
Band structure calculations on Ti-enhanced NaAlH4 - HZG
37
First principles modelling for predicting and understanding novel complex hydrides Ole M. Løvvik Centre for Materials Science and Nanotechnology, University of Oslo, Norway Institute for Energy Technology, Kjeller, Norway FuncHy 2006:
Transcript of Band structure calculations on Ti-enhanced NaAlH4 - HZG

First principles modellingfor predicting and
understanding novel complex hydrides
Ole M. LøvvikCentre for Materials Science and Nanotechnology, University of Oslo, Norway
Institute for Energy Technology, Kjeller, Norway
FuncHy 2006:
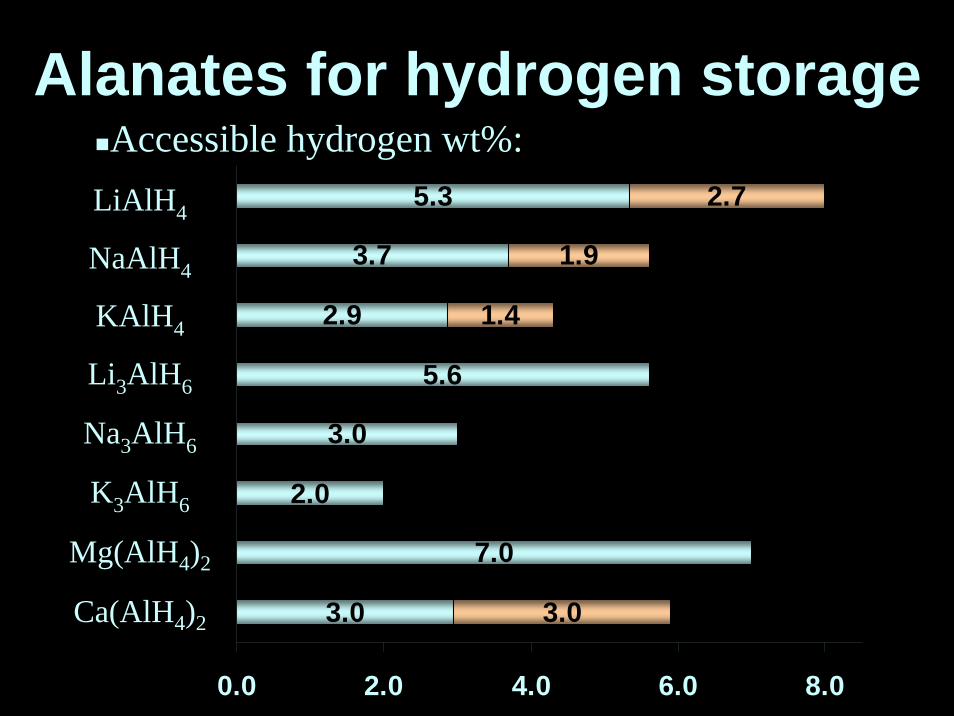
5.3
3.7
2.9
5.6
3.0
2.0
7.0
3.0
2.7
1.9
1.4
3.0
0.0 2.0 4.0 6.0 8.0
LiAlH4
NaAlH4
KAlH4
Li3AlH6
Na3AlH6
K3AlH6
Mg(AlH4)2
Ca(AlH4)2
LiAlH4
NaAlH4
KAlH4
Li3AlH6
Na3AlH6
K3AlH6
Mg(AlH4)2
Ca(AlH4)2
Accessible hydrogen wt%:Alanates for hydrogen storage
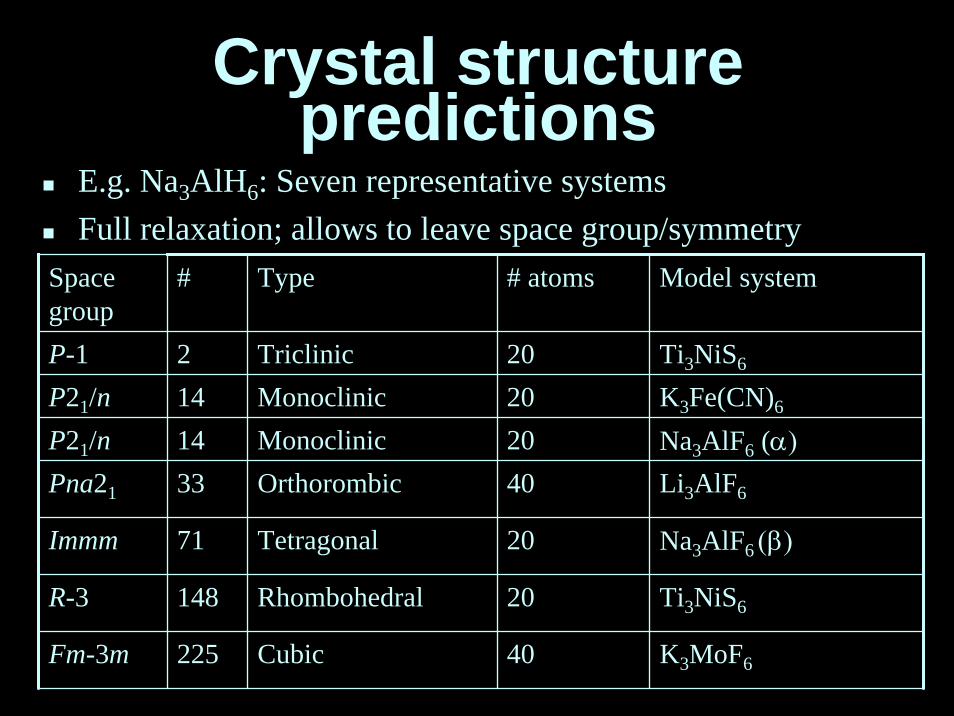
Crystal structurepredictions
Spacegroup
# Type # atoms Model system
Ti3NiS6
K3Fe(CN)6
P21/n 14 Monoclinic 20 Na3AlF6 (α)Li3AlF6
Na3AlF6 (β)
Ti3NiS6
Fm-3m 225 Cubic 40 K3MoF6
P-1 2 Triclinic 20P21/n 14 Monoclinic 20
Pna21 33 Orthorombic 40
Immm 71 Tetragonal 20
R-3 148 Rhombohedral 20
E.g. Na3AlH6: Seven representative systemsFull relaxation; allows to leave space group/symmetry

Calculation detailsBand-structure DFTVASPGeneralized gradient appr. (GGA) PW91Projector augmented wave (PAW) methodSpin polarization allowedCut-off energy 780 eVOverall convergence: ~1 meV (0.01 kJ/mol)/unit cell

Predicted crystal structuresCompound Predicted
structureExpt. structure
LiAlH4 P21/c P21/cNaAlH4 I41 /a I41 /aKAlH4 Pnma PnmaLi3AlH6 R–3 R–3Na3AlH6 P21/n P21/nK3AlH6 P21/nMg(AlH4)2 P–3m1 P–3m1Ca(AlH4)2 Pbca
O. M. Løvvik, S. M. Opalka, H. W. Brinks, B. C. Hauback, Phys. Rev. B 69 (2004) 134117)O. M. Løvvik, O. Swang, Europhys. Lett. 67 (2004) 607.O. M. Løvvik, Phys. Rev. B. 71(2005) 144111.O. M. Løvvik, O. Swang, J. Alloys Comp. 404-406 (2005) 757-761.O. M. Løvvik, P. N. Molin, Phys. Rev. B. 72 (2005) 073201.O. M. Løvvik, O. Swang, S. M. Opalka, J. Mater. Res. 20 (2005) 3199 (Invited paper).

