLR4 andinflammasomes and is essential for the maturation of IL-1β viaa caspase-1–dependent, NLRP1/NLRP3-mediated pathway and
a caspase-1–independent pathway. Our study provides insightinto the role of inflammatory response in IOP-induced RGCdeath and indicates that suppression of the TLR4/caspase-8/IL-1β axis is a potential therapeutic strategy for treatment of RGCdeath and glaucoma.
MethodsAnimal Model of Acute Glaucoma. Adult male C57BL/6 mice, TLR4−/− mice, andfemale Sprague–Dawley rats were purchased from the model animal re-search center of Zhongshan University and Nanjing University. Animal careand use strictly conformed to the Association for Research in Vision andOphthalmology Statement for the Use of Animals in Ophthalmic and VisionResearch, and the study was approved by the Committee of Animal Care ofthe Zhongshan Ophthalmic Center (2011-038).
The mice and rats were anesthetized by using an i.p. injection of a mixtureof 100 mg/kg ketamine and 10 mg/kg xylazine. The corneas were topicallyanesthetizedwith 0.5% tetracaine hydrochloride, and the pupils were dilatedwith 1% tropicamide. The anterior chamber of the right eye was cannulatedwith a 30-gauge infusion needle connected to a normal saline reservoir,which was elevated to maintain an intraocular pressure of 110 mmHg(Tono-Pen; Medtronic Solan) for 60 min. Retinal ischemia was confirmed bywhitening of the iris and loss of the red reflex, and subsequent reperfusionwasevident by the return of the red reflex. A sham procedure performed withoutelevating the pressure in the contralateral left eye served as the control. After1 h, the needle was withdrawn, and intraocular pressure was normalized. Theanimals were allowed to recover for 6, 24, 48, or 72 h before sacrifice.
Intravitreal Injections. The experimental eyes were injectedwith the caspase-8inhibitor Z-IETD-fmk, the caspase-1 inhibitor Z-YVAD-fmk (20 μM/2 μL; Cal-biochem), or 2 μL of control vehicle into the vitreous cavity before the onsetof reperfusion.
Histological Examination. At designated time points, the eyes were fixed informalin and embedded in paraffin. Three sections (thickness: 5 μm) throughthe optic disk of each eye were prepared in a standard manner and stained
Fig. 5. Caspase-8 signaling was required for processing of IL-1β only par-tially through caspase-1–dependent NLRP1 and NLRP3 inflammasome acti-vation. Gene expression (A) (n = 10) and protein expression of IL-1β (B) (n =15) were elevated in ischemic retina at different time points after reperfu-sion. (C) The intravitreous injection of caspase-8 inhibitor, Z-IETD-fmk (20 μM),completely inhibited the processing of IL-1β (n = 9). (D) The intravitreousinjection of caspase-1 inhibitor, Z-YVAD-fmk (20 μM), partially reducedthe processing of IL-1β (n = 9). (E and F ) Inhibition of caspase-1 attenu-ated the damage and RGC death from retinal ischemia (n = 12). Repre-sentative images are shown. Data are presented as mean ± SD; *P < 0.05,**P < 0.001.
Fig. 6. Diagram illustrating the proposed pathway by which TLR4/caspase-8/inflammasome up-regulation induces the processing of IL-1β and promotesthe inflammatory response in acute glaucoma.
Chi et al. PNAS | July 29, 2014 | vol. 111 | no. 30 | 11185
NEU
ROSC
IENCE

with H&E. Morphometric analysis was performed to quantify ischemic injury.The total retinal thickness (from the internal to the outer limiting mem-brane) was measured in four adjacent areas of the inferior retina within1 mm of the optic nerve by using Axiovision software (Carl Zeiss), and themean value was calculated.
RGC Labeling and Quantification. Mice were anesthetized with a mixture of100 mg/kg ketamine and 10 mg/kg xylazine by i.p. injection and placed ina stereotactic apparatus (Stoelting). FG [1 μL per injection of 4% (wt/vol);Invitrogen] diluted in saline was injected into both superior colliculi. Toensure proper RGC labeling, the animals were allowed 7 d for retrogradetransport of FG before sacrifice. At 24, 48, and 72 h after reperfusion, FG-positive RGCs were identified with a fluorescent microscope (AxioImager;Carl Zeiss) after retinal flat mount preparation. Surviving RGCs (gold dots)were counted by using Image Pro Plus (Version 6.0; Media Cybernetics).
