ASTX660, a novel non-peptidomimetic antagonist of cIAP1/2 ... · by fragment-based screening and...
30
1 ASTX660, a novel non-peptidomimetic antagonist of cIAP1/2 and XIAP, potently induces TNF-α dependent apoptosis in cancer cell lines and inhibits tumor growth Authors: George A. Ward, Edward J. Lewis, Jong Sook Ahn, Christopher N. Johnson, John F. Lyons, Vanessa Martins, Joanne M. Munck, Sharna J. Rich, Tomoko Smyth, Neil T. Thompson, Pamela A. Williams, Nicola E. Wilsher, Nicola G. Wallis, Gianni Chessari. Affiliation: Astex Pharmaceuticals, 436 Cambridge Science Park, Milton Road, Cambridge, CB4 0QA, UK. Running title: ASTX660 is an antagonist of cIAP1/2 and XIAP Key Words: XIAP, cIAP1, cIAP2, apoptosis, ASTX660, antagonist Authors to whom correspondence should be addressed: George A Ward, Astex Pharmaceuticals, 436 Cambridge Science Park, Milton Road, Cambridge, CB4 0QA, UK. Tel: +44 1223 435068, Fax: +44 1223 226201, Email: [email protected] Conflict of Interest Statement: GAW, EJL, JSA, CNJ, JFL, VM, JMM, SJR, TS, NTT, PAW, NEW, NGW, GC are employees of Astex Pharmaceuticals Total word count: 4902, Total Figures and Tables: 6 on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848
Transcript of ASTX660, a novel non-peptidomimetic antagonist of cIAP1/2 ... · by fragment-based screening and...

1
ASTX660, a novel non-peptidomimetic antagonist of cIAP1/2 and
XIAP, potently induces TNF-α dependent apoptosis in cancer cell lines
and inhibits tumor growth
Authors: George A. Ward, Edward J. Lewis, Jong Sook Ahn, Christopher N. Johnson, John F. Lyons,
Vanessa Martins, Joanne M. Munck, Sharna J. Rich, Tomoko Smyth, Neil T. Thompson, Pamela A.
Williams, Nicola E. Wilsher, Nicola G. Wallis, Gianni Chessari.
Affiliation: Astex Pharmaceuