
Case 14 2006 - Correlación Clínico-Patológica - Desarrollo
69
ΒETA-TALASEMIA Y ANEMIA DE DIAMOND- BLACKFAN The New England Journal of Medicine CASE 14-2006: A 25-YEAR-OLD WOMAN WITH ANEMIA AND IRON OVERLOAD CASE RECORDS of the MASSACHUSETTS GENERAL HOSPITAL
-
Upload
daniel-marcelo -
Category
Health & Medicine
-
view
81 -
download
2
Transcript of Case 14 2006 - Correlación Clínico-Patológica - Desarrollo

ΒETA-TALASEMIA Y
ANEMIA DE DIAMOND-
BLACKFAN
The New England Journal of Medicine
CASE 14-2006: A 25-YEAR-OLD WOMAN WITH ANEMIA AND IRON
OVERLOAD
CASE RECORDS of the MASSACHUSETTS GENERAL HOSPITAL

PRESENTACION DEL CASO
Una mujer de
25 años de
edad con
anemia y
reservas de
hierro
aumentadas.
METODO DE
WEED

DATOS DE LA PACIENTE
Paciente Femenina 25 años de edad
Raza: Caucásica
Procedencia: Sicilia, Italia
Consulta Por
Anemia y evidencias de laboratorio que demostraban
un incremento en las reservas de hierro.
Presenta Dolor en CII del abdomen
Litiasis ureteral izquierda
Anemia normocítica, normocrómica
Trombosis de vena ovárica izquierda
Ingesta de anticonceptivos (4 meses)
Base de datos
ANAMNESIS
PATOLOGICOS
ANTECEDENTES
MEDICOS
Meningitis neumococcica y onfalitis a los 2
meses de edad.
Desarrollo ascitis durante el proceso de la
enfermedad.
Sangrado digestivo alto y signos de
hipertensión portal secundarios a una
trombosis de la vena porta a los 2 años de

Base de datos
ANAMNESIS
PATOLOGICOS
ANTECEDENTES
QUIRURGICOS
Derivación esplenorrenal y posteriormente
una esplenectomía a los 5 años de edad.
OTROS: Recibió varias transfusiones de
sangre entre los 3 y 5 años de edad.

Base de datos
ANAMNESIS
FAMILIARES
ANTECEDENTES
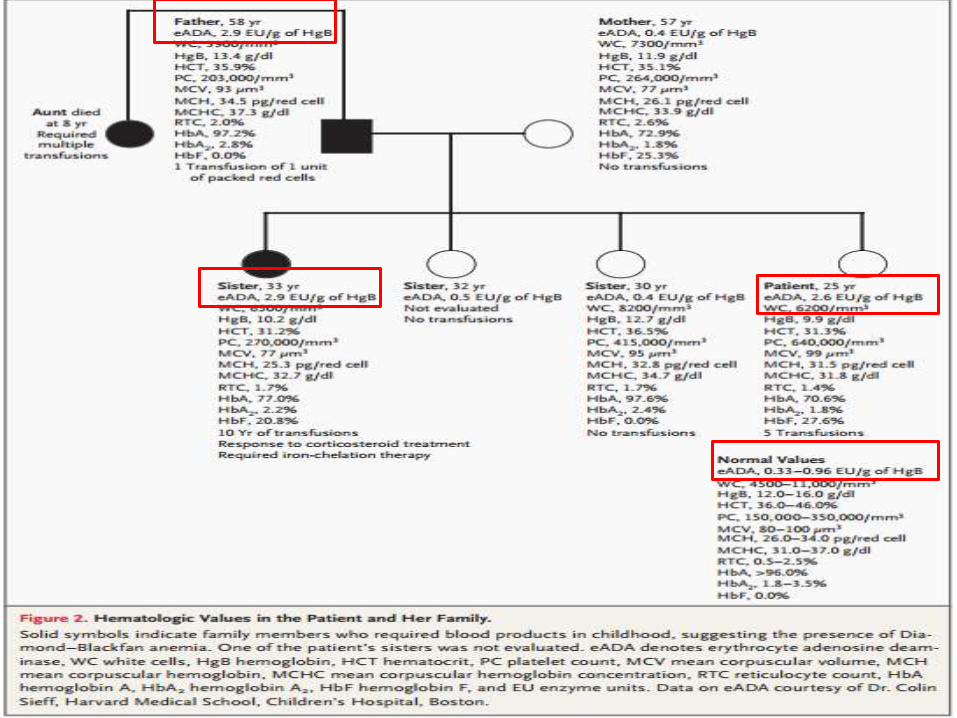
Abuela PATERNA: Anemia de Diamond-Blackfan
PADRE: Anemia de Diamond-Blackfan
MADRE: Anemia desde la infancia.
Portadora de rasgo Beta-Talasémico.
TIA: Anemia Crónica desde la infancia
Base de datos
ANAMNESIS
FAMILIARES
ANTECEDENTES
HERMANOS:
1. Femenino. Anemia Crónica desde los 3
años. Diamond-Blackfan. Fallecida.
2. Femenino. 33 años. Tx. Con quelantes de
hierro. Anemia remitida a los 13 años.
3. y 4. Sin historia de Anemia

Base de datos
ANAMNESIS REVISION POR
SISTEMAS
Síntomas generales: debilidad generalizada
Ojos: dolor y sensación de decaimiento.
Cardiovascular: Palpitaciones, cansancio, ortopnea.
Respiratorio: disnea
EXAMEN FISICO
Edad aparente concuerda
con edad cronológica
Mal estado general
COTEP

Base de datos
ANAMNESIS SIGNOS VITALES
Peso: 115 lb
Talla: 1.60 cm
T: 37.7 °C
P/A: 110/65 mmHg
F. C.: 100 latidos por
minuto
INSPECCION GENERAL
Piel y fanéras
Aparato respiratorio
Corazón

Base de datos
ANAMNESIS EVOLUCION
Aspirado-biopsia de médula ósea le fue realizada al cuarto día de estancia.
Alta otorgada
Anticogulante
Suspensión de anticonceptivos
Eliminar tabaquismo
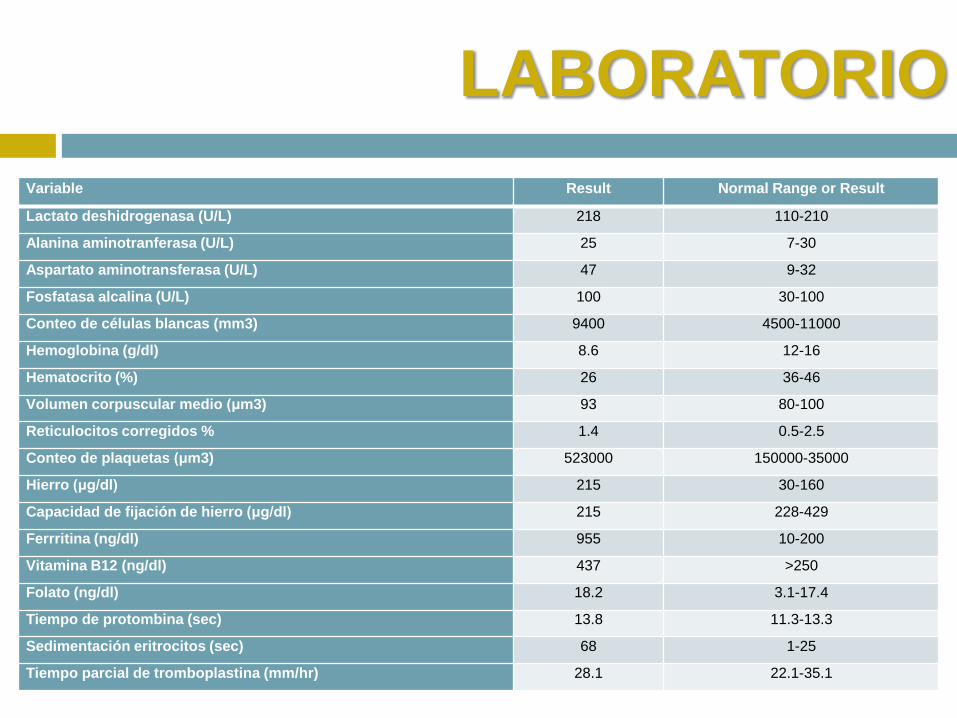
LABORATORIO
Variable Result Normal Range or Result
Lactato deshidrogenasa (U/L) 218 110-210
Alanina aminotranferasa (U/L) 25 7-30
Aspartato aminotransferasa (U/L) 47 9-32
Fosfatasa alcalina (U/L) 100 30-100
Conteo de células blancas (mm3) 9400 4500-11000
Hemoglobina (g/dl) 8.6 12-16
Hematocrito (%) 26 36-46
Volumen corpuscular medio (μm3) 93 80-100
Reticulocitos corregidos % 1.4 0.5-2.5
Conteo de plaquetas (μm3) 523000 150000-35000
Hierro (μg/dl) 215 30-160
Capacidad de fijación de hierro (μg/dl) 215 228-429
Ferrritina (ng/dl) 955 10-200
Vitamina B12 (ng/dl) 437 >250
Folato (ng/dl) 18.2 3.1-17.4
Tiempo de protombina (sec) 13.8 11.3-13.3
Sedimentación eritrocitos (sec) 68 1-25
Tiempo parcial de tromboplastina (mm/hr) 28.1 22.1-35.1

DESARROLLO DE PROBLEMAS
11.05.2006
Eyal Attar, MD.
Hassejian, MD.
22:30 hrs.
No. 1 Ferremia alta
No. 2 Esplenectomía
No. 3 Predisposición genética a
Anemia y beta-talasemia
No. 4 Tabaquismo

PROBLEMA NO. 1
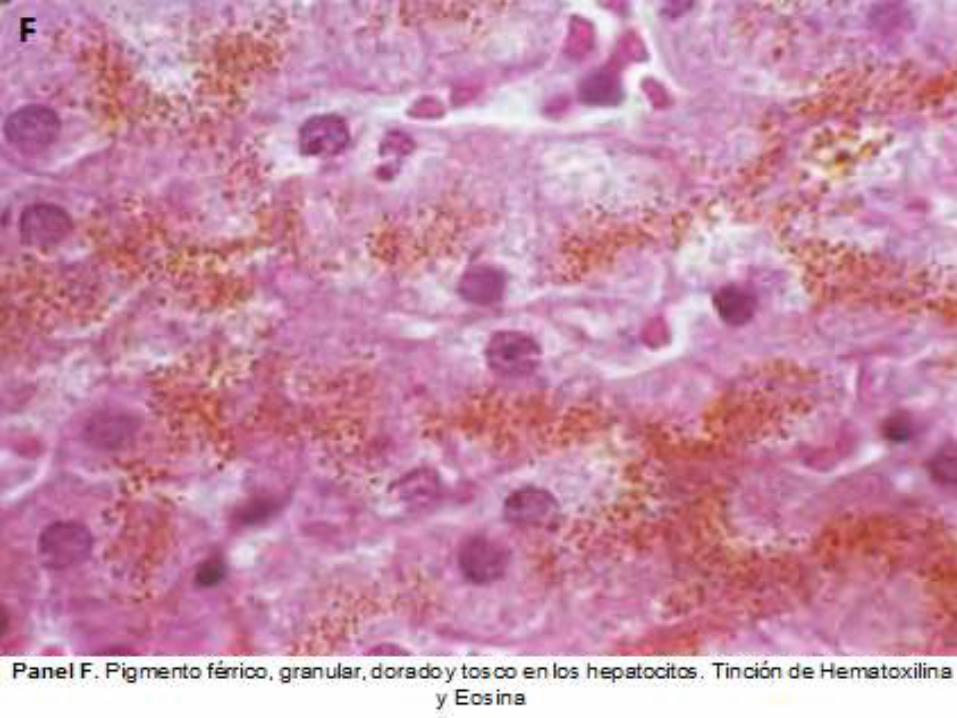
FERREMIA ALTA
Datos Objetivos
Ferritina: 955 ng/dl Normal: 10-200
Hemoglobina: 8.6 g/dl Normal: 12-16
(Saturación de
transferrina)
Datos Subjetivos
Historia familiar de
Anemia a lo largo de
varias generaciones
DESARROLLO DE PROBLEMAS

PROBLEMA NO. 2
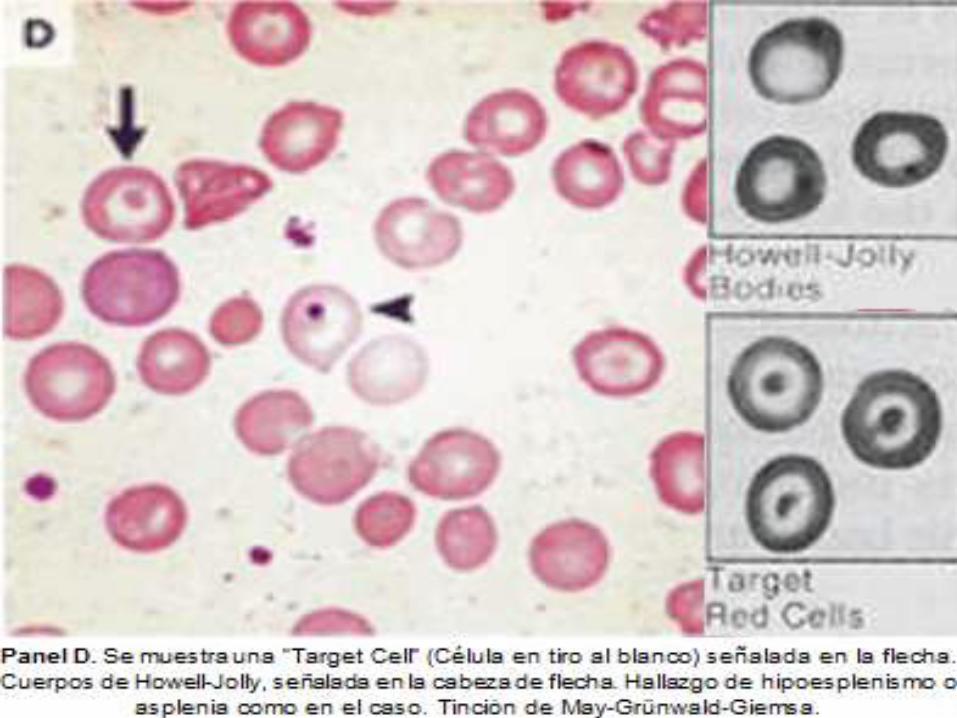
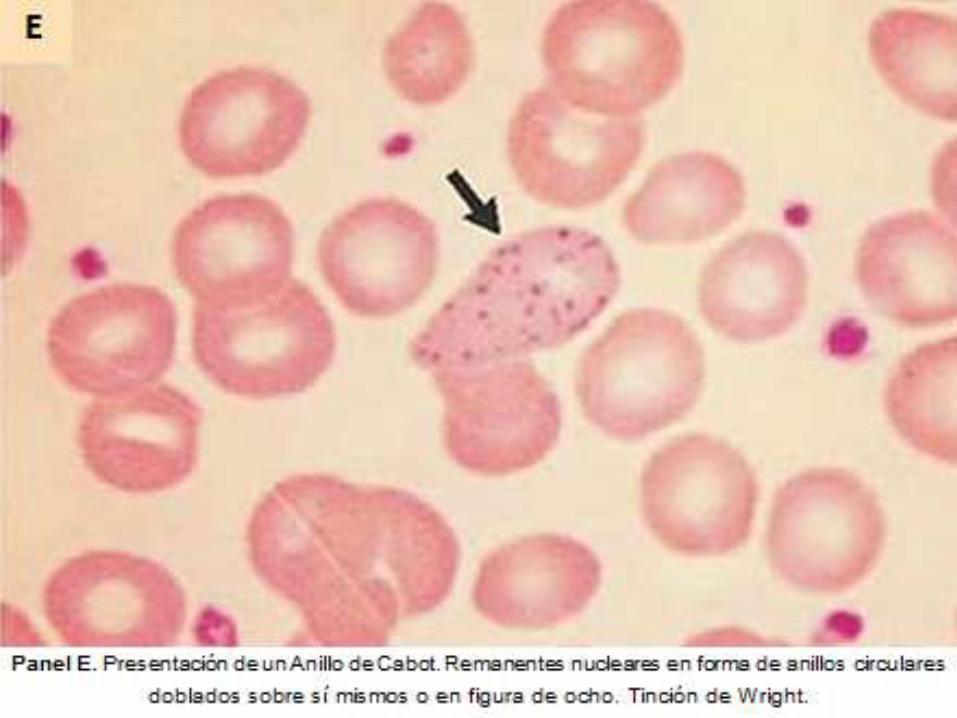
ASPLENIA
Datos Objetivos
Hallazgos en frotis
periférico de: Células
en tiro al blanco,
Cuerpos de Howell-
Jolly, Anillos de
Cabot y Cuerpos de
Pappenheimer
Datos Subjetivos
Antecedente de
esplenectomía de 20
años de evolución
DESARROLLO DE PROBLEMAS
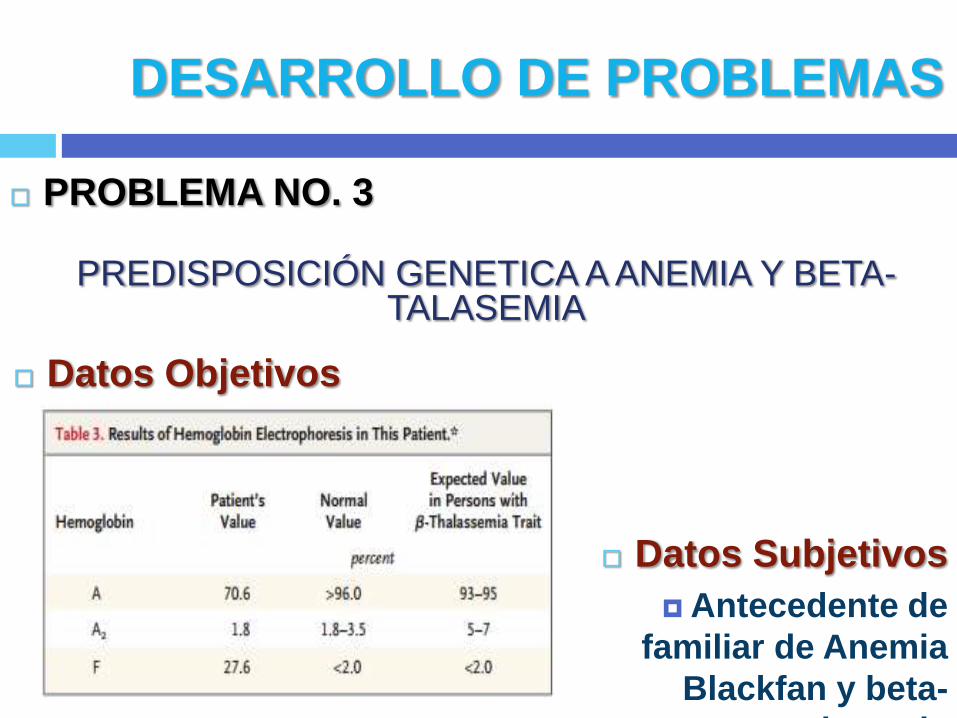
PROBLEMA NO. 3
PREDISPOSICIÓN GENETICA A ANEMIA Y BETA-TALASEMIA
Datos Objetivos
Datos Subjetivos
Antecedente de
familiar de Anemia
Blackfan y beta-
talasemia
DESARROLLO DE PROBLEMAS

PROBLEMA NO. 4
TABAQUISMO
Datos Objetivos
TAC. Trombosis
Venosa Profunda.
Estado
Hipercoagulable
secundario al
tabaquismo
Datos Subjetivos
Antecedente de
tabaquismo (5-10
cigarrillos) de varios
años de evolución.
DESARROLLO DE PROBLEMAS

ANALISIS
Anemia con déficit de producción de hematíes
Anemia por destrucción de glóbulos rojos
Anemia por pérdida de sangre

ANALISIS

PLAN INICIAL
PLAN
DIAGNOSTICO
DIAGNOSTICO
DIFERENCIAL Eritropoyesis inefectiva por o
talasemia
Púrpura trombocitopénica
Sangrado genitourinario
PLAN TERAPEUTICO
ACTIVIDAD
DIETA
CONTROLES ESPECIFICOS
Hemograma y electroforesis de hemoglobinas
MEDICAMENTOS
OTRAS TERAPIAS

PLAN EDUCACIONAL
Se le explica a la paciente la importancia de
continuar el monitoreo de sus análisis de
sangre así como estudio electroforéticos
constantes.
Se le explica también la importancia de
mantener adecuados estilos de vida
saludable para evitar complicaciones
consecuentes a la enfermedad.
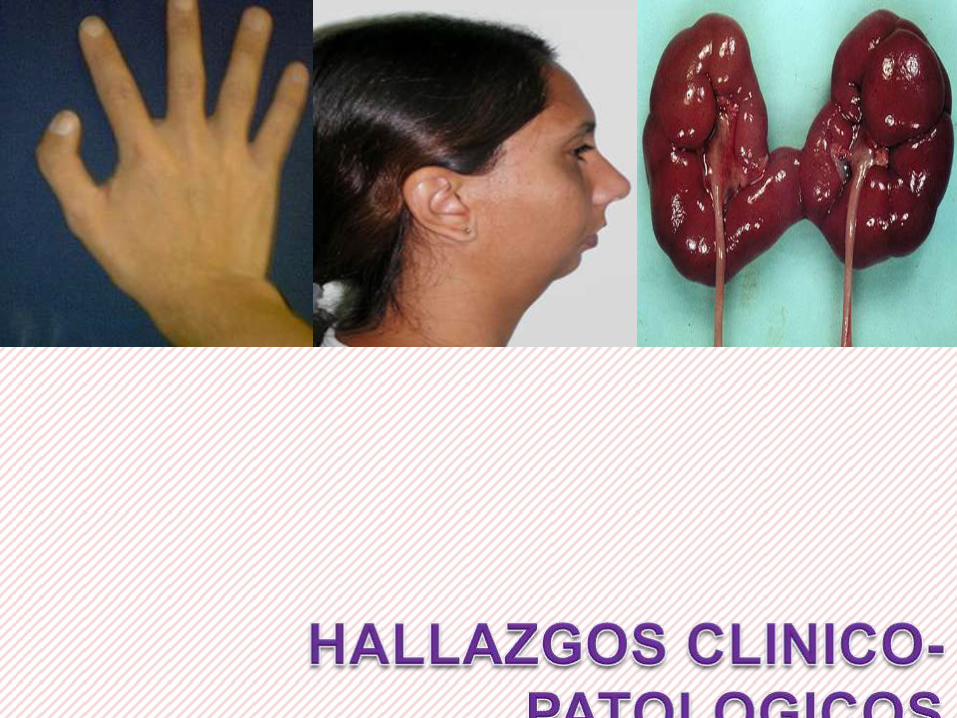