Thermodynamic stabilityFormation enthalpy:Hform(MnAlH(n+3)) =
E(MnAlH(n+3)) – n E(M) – E(Al) – (n+3)/2 E(H2)
Compound LiAlH4 NaAlH4 KAlH4 Li3AlH6 Na3AlH6 K3AlH6
Formation enthalpy (kJ/mol H2) -55.5 -54.9 -70.0 -102.8 -69.9 -78.5
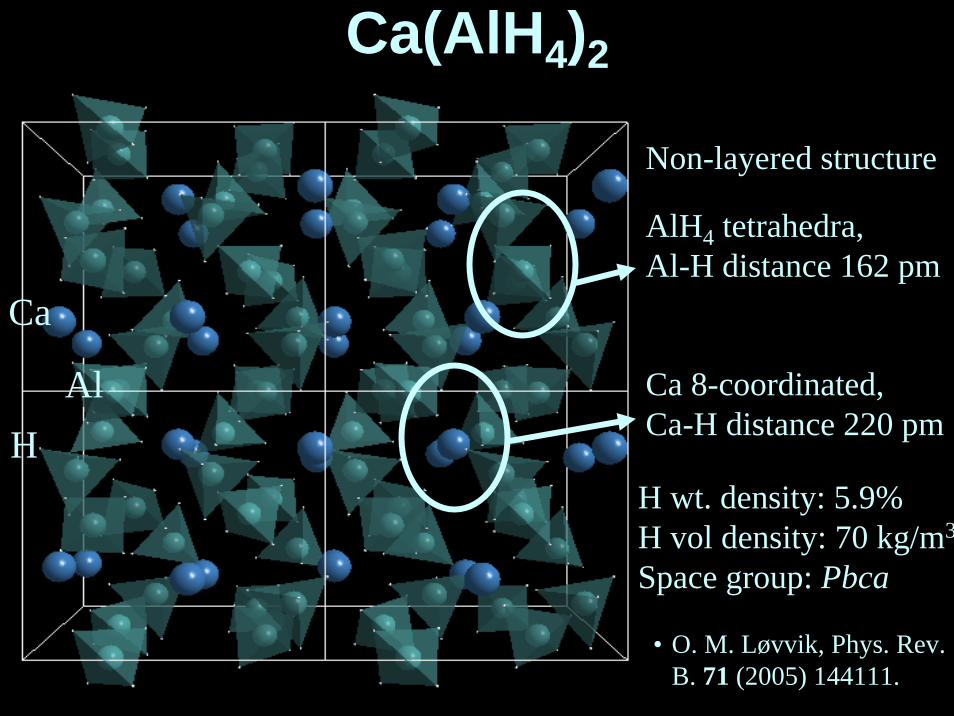
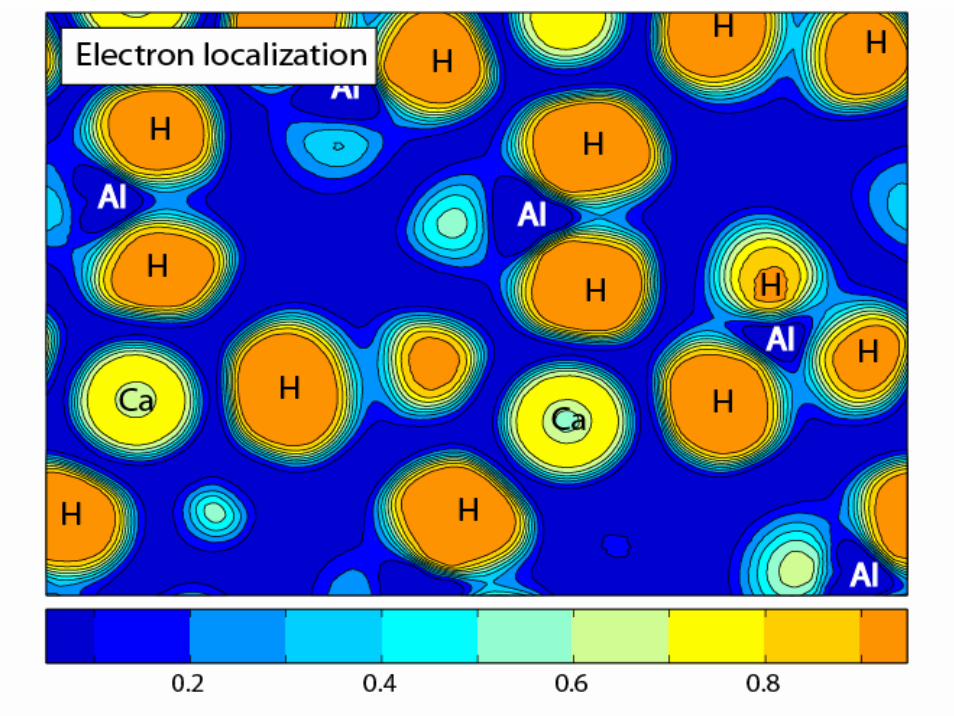
Ca(AlH4)2
Ca
AlH
Non-layered structure
AlH4 tetrahedra,Al-H distance 162 pm
Ca 8-coordinated,Ca-H distance 220 pm
H wt. density: 5.9%H vol density: 70 kg/m3
Space group: Pbca
• O. M. Løvvik, Phys. Rev. B. 71 (2005) 144111.
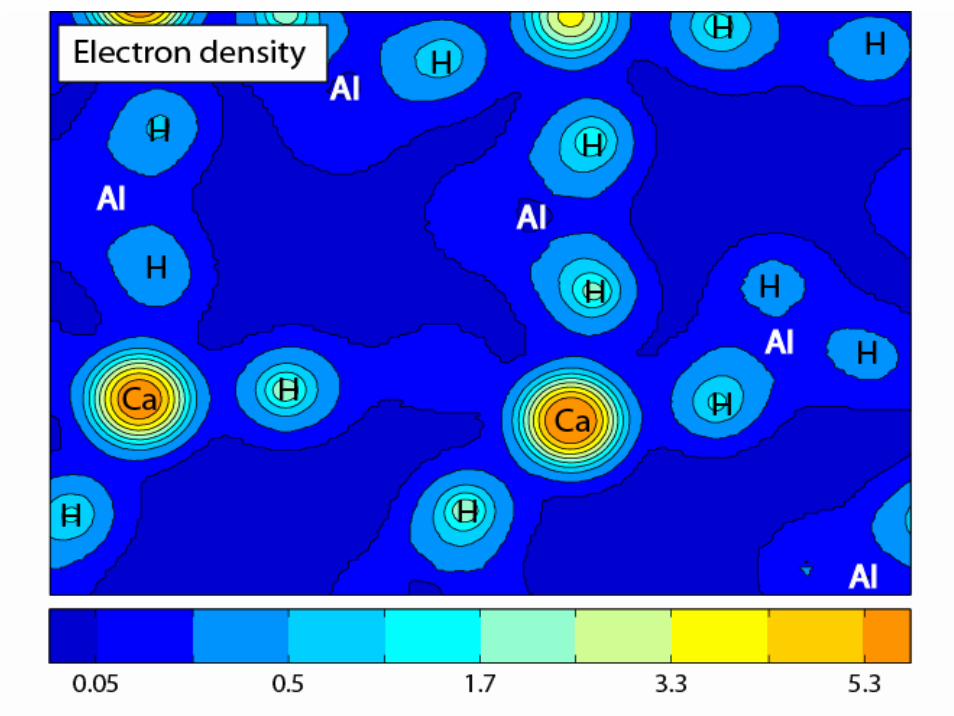
-5 0 50
0.1
0.2
E (eV)
DO
S
H
-5 0 50
50
100
E (eV)
Total DOS
0
1
2
3Ca
0
0.5
1
Al
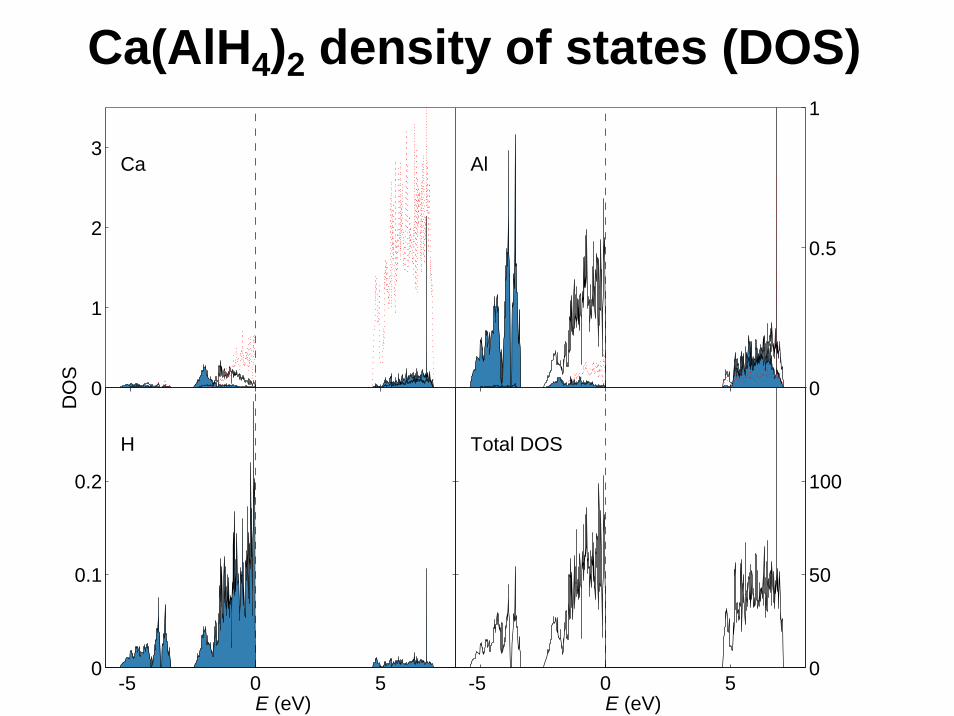
Ca(AlH4)2 density of states (DOS)