Immunofluorescence and Evaluation of Microglia Activation. Mice were per-fused by 4% (wt/vol) paraformaldehyde transcardially through the ascendingaorta after being anesthetized, and their eyes were enucleated and bisectedequatorially. The posterior halves were immersed in fixative (4% para-formaldehyde) for 2 h and infiltrated with increasing concentrations of su-crose. Tissue was then embedded in optimum cutting temperature (OCT)medium, frozen, and sectioned to a thickness of 6 μm. The presence ofionized calcium binding adaptor molecule 1 (Iba1) was detected to evaluatemicroglia activation by using polyclonal rabbit anti-Iba1 (1:300; WakoChemicals). Antibody binding was visualized by incubation with Alexa Fluor555 donkey anti-rabbit IgG (1:500; Invitrogen). The nuclei of cells in theretina were stained with DAPI (Sigma). Images were obtained by usinga Zeiss LSM 710 META confocal laser scanning microscope (Carl Zeiss).
sqRT-PCR and qPCR. Total RNA was isolated from the retina by using TRIzolreagent (Invitrogen), and cDNA was synthesized with PrimeScript RT MasterMix (TaKaRa). The mouse and rat primers and annealing temperatures arelisted in Tables S1 and S2. SqRT-PCR was carried out by using Premix EX Taq
Kit (TaKaRa) according to the standard protocol. The reaction was per-formed with 25 cycles for GAPDH and 30–32 cycles for the other transcripts.PCR products were run on 2% agarose gel, and semiquantification of geneexpression was determined by relative pixel densitometry using ImageJsoftware after normalization to GAPDH. Amplification and qPCR measure-ments were performed by using the Light Cycler 480 Real-Time PCR System,with version LCS480 1.5.1.62 software. Quantitative analysis was performedby real-time qPCR using SYBR Advantage qPCR Premix Master Mix (TaKaRa)following the standard protocol. The amount of target mRNA in test sam-ples was measured and normalized to GAPDH. The comparative Cp methodwas used to evaluate expression.
Western Blot Analysis. For analysis of protein expression, retinal tissue washarvested at 6, 24, 48, and 72 h after temporary elevation of IOP. Total proteinwas extracted and processed for Western blot. The membranes were blockedwith 5% (wt/vol) BSA in Tween 20/PBS and incubated at 4 °C overnight inprimary antibodies, which were diluted in blocking solution. Primary antibodiesconsisted of TLR4 (1:500; Abgent), cleaved caspase-8 (1:500; Cell SignalingTechnology), cleaved caspase-3 (1:1,000; Cell Signaling Technology), NLRP1(1:500; Aviva), NLRP3 (1:1,000; Novus), ASC (1:100; Millipore), IL-1β (1:500; CellSignaling Technology), β-actin (1:3,000; Multi Sciences Biotech), and GAPDH(1:1,000; Cell Signaling Technology). Relative changes in protein expressionwere calculated in relation to nonischemic retinas and expressed as the “x-fold change”.
Statistical Analysis. Data were presented as mean ± SD. One-way analysis ofvariance (ANOVA) was performed, followed by the Bonferroni multiplecomparison tests and two-way ANOVA using GraphPad Prism data analysissoftware (version 5.0; GraphPad Software). A P value less than 0.05 wasconsidered statistically significant.
ACKNOWLEDGMENTS. We thank Dr. Changyou Wu and Dr. Qiang Liu fordata analysis. This study was supported in part by National Natural ScienceFoundation of China Grant 81270992 and 973 Program Grant 2013CB967504.
1. Weinreb RN, Aung T, Medeiros FA (2014) The pathophysiology and treatment of
glaucoma: A review. JAMA 311(18):1901–1911.2. Wang N, Wu H, Fan Z (2002) Primary angle closure glaucoma in Chinese and Western
populations. Chin Med J (Engl) 115(11):1706–1715.3. Ang LP, Ang LP (2008) Current understanding of the treatment and outcome of acute
primary angle-closure glaucoma: An Asian perspective. Ann Acad Med Singapore
37(3):210–215.4. Quek DT, et al. (2011) Blindness and long-term progression of visual field defects in chinese
patients with primary angle-closure glaucoma. Am J Ophthalmol 152(3):463–469.5. Ousman SS, Kubes P (2012) Immune surveillance in the central nervous system. Nat
Neurosci 15(8):1096–1101.6. Zhang C, Lam TT, Tso MO (2005) Heterogeneous populations of microglia/macro-
phages in the retina and their activation after retinal ischemia and reperfusion injury.