PALIDEZ, LETARGIA, IRRITABILIDAD EN
GENERAL
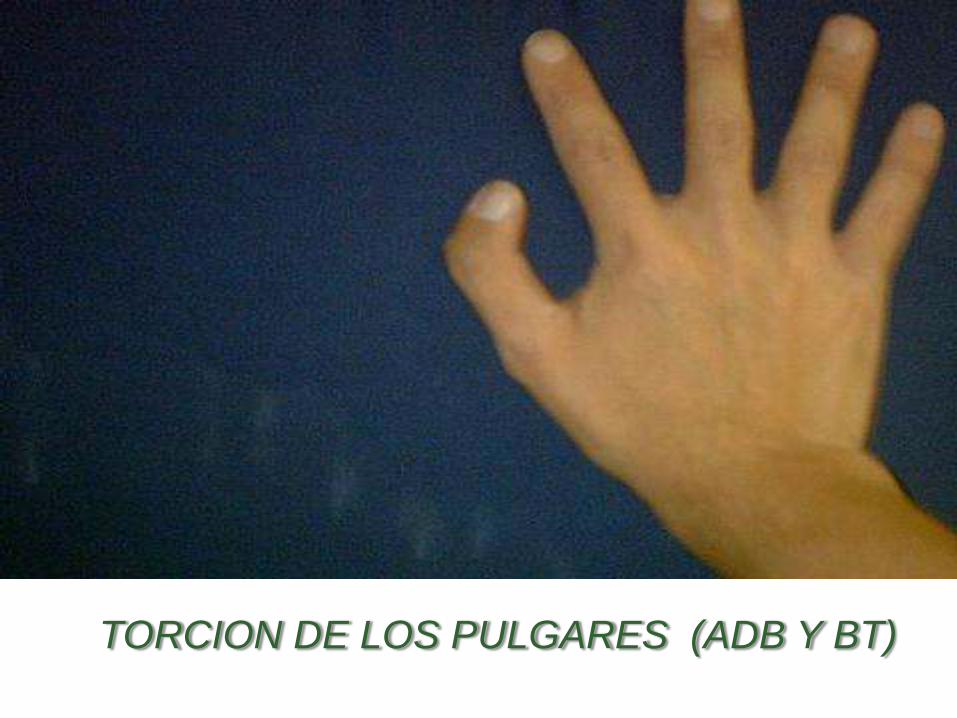
TORCION DE LOS PULGARES (ADB Y BT)