Thermodynamic stabilityFormation enthalpy:
Reaction enthalpy:
kJ/mol H2 Formationenthalpy
Reactionenthalpy
Mg(AlH4)2 -21.1 -6.2
Ca(AlH4)2 -59.4 -20.7
This is whyMg(AlH4)2is not reversible ...

Why are not Li and Mg alanates reversible?
Na and K alanates exhibit reversible hydrogenation, Li and Mg alanates not.Indicators of reversibility from calculations?Electronic structure: no clear signs.Crystal structure: Li3AlH6 stands out.Thermodynamic stability: Li3AlH6 stands out.The reason: Li small enough for Li3AlH6 to attain the R–3structure, making Li3AlH6 too stable.Similarly: Mg alanate too unstable.No easy generalization to other materials.

O. M. Løvvik, O. Swang, S. M. Opalka, J. Mater. Res. 20 (2005) 3199
O. M. Løvvik, O. Swang, J. Alloys Comp. 404-406(2005) 757-761
Prediction of new mixedalanate phases
LiAlH4
KAlH4NaAlH4
Li0.75Na0.25AlH4
Li0.5Na0.5AlH4
Li0.25Na0.75AlH4
K0.25Li0.75AlH4
K0.5Li0.5AlH4
K0.75Li0.25AlH4
K0.75Na0.25AlH4K 0.5Na 0.5
AlH 4
K 0.25Na 0.75A
lH 4
”Tetrahydrides”• O. M. Løvvik, O. Swang, Europhys. Lett. 67 (2004) 607.
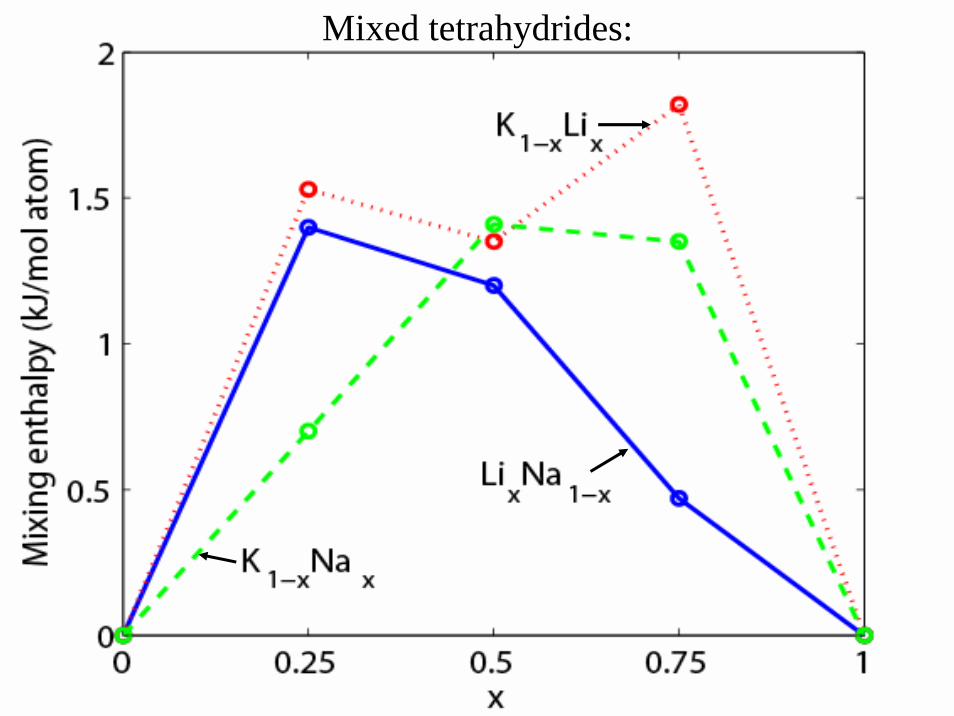
Mixed tetrahydrides:

Prediction of new mixedalanate phases
Li3AlH6
K3AlH6Na3AlH6
Li2NaAlH6
LiNa2AlH6
KLi2AlH6
K2LiAlH6
Li2.5Na0.5AlH6
Li1.5Na1.5AlH6
Li0.5Na2.5AlH6
K2.5Li0.5AlH6
K1.5Li1.5AlH6
K0.5Li2.5AlH6
K2.5Na0.5AlH6K 2NaA
lH 6
K 1.5Na 1.5
AlH 6
KNa 2AlH 6
K 0.5Na 2.5
AlH 6
”Hexahydrides”
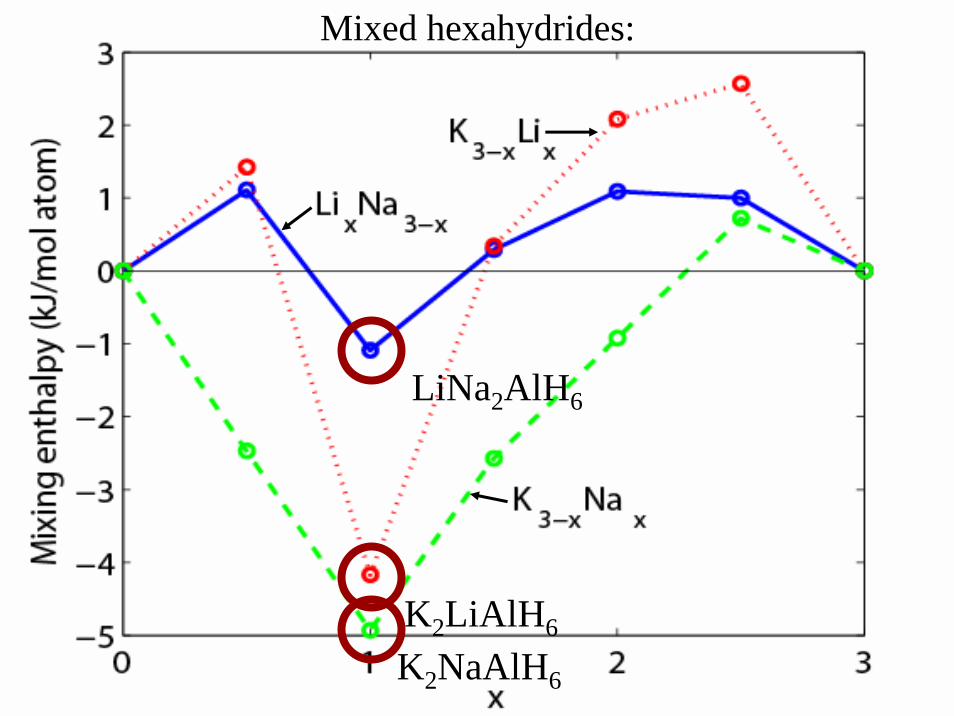
Mixed hexahydrides:
K2NaAlH6
K2LiAlH6
LiNa2AlH6

Phonon calculationsVibration frequencies from displacementcalculationsPhonon spectrum and partition functionIntegrated phonon density → Thermodynamics
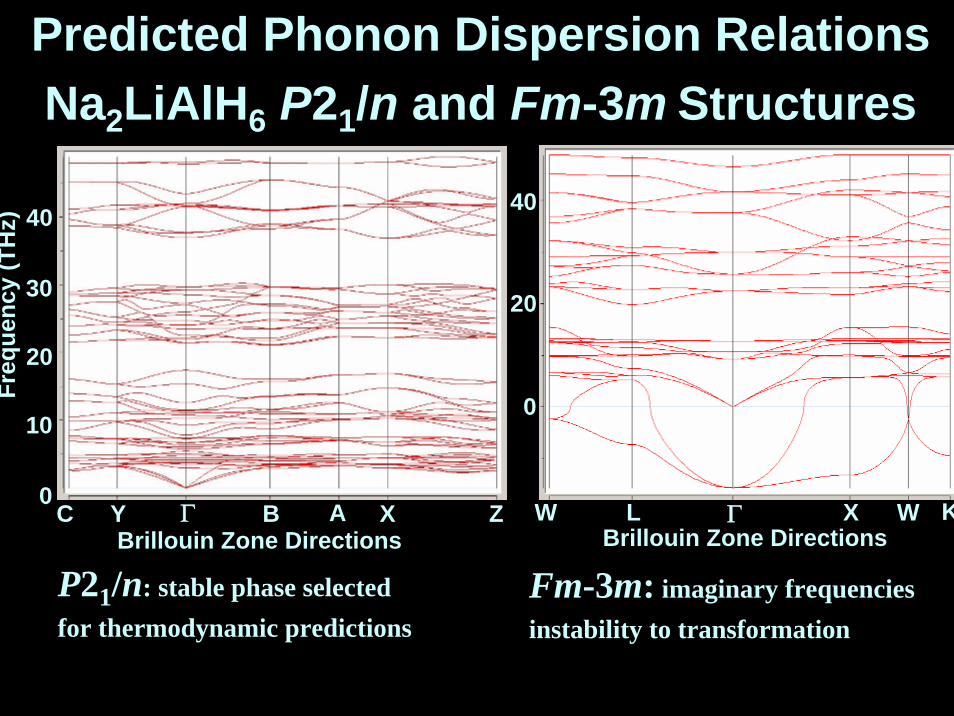
P21/n: stable phase selectedfor thermodynamic predictions
Fm-3m: imaginary frequenciesinstability to transformation
Predicted Phonon Dispersion RelationsNa2LiAlH6 P21/n and Fm-3m Structures
Freq
uenc
y (T
Hz)
40
20
0
W WL Γ X KC BΓ X ZY A
40
20
0
10
30
Brillouin Zone Directions Brillouin Zone Directions
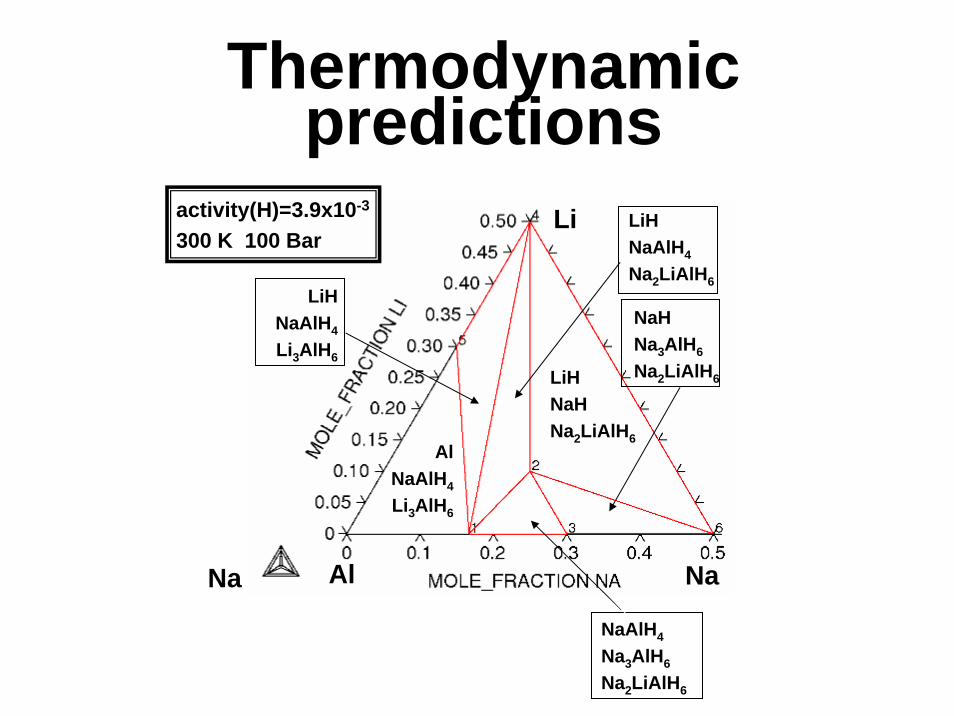
Thermodynamicpredictions
Na
Li
NaAl
activity(H)=3.9x10-3
300 K 100 Bar
AlNaAlH4
Li3AlH6
LiHNaAlH4
Li3AlH6
LiHNaAlH4
Na2LiAlH6
LiHNaHNa2LiAlH6
NaHNa3AlH6
Na2LiAlH6
NaAlH4
Na3AlH6
Na2LiAlH6