Exp Eye Res 81(6):700–709.7. Yamasaki Y, et al. (1995) Interleukin-1 as a pathogenetic mediator of ischemic brain
damage in rats. Stroke 26(4):676–680, discussion 681.8. Hanamsagar R, Hanke ML, Kielian T (2012) Toll-like receptor (TLR) and inflammasome
actions in the central nervous system. Trends Immunol 33(7):333–342.9. Lehnardt S (2010) Innate immunity and neuroinflammation in the CNS: The role of
microglia in Toll-like receptor-mediated neuronal injury. Glia 58(3):253–263.10. Saijo K, Crotti A, Glass CK (2013) Regulation of microglia activation and deactivation
by nuclear receptors. Glia 61(1):104–111.11. Dvoriantchikova G, Barakat DJ, Hernandez E, Shestopalov VI, Ivanov D (2010) Toll-
like receptor 4 contributes to retinal ischemia/reperfusion injury. Mol Vis 16:
1907–1912.12. Marsh BJ, Williams-Karnesky RL, Stenzel-Poore MP (2009) Toll-like receptor signaling
in endogenous neuroprotection and stroke. Neuroscience 158(3):1007–1020.13. Crack PJ, Bray PJ (2007) Toll-like receptors in the brain and their potential roles in
neuropathology. Immunol Cell Biol 85(6):476–480.14. Tang SC, et al. (2007) Pivotal role for neuronal Toll-like receptors in ischemic brain
injury and functional deficits. Proc Natl Acad Sci USA 104(34):13798–13803.15. Hyakkoku K, et al. (2010) Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out
mice have neuroprotective effects against focal cerebral ischemia. Neuroscience
171(1):258–267.16. Netea MG, van de Veerdonk FL, Kullberg BJ, Van der Meer JW, Joosten LA (2008) The
role of NLRs and TLRs in the activation of the inflammasome. Expert Opin Biol Ther
8(12):1867–1872.17. Schroder K, Tschopp J (2010) The inflammasomes. Cell 140(6):821–832.18. Abulafia DP, et al. (2009) Inhibition of the inflammasome complex reduces the in-
flammatory response after thromboembolic stroke in mice. J Cereb Blood Flow
Metab 29(3):534–544.
19. de Rivero Vaccari JP, et al. (2009) Therapeutic neutralization of the NLRP1 in-flammasome reduces the innate immune response and improves histopathology aftertraumatic brain injury. J Cereb Blood Flow Metab 29(7):1251–1261.
20. Crawford ED, Wells JA (2011) Caspase substrates and cellular remodeling. Annu RevBiochem 80:1055–1087.
21. Crowder RN, El-Deiry WS (2012) Caspase-8 regulation of TRAIL-mediated cell death.Exp Oncol 34(3):160–164.
22. Burguillos MA, et al. (2011) Caspase signalling controls microglia activation andneurotoxicity. Nature 472(7343):319–324.
23. Maelfait J, Beyaert R (2008) Non-apoptotic functions of caspase-8. Biochem Phar-macol 76(11):1365–1373.
24. Cao CX, et al. (2007) Reduced cerebral ischemia-reperfusion injury in Toll-like receptor4 deficient mice. Biochem Biophys Res Commun 353(2):509–514.
25. Hamanaka J, Hara H (2011) Involvement of Toll-like receptors in ischemia-inducedneuronal damage. Cent Nerv Syst Agents Med Chem 11(2):107–113.
26. Chakraborty S, Kaushik DK, Gupta M, Basu A (2010) Inflammasome signaling at theheart of central nervous system pathology. J Neurosci Res 88(8):1615–1631.
27. Savage CD, Lopez-Castejon G, Denes A, Brough D (2012) NLRP3-inflammasome acti-vating DAMPs stimulate an inflammatory response in glia in the absence of primingwhich contributes to brain inflammation after injury. Front Immunol 3:288.
28. Yoneda S, et al. (2001) Interleukin-1beta mediates ischemic injury in the rat retina.Exp Eye Res 73(5):661–667.
29. Gringhuis SI, et al. (2012) Dectin-1 is an extracellular pathogen sensor for the in-duction and processing of IL-1β via a noncanonical caspase-8 inflammasome. NatImmunol 13(3):246–254.
30. Maelfait J, et al. (2008) Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J Exp Med 205(9):1967–1973.
31. Lemmers B, et al. (2007) Essential role for caspase-8 in Toll-like receptors andNFkappaB signaling. J Biol Chem 282(10):7416–7423.
32. Minckler DS, Bunt AH, Johanson GW (1977) Orthograde and retrograde axoplasmictransport during acute ocular hypertension in the monkey. Invest Ophthalmol Vis Sci16(5):426–441.
33. Wallach D, et al. (2002) How are the regulators regulated? The search for mechanismsthat impose specificity on induction of cell death and NF-kappaB activation bymembers of the TNF/NGF receptor family. Arthritis Res 4(Suppl 3):S189–S196.
34. Hanisch UK, Kettenmann H (2007) Microglia: Active sensor and versatile effector cellsin the normal and pathologic brain. Nat Neurosci 10(11):1387–1394.
35. Gurung P, et al. (2014) FADD and caspase-8 mediate priming and activation of thecanonical and noncanonical Nlrp3 inflammasomes. J Immunol 192(4):1835–1846.
36. Kanneganti TD, et al. (2006) Critical role for Cryopyrin/Nalp3 in activation of caspase-1 inresponse to viral infection and double-stranded RNA. J Biol Chem 281(48):36560–36568.
37. Guma M, et al. (2009) Caspase 1-independent activation of interleukin-1beta inneutrophil-predominant inflammation. Arthritis Rheum 60(12):3642–3650.
11186 | www.pnas.org/cgi/doi/10.1073/pnas.1402819111 Chi et al.