ANOMALIAS CRANEOFACIALES –
MICROGNATIA (BT)
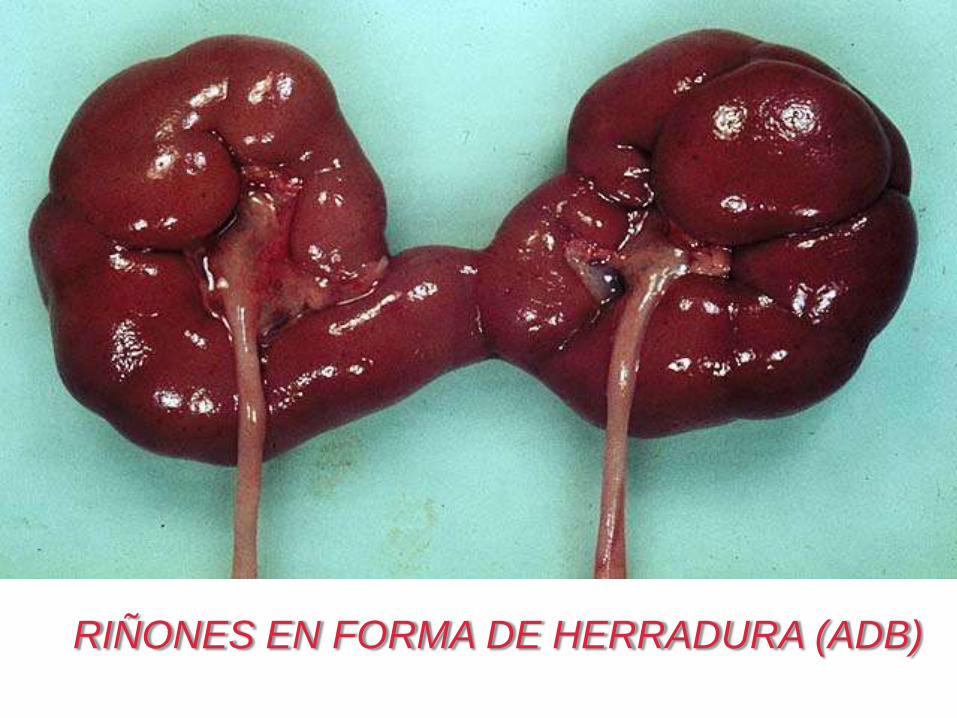
RIÑONES EN FORMA DE HERRADURA (ADB)
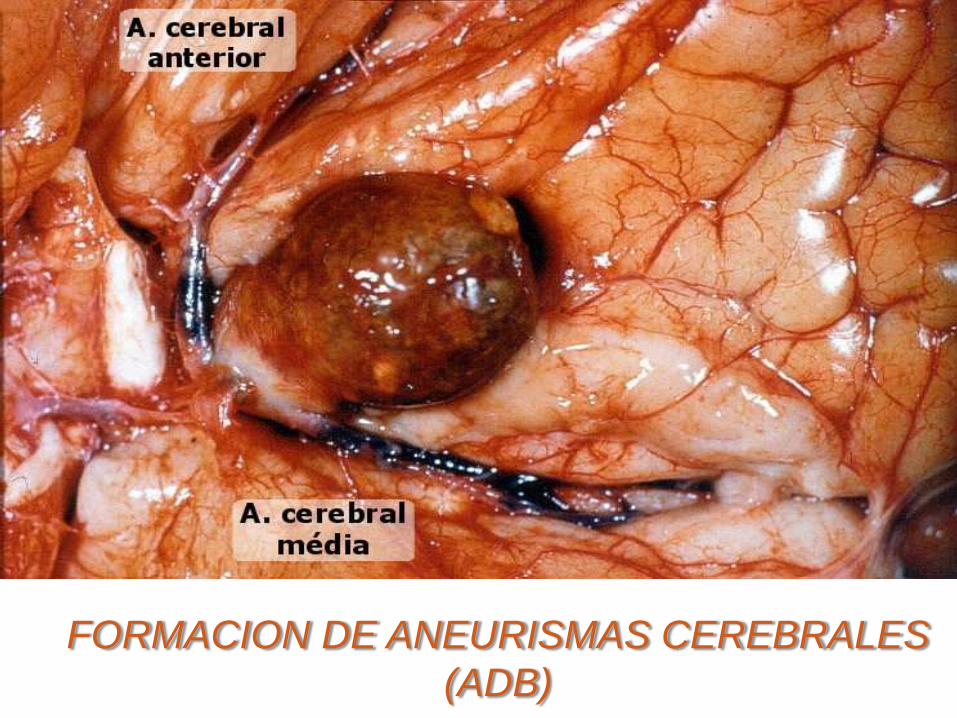
FORMACION DE ANEURISMAS CEREBRALES
(ADB)
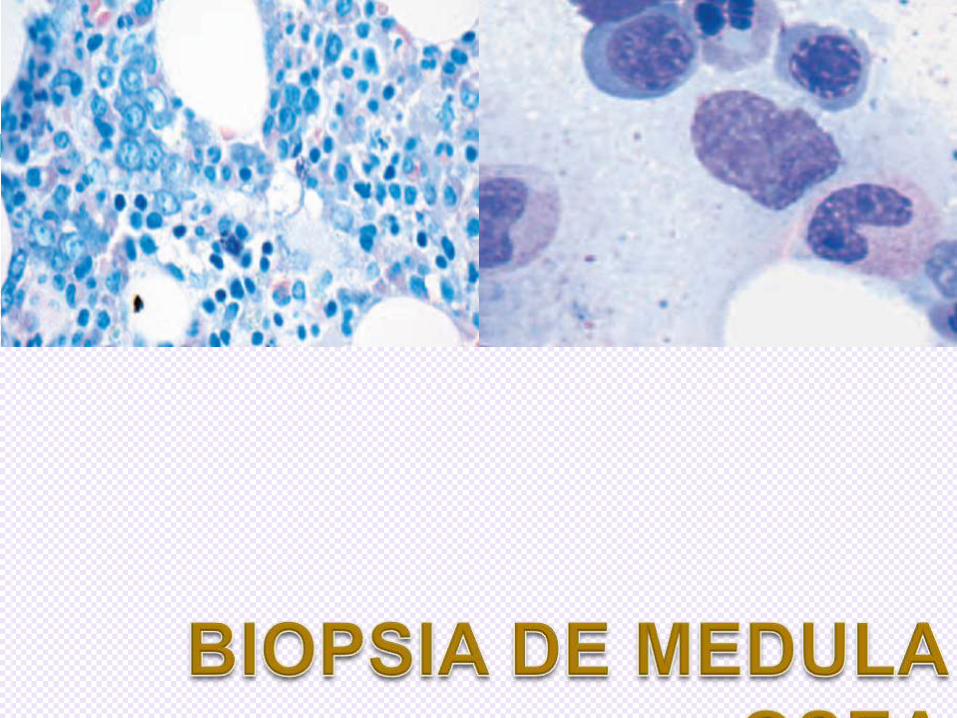
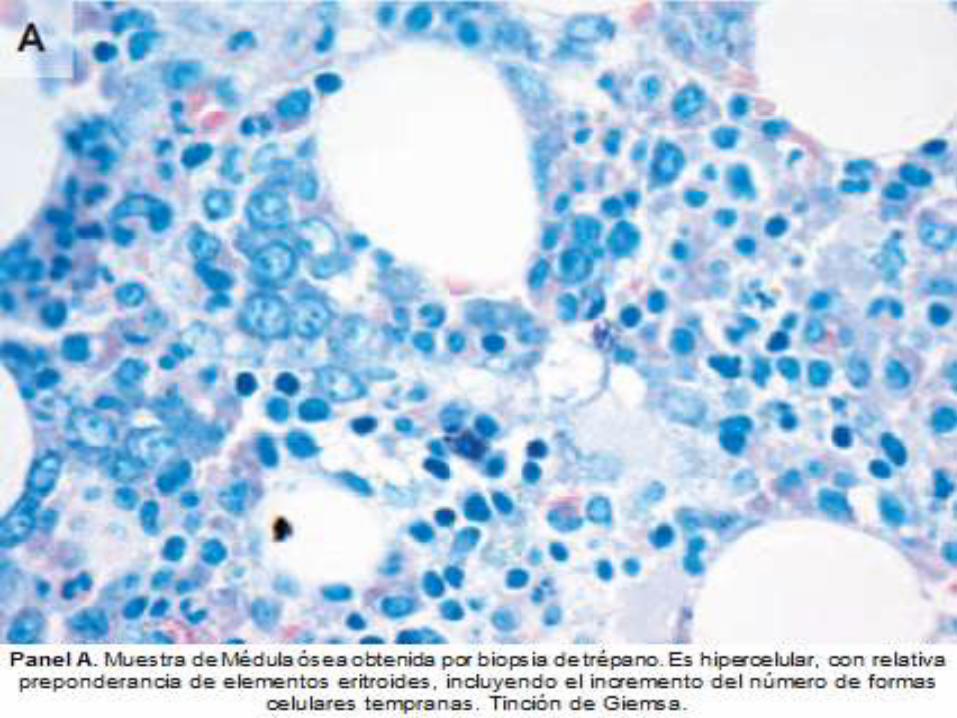
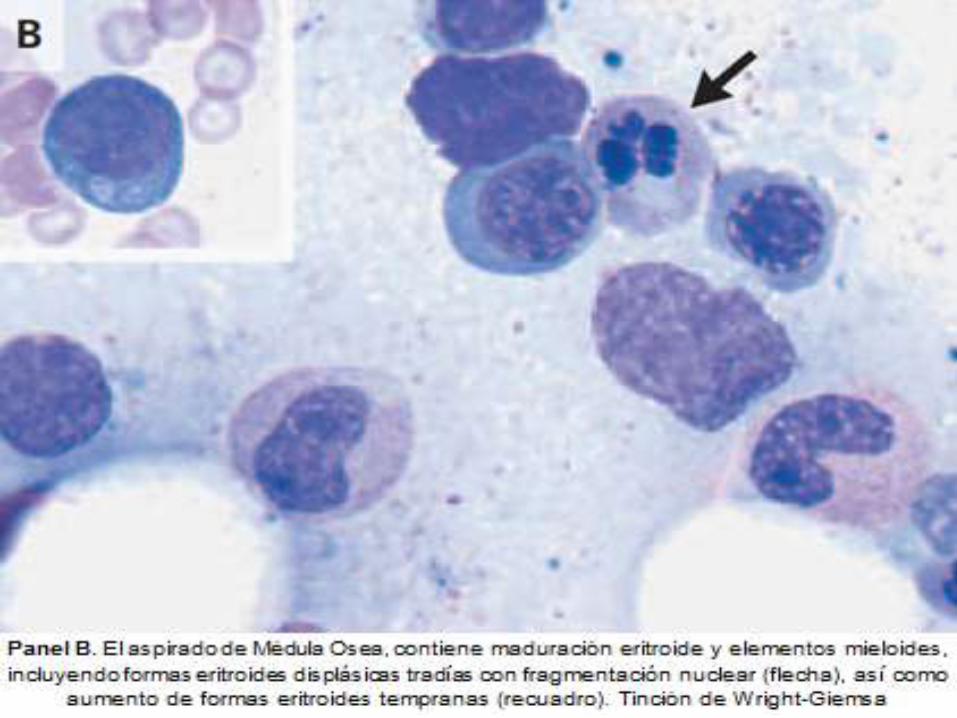
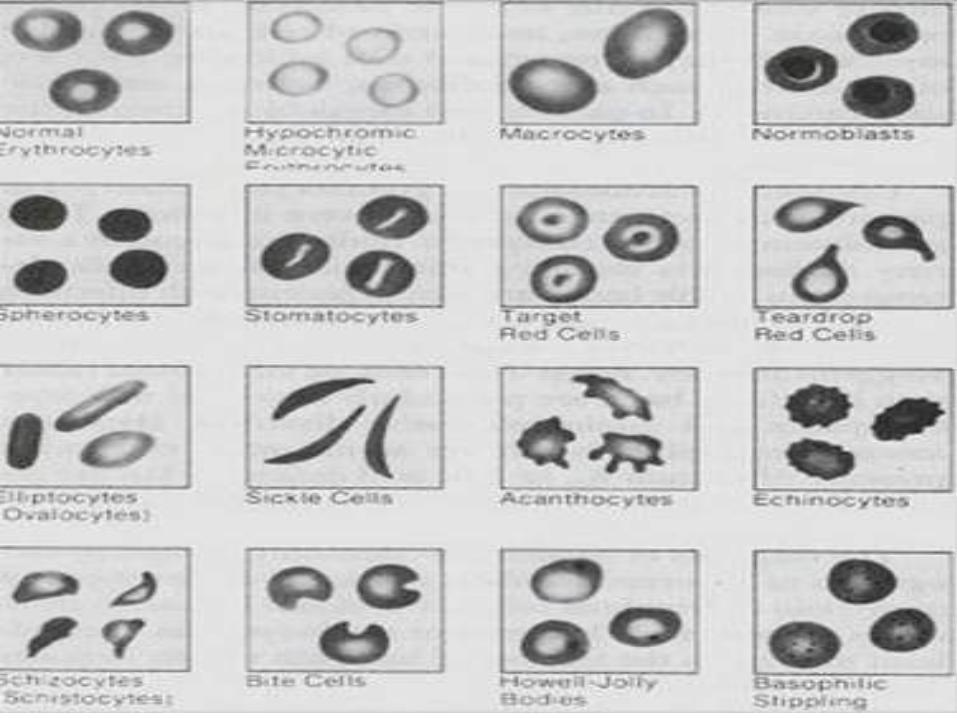
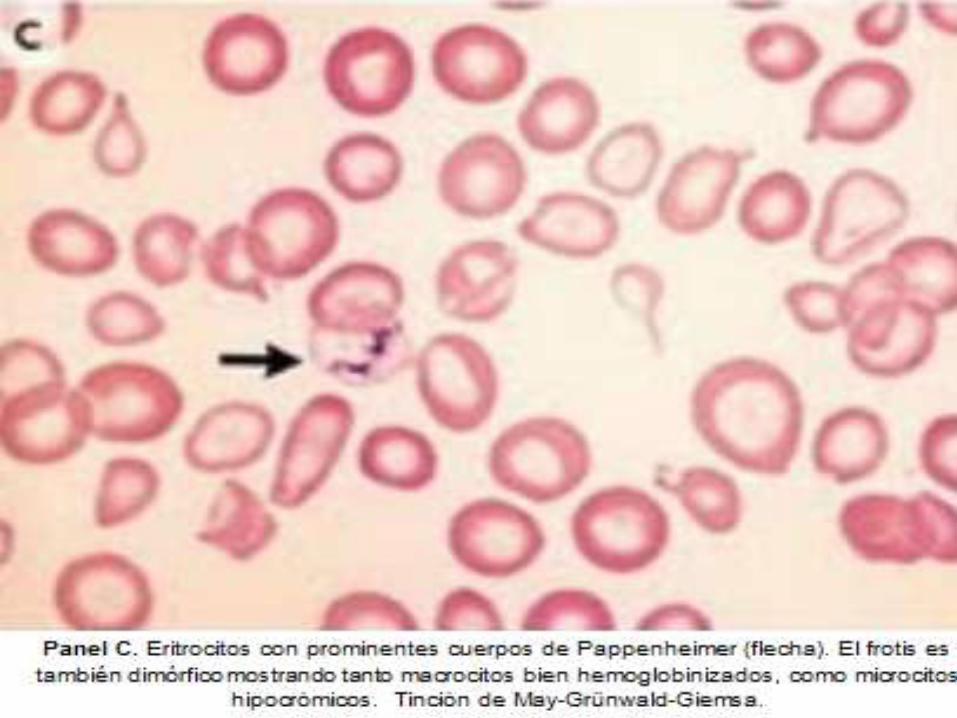
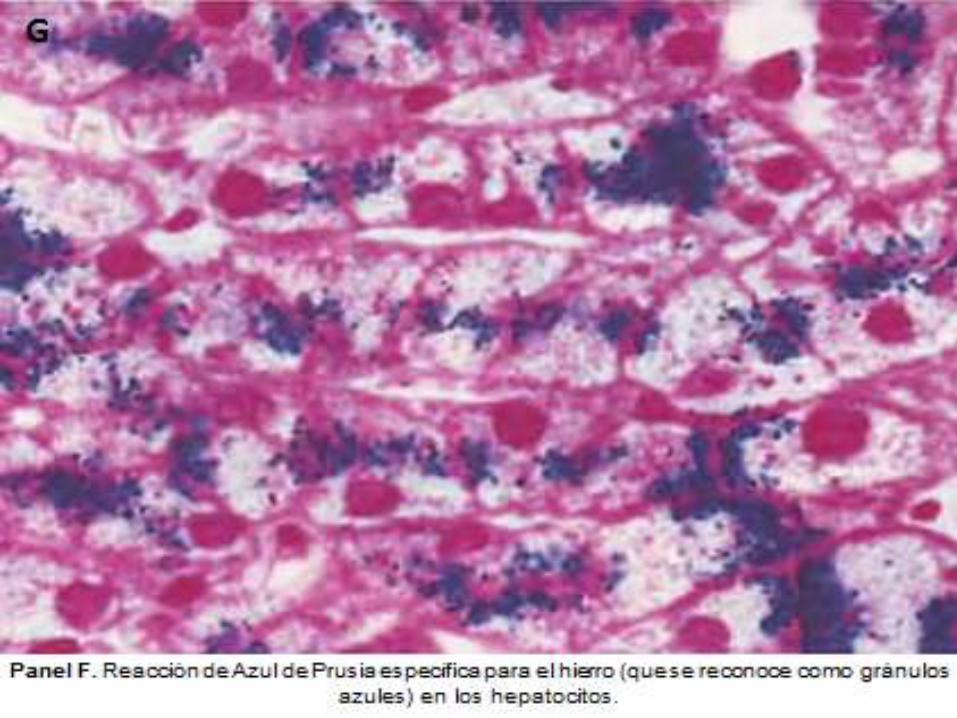