Optimum H reversibility{2Na : 1Li : 2Al : 9H}:
2NaAlH4+LiH300 K /100 Bar
Na2LiAlH6+Al300 K / 1 Bar
3/2 H2
2.6 wt%
2NaH+LiH+2Al400 K / 1 Bar
3/2 H2
2.6 wt%
Potential for 5.2 wt% Reversible H
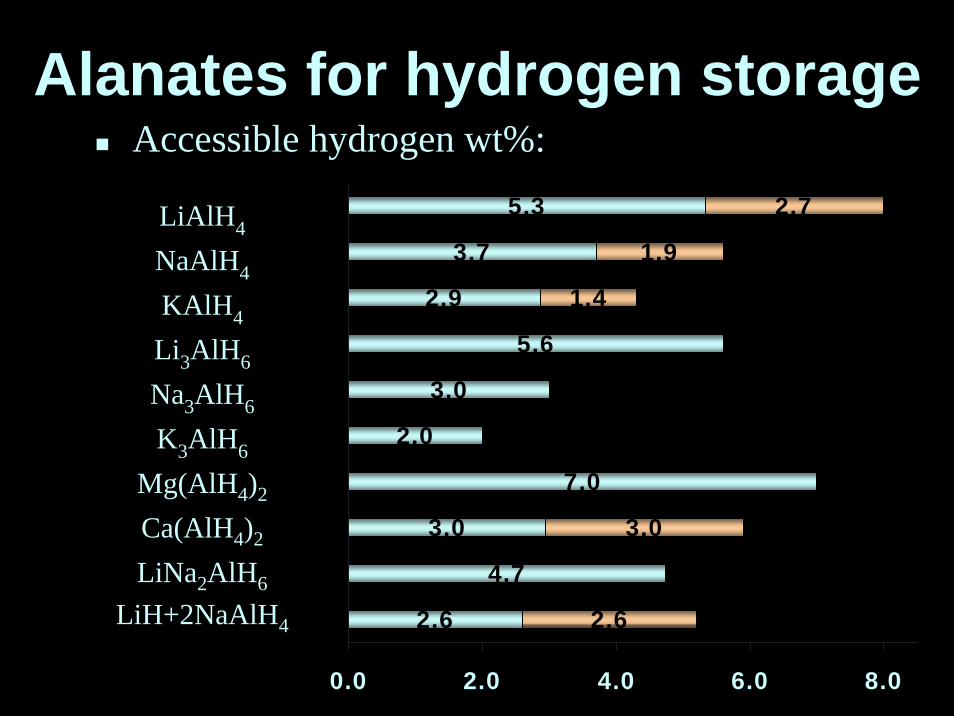
5.3
3.7
2.9
5.6
3.0
2.0
7.0
3.0
4.7
2.6
2.7
1.9
1.4
3.0
2.6
0.0 2.0 4.0 6.0 8.0
LiAlH4
NaAlH4
KAlH4
Li3AlH6
Na3AlH6
K3AlH6
Mg(AlH4)2
Ca(AlH4)2
LiNa2AlH6
LiH+2NaAlH4
LiAlH4
NaAlH4
KAlH4
Li3AlH6
Na3AlH6
K3AlH6
Mg(AlH4)2
Ca(AlH4)2
LiNa2AlH6
LiH+2NaAlH4
Accessible hydrogen wt%:Alanates for hydrogen storage