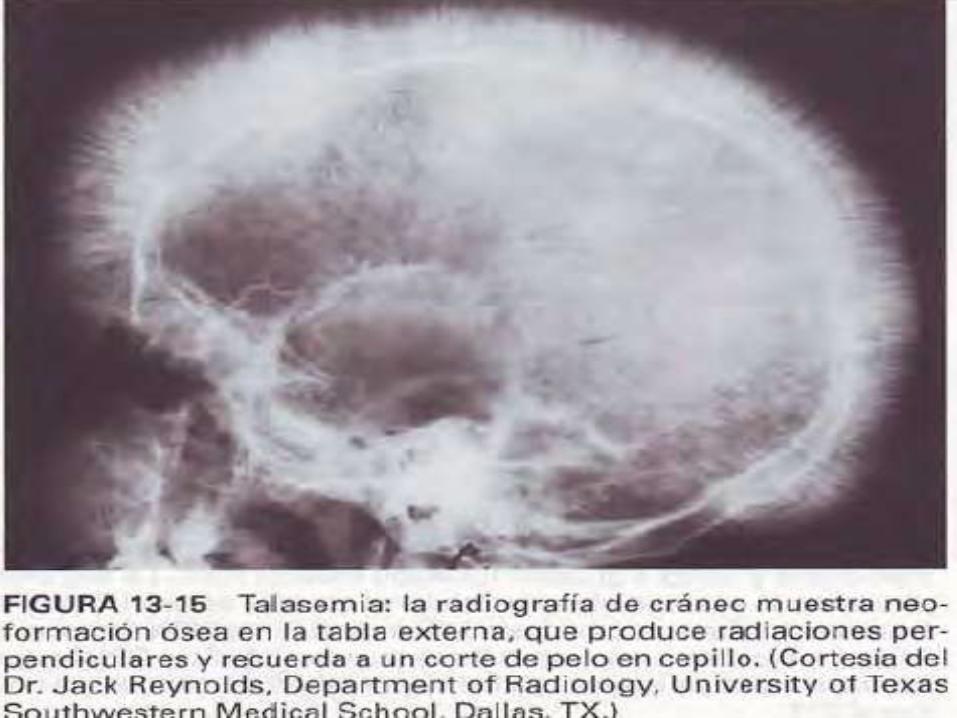
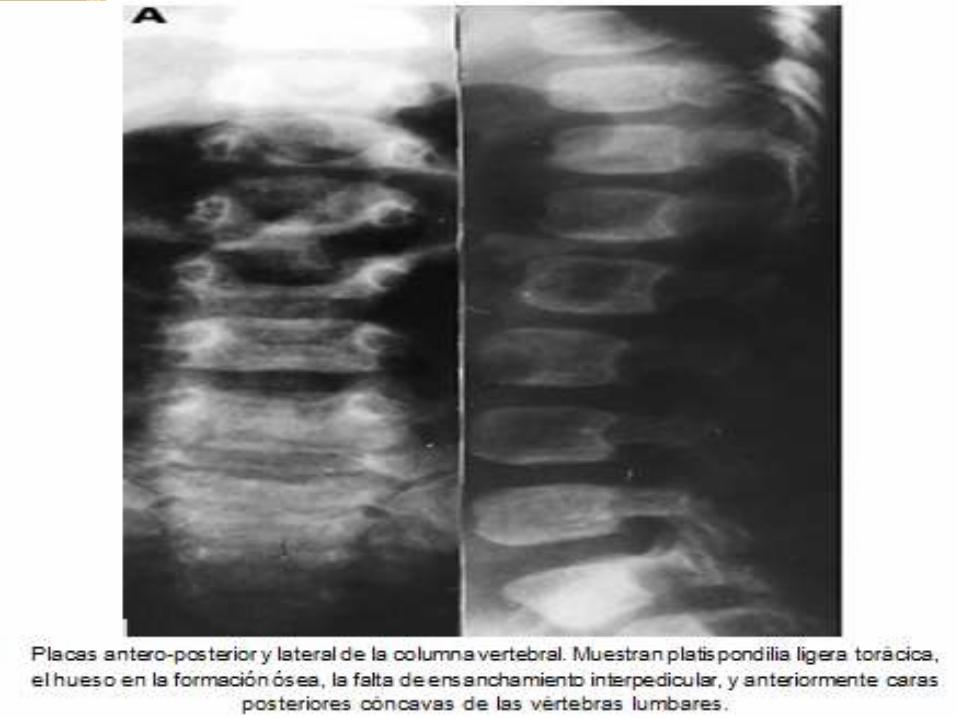
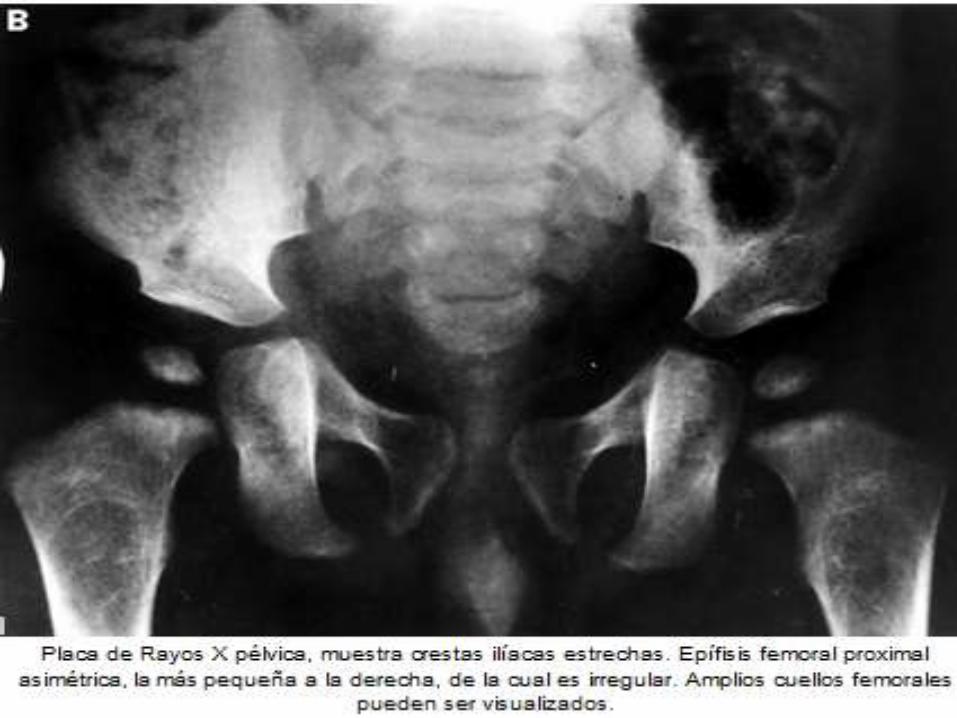
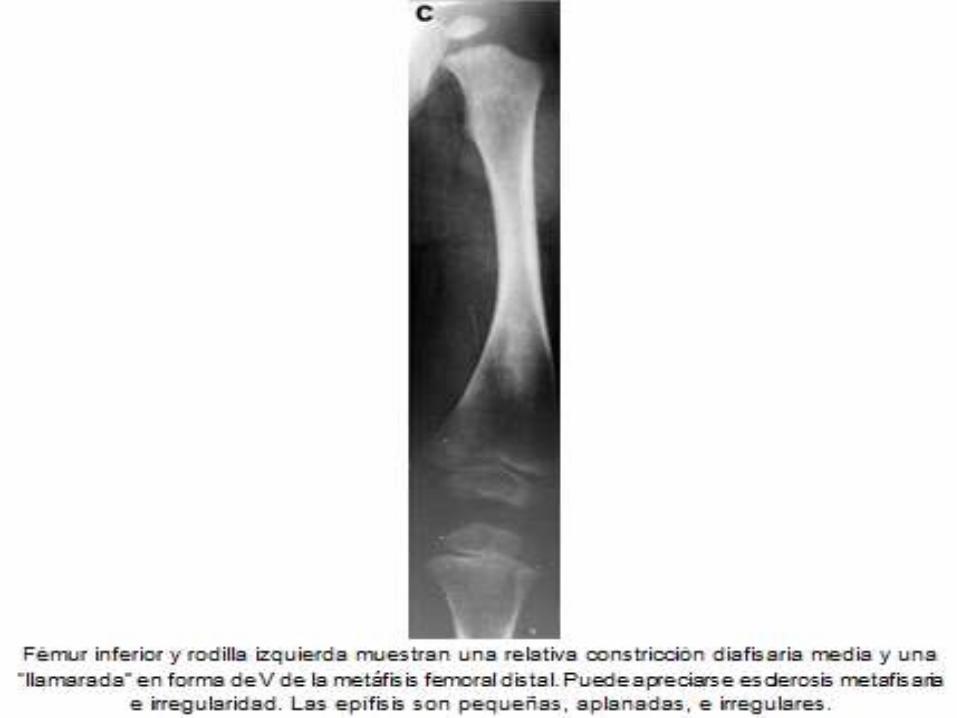

DIAGNOSTICO ANATOMOPATOLOGICO
BETA TALASEMIA
ANEMIA DE DIAMOND-BLACKFAN
Principalmente determinado por los factores genéticos predisponentes así como a diversas entidades clínicas y paraclínicas expuestas anteriormente

*

La molécula de hemoglobina es un tetrámero compuesto de dos cadenas de α-globina y dos de β-globina ligadas al heme (hierro y protoporfirina); se une de una manera reversible con una molécula de oxígeno.

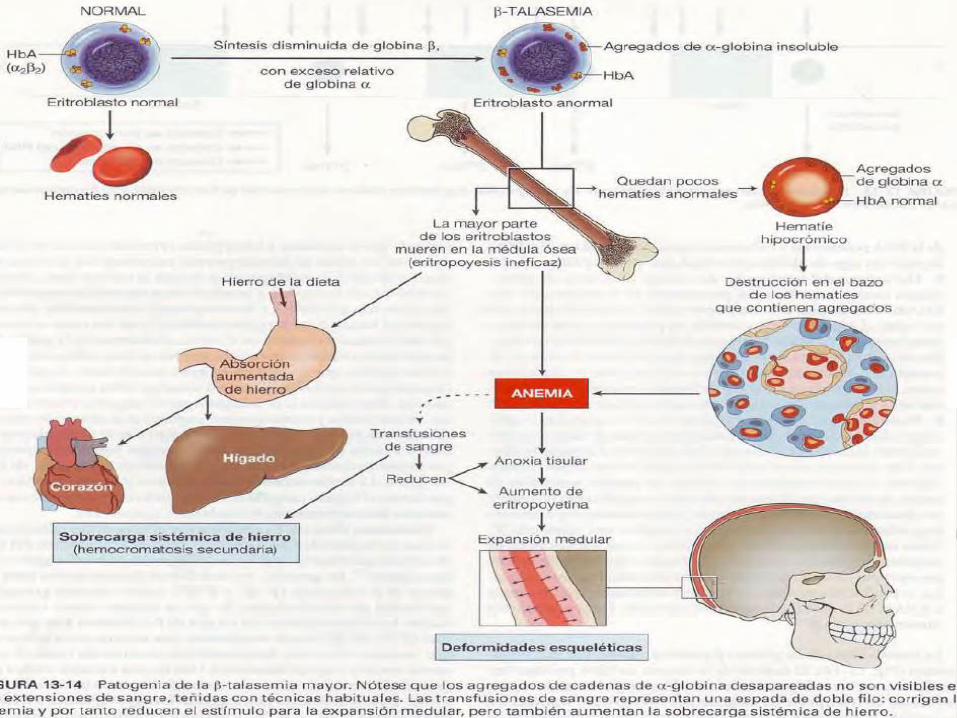
Los síndromes talasémicos ocurren
como resultado de defectos en la
síntesis de las cadenas α y β,
promoviendo una eritropoyesis
inefectiva con hemólisis.