Ti in NaAlH4 –experimental status
Nature of Ti additive not important (TiCl3, TiF3, Ti powder, Ti(OBu)4, etc.)Three different majority phases found:
Al3TiDispersed, amorphous Al1-uTiu (u = 0.07 – 0.15)TiH2
Samples with different Ti majority phases have comparablekineticsIs there an active minority phase? Doping or catalysis?Evidence for point defects (hydrogen vacancies), alanes(AlH3), and heterogeneous catalysis.
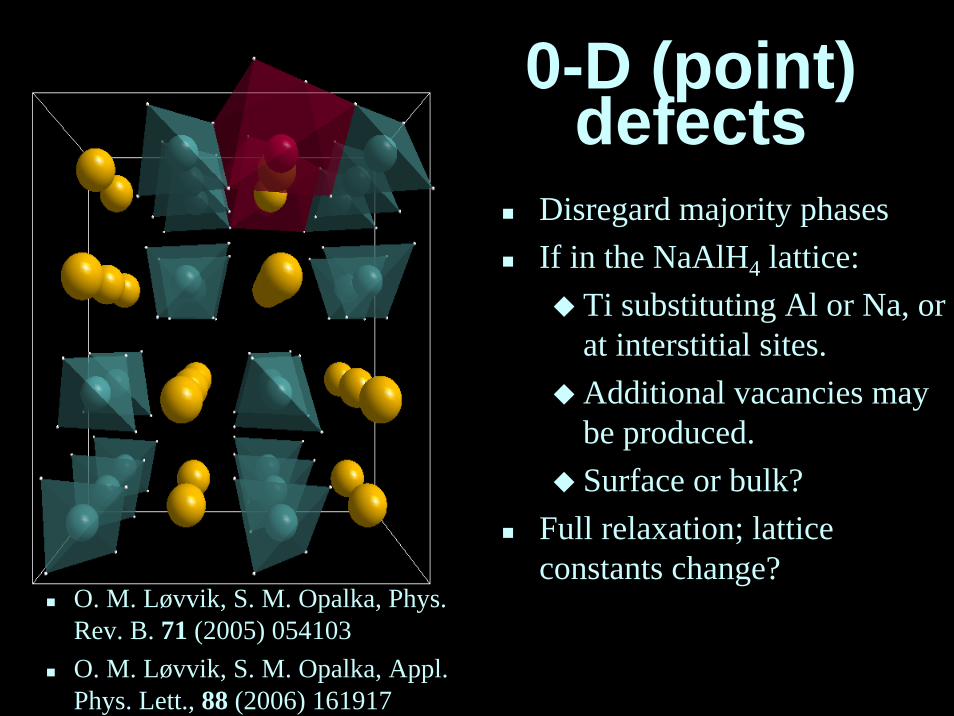
0-D (point) defects
Disregard majority phasesIf in the NaAlH4 lattice:
Ti substituting Al or Na, or at interstitial sites.Additional vacancies maybe produced.Surface or bulk?
Full relaxation; latticeconstants change?
O. M. Løvvik, S. M. Opalka, Phys. Rev. B. 71 (2005) 054103O. M. Løvvik, S. M. Opalka, Appl. Phys. Lett., 88 (2006) 161917
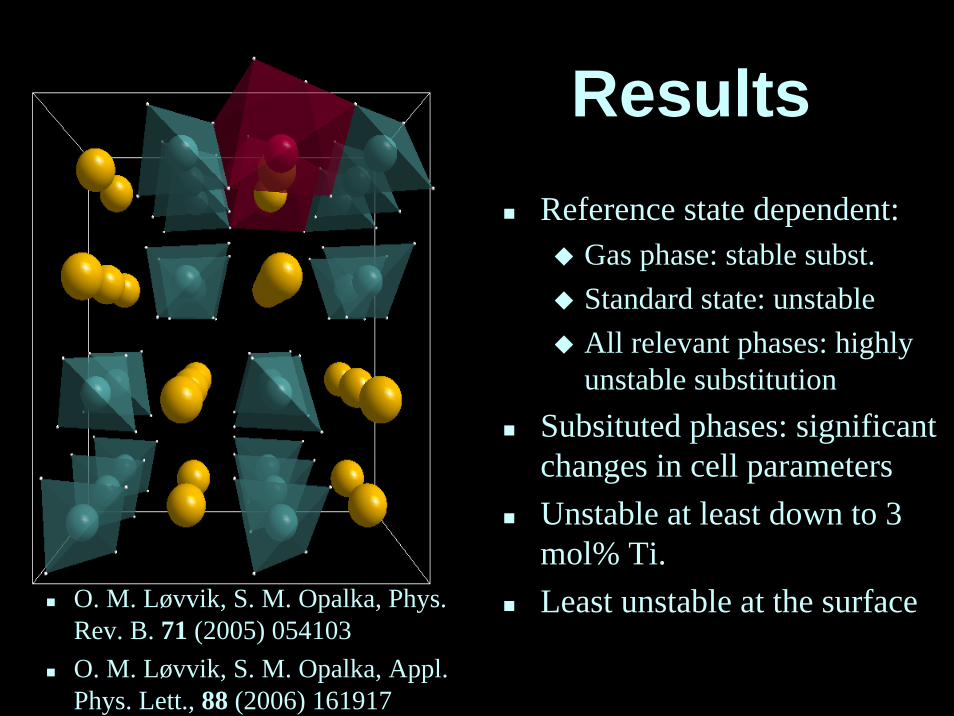
ResultsReference state dependent:
Gas phase: stable subst.Standard state: unstableAll relevant phases: highlyunstable substitution
Subsituted phases: significantchanges in cell parameters Unstable at least down to 3 mol% Ti.Least unstable at the surfaceO. M. Løvvik, S. M. Opalka, Phys.
Rev. B. 71 (2005) 054103O. M. Løvvik, S. M. Opalka, Appl. Phys. Lett., 88 (2006) 161917
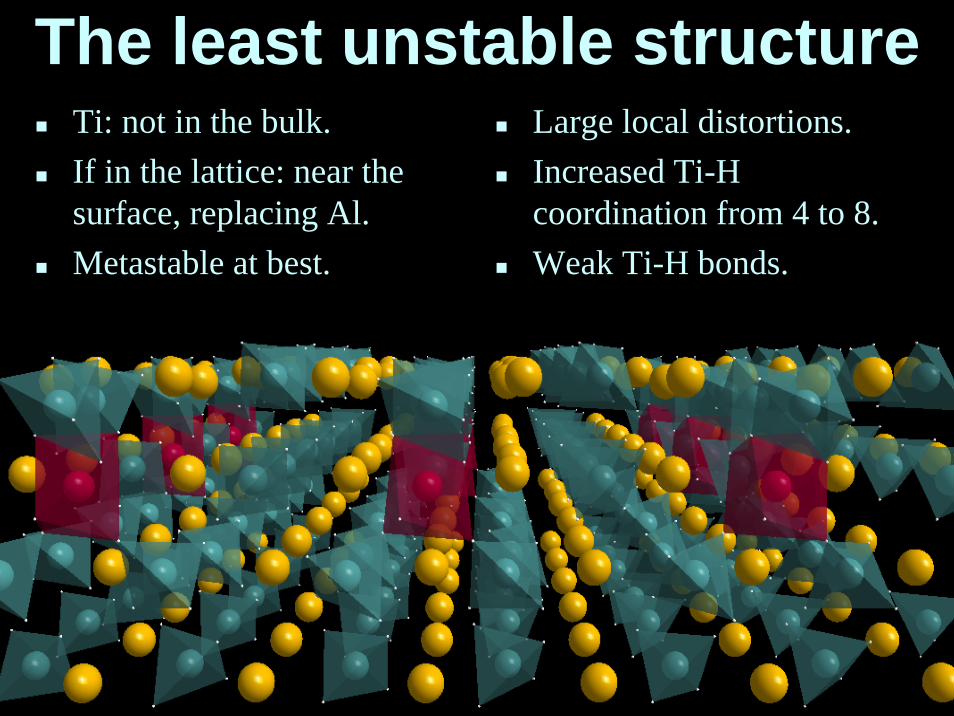
The least unstable structureTi: not in the bulk.If in the lattice: near thesurface, replacing Al.Metastable at best.
Large local distortions.Increased Ti-H coordination from 4 to 8.Weak Ti-H bonds.
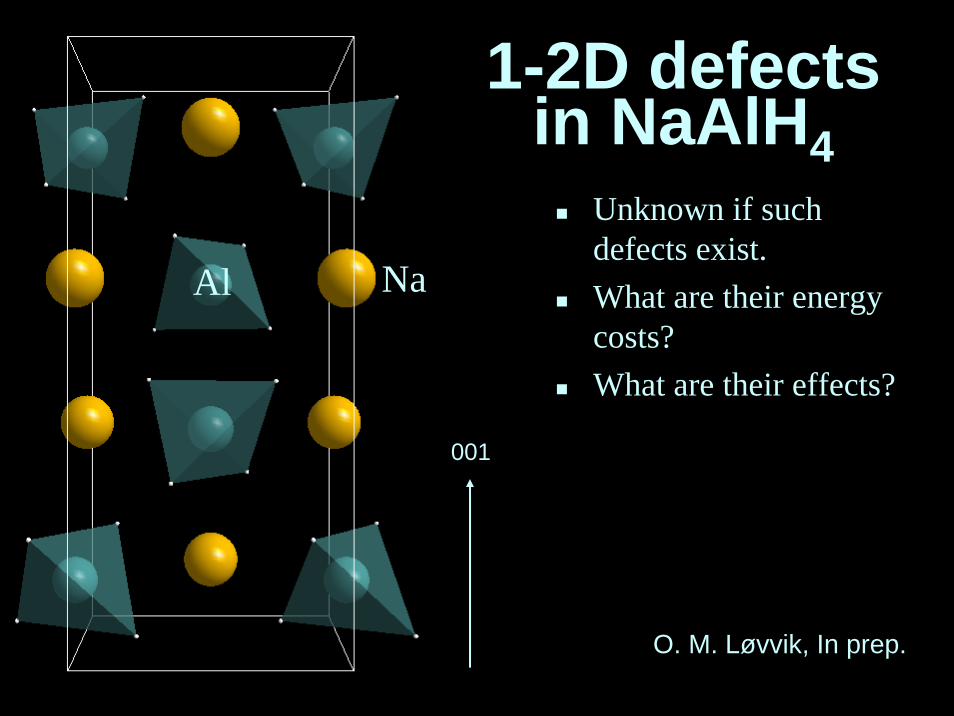
1-2D defectsin NaAlH4
Unknown if suchdefects exist.What are their energycosts?What are their effects?
NaAl
001
O. M. Løvvik, In prep.

1D: dislocations.2D: stacking faults, twins, anti phaseboundaries, grainboundaries.Unknown if suchdefects exist.What are their energycosts?What are their effects?
1-2D defects in NaAlH4
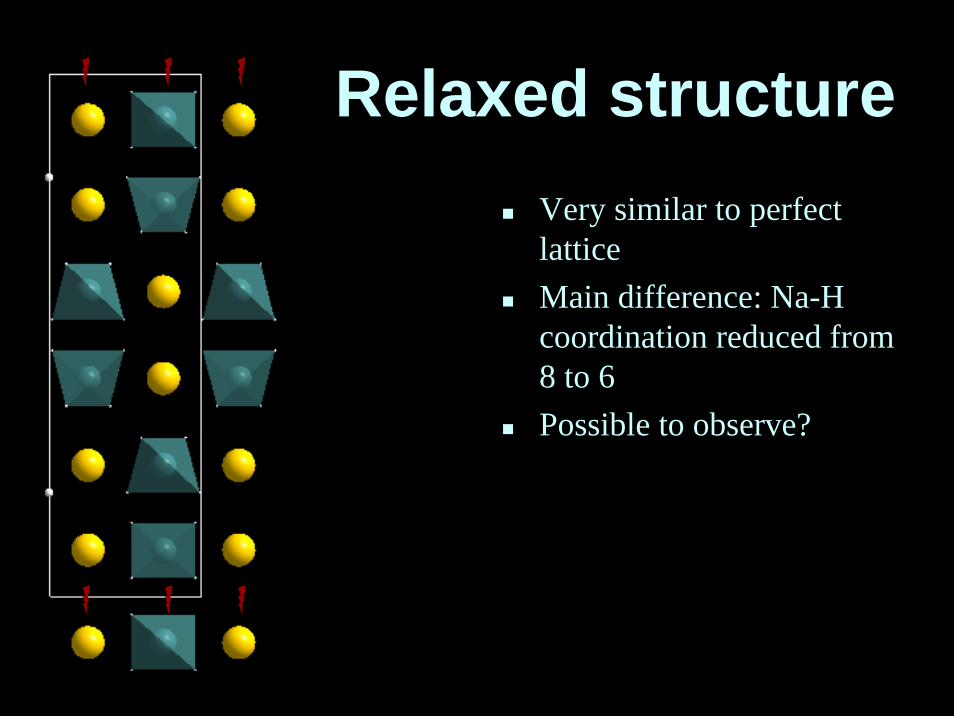
Relaxed structureVery similar to perfectlatticeMain difference: Na-Hcoordination reduced from 8 to 6Possible to observe?
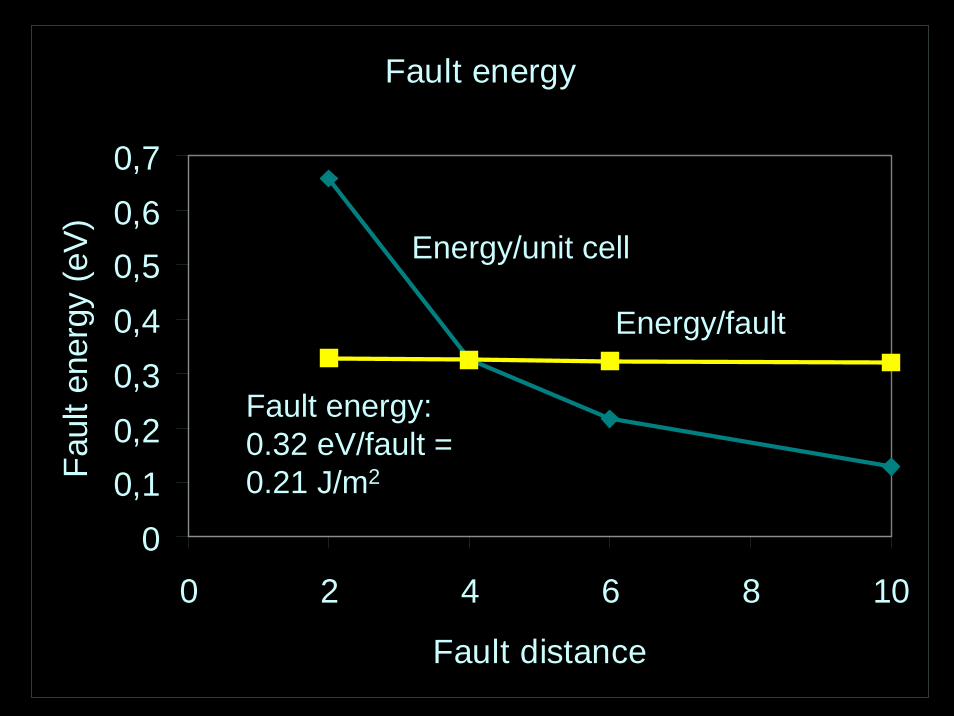
Fault energy
00,10,20,30,40,50,60,7
0 2 4 6 8 10
Fault distance
Faul
t ene
rgy
(eV
)
Energy/unit cell
Energy/fault
Fault energy: 0.32 eV/fault = 0.21 J/m2
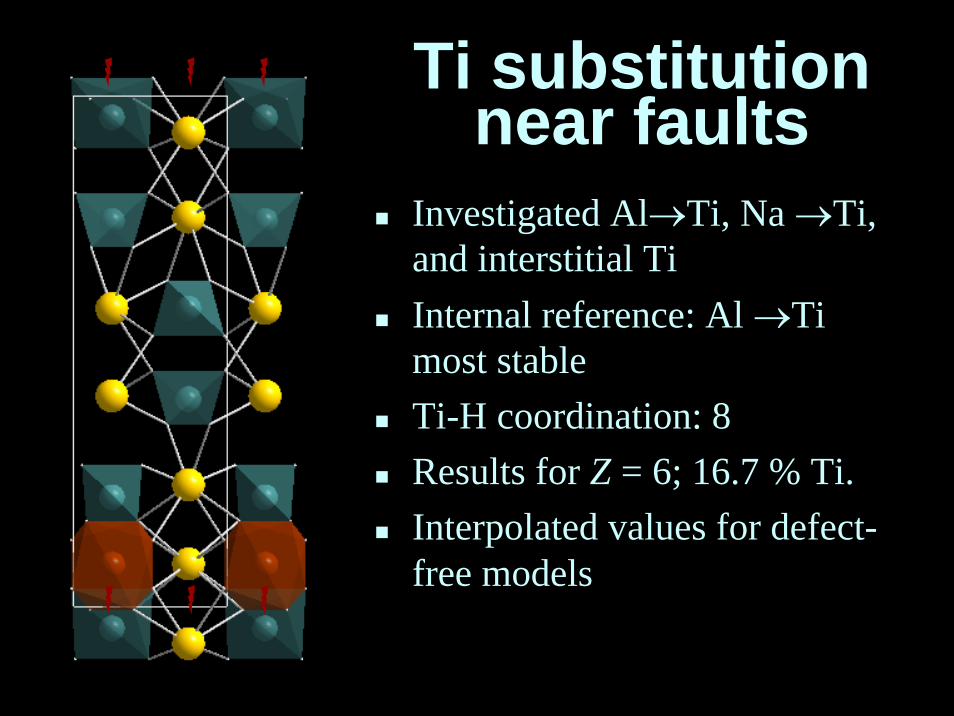
Ti substitutionnear faults
Investigated Al→Ti, Na →Ti, and interstitial TiInternal reference: Al →Ti most stableTi-H coordination: 8Results for Z = 6; 16.7 % Ti.Interpolated values for defect-free models
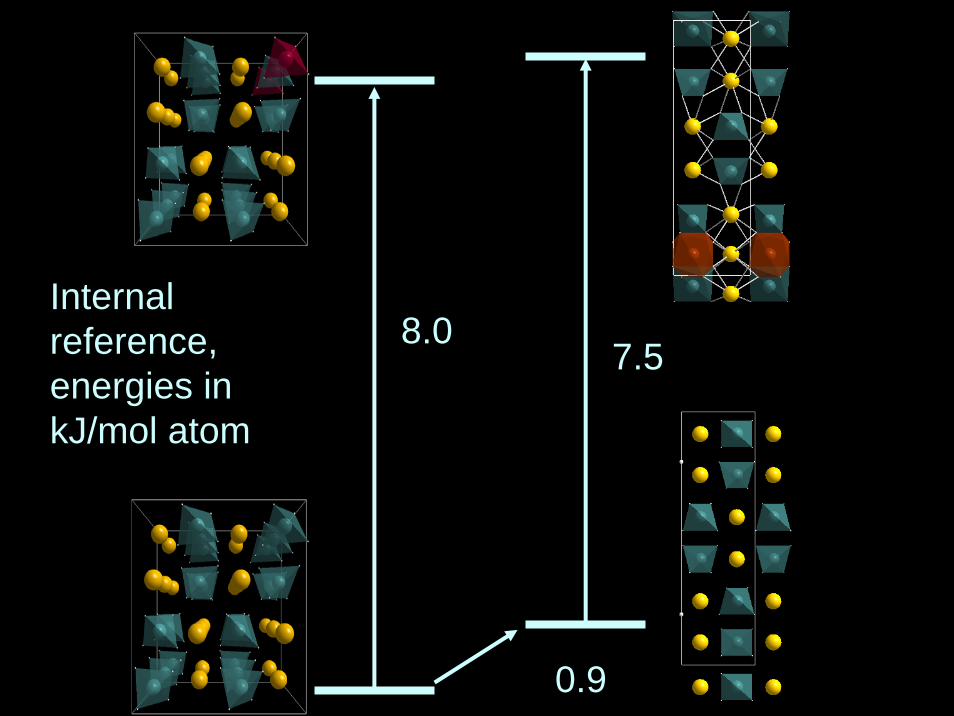
8.0
0.9
7.5Internalreference,energies in kJ/mol atom
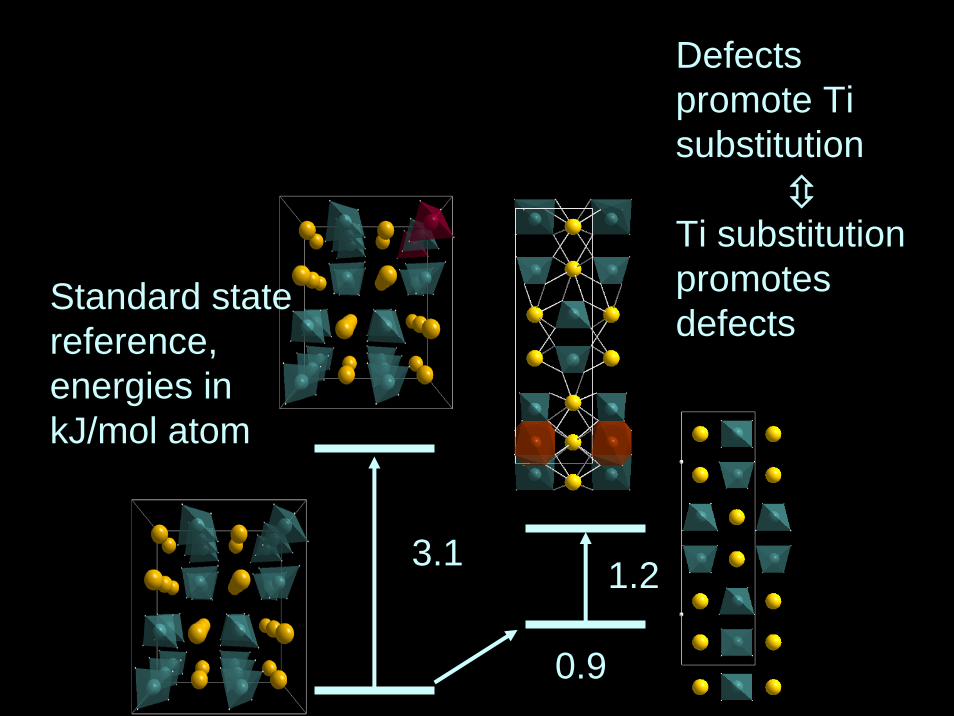
3.1
0.9
1.2
Standard statereference,energies in kJ/mol atom
Defectspromote Ti substitution
Ti substitutionpromotesdefects