Los defectos son heterogéneos e
incluyen mutaciones genéticas, mRNA
inestable, alteraciones en regiones
promotoras y diversas alteraciones
genéticas.

Típicamente cursan con microcitosis y
células “en tiro al blanco” en el frotis
de sangre periférica. La mayoría
presentan esplenomegalia y tendencia
a desarrollar colelitiasis pigmentadas
(por la hemólisis).

La α-talasemia es prevalente en el
África, el Mediterráneo y en Asia.
Diversos genotipos son posibles
induciendo grados variables de
anemia.
La β-talasemia es común en el
Mediterráneo, Sureste Asiático, la
India y Pakistán.

La disminución en la síntesis de las
cadenas de β-globina, induce a un
incremento en la producción de
hemoglobina A, A2 y/o F.

Las β-talasemias son causadas por
sustituciones de un nucleótido en la
secuencia del gen HBB (codifica la
cadena β) localizado en el
cromosoma 11.

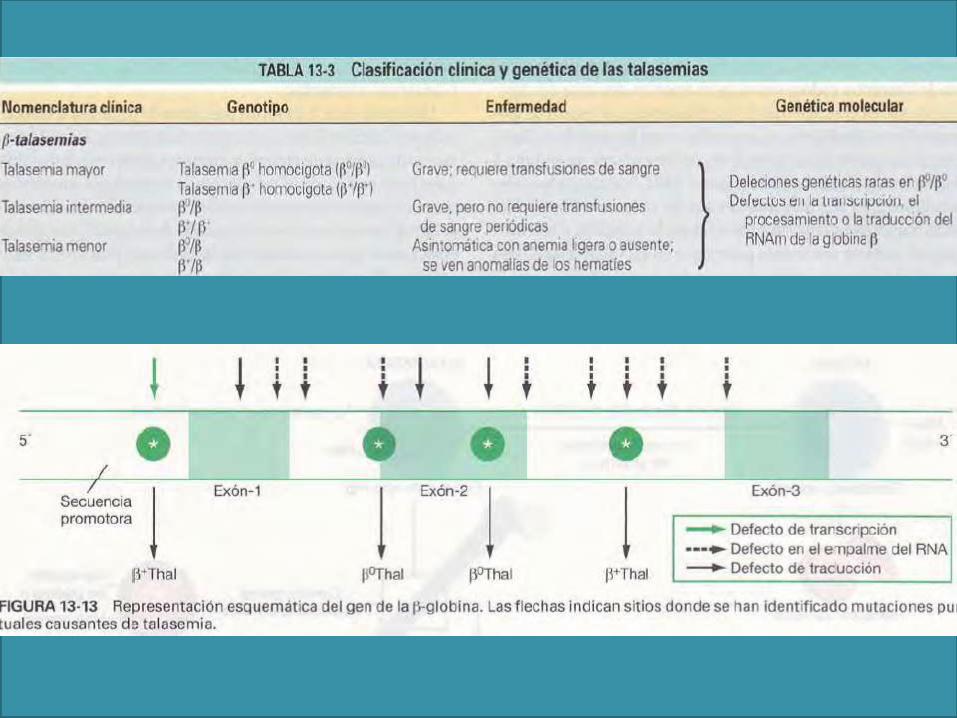
La talasemia mayor, resultado de una ausencia total de las cadenas β, se asocia con cuadros severos de retardo en el crecimiento, al menos que los niños sean transfundidos precozmente (anemia de Cooley).

La mayoría de los pacientes con
talasemia intermedia ameritan
transfusiones, pero tienden a
desarrollar sobrecargas de hierro
(disfunción de hepcidina)

Los pacientes con rasgo talasémico
cursan asintomáticos a pesar de
presentar anemia microcítica leve.
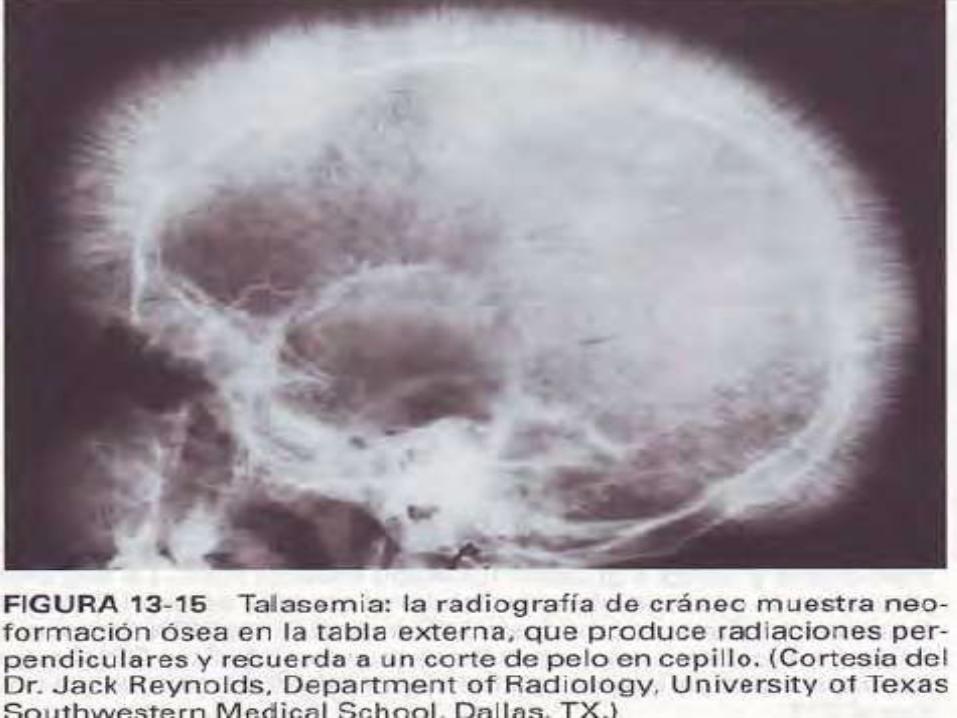

*

Es una anemia rara, en general
descubierta en la primera infancia. El
resultado de una incapacidad de la
médula ósea para producir glóbulos
rojos.

Es una eritroblastopenia (recuento
bajo de globulos rojos) congénita, de
origen autosómico
recesivo, dominante, o esporádico

“anemia hipoplásica congénita”,
“anemia congénita crónica” o
“eritropoyesis imperfecta”.

Podemos encontrar las anomalías
biológicas sin anemia entre los padres
y hermanos de los enfermos (aumento
de la hemoglobina F, macrocitosis,
aumento de la adenosindeaminasa
eritropoyética).

Para tener el diagnóstico, es necesario eliminar todas las otras causas de eritoblastopenia crónica, en particular un virus (el parvovirus B19) que puede dar un cuadro análogo en el bebé, o excepcionalmente, una insuficiencia renal crónica desconocida (el riñón secreta una hormona, la eritropoyetina, indispensable para la eritropoyesis).

Las personas con ADB tienen rasgos
faciales característicos, con cabezas
más pequeñas, ojos almendrados y
sonrisa invertida.

Existen esencialmente tres tipos de
tratamiento:
◦ - La cortisona y sus derivados
(Prednisona)
◦ - las transfusiones de sangre Terapia de Quelación
◦ - el transplante de médula ósea

Los efectos clínicos de la sobrecarga de hierro incluyen:◦ Diabetes mellitus
◦ Cardiopatía
◦ Hipoparatiroidismo
◦ Hipogonadismo

Attar EC, Hasserjian RP, et al. A 25-year-old woman with anemia and iron overload. N Engl J Med 2006;354:2047-56


