What about non-equilibrium structures?
Suppose that mono-dispersedTi is available.Investigated clusters withconstant number of atoms.⇒ reference is NaAlH4 + Ti.
A. Marashdeh, R. A. Olsen, O. M. Løvvik, G.-J. Kroes, Chem. Phys. Lett. 426 (2006) 180–186
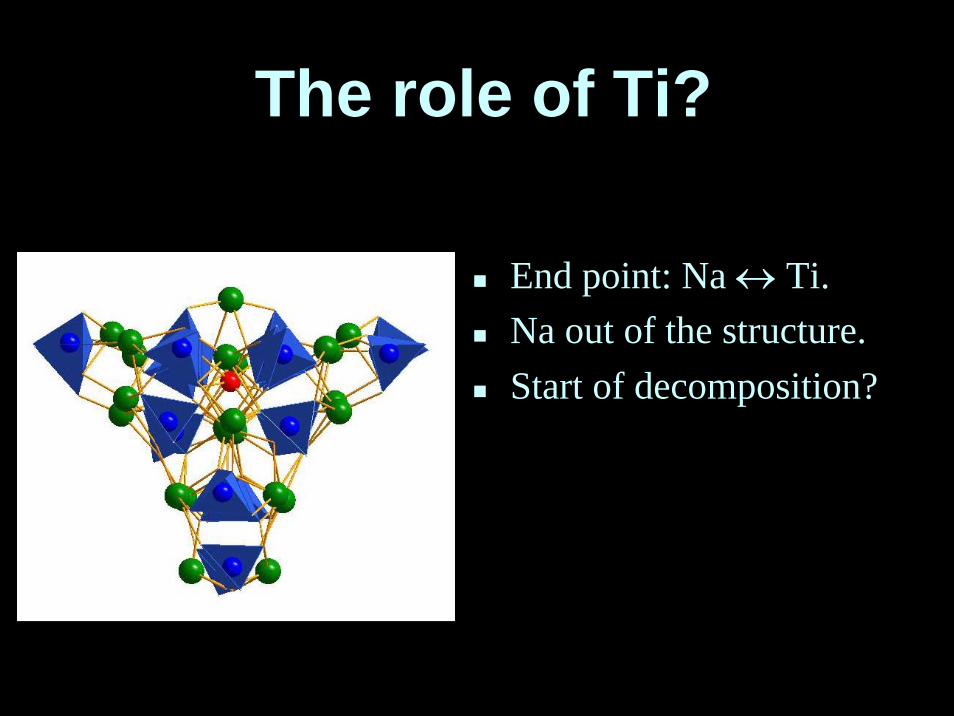
The role of Ti?
End point: Na ↔ Ti.Na out of the structure.Start of decomposition?

The role of Ti?The ”zipper model”:
Ti acts as a slider.Catalysis, but not traditional.May be combined with H2splitting in the absorptionprocess.Ti substitution promoted by defects.

SummaryProbably no better alanate than NaAlH4.The LiH + NaAlH4 mixture interesting.Minority phase of Ti probably important in NaAlH4(de-)hydrogenation:
Ti substitution in bulk ruled out.Ti substitution promoted by defects.Non-equilibrium: Ti may enter lattice.Proposed model: zipper model with Ti-slider.

AcknowledgementsCoworkers:
Susanne Opalka, UTRCPeter Molin, UiOBjørn Hauback et al., IFERoar Olsen et al., Leiden UniversityOle Swang et al., Sintef
Funding: Nanomat, Norwegian Research Council






![Index [application.wiley-vch.de] · Index a Abbasov/Romo’s Diels–Alder lactonization 628 ab initio – calculations 1159 – molecular orbital calculations 349 – wavefunction](https://static.fdocument.org/doc/165x107/5b8ea6bc09d3f2a0138dd0b3/index-index-a-abbasovromos-dielsalder-lactonization-628-ab-initio.jpg)











![Index [] a Abbasov/Romo’s Diels–Alder lactonization 628 ab initio – calculations 1159 – molecular orbital calculations 349 – wavefunction 209](https://static.fdocument.org/doc/165x107/5aad6f3f7f8b9aa9488e42ac/index-a-abbasovromos-dielsalder-lactonization-628-ab-initio-calculations.jpg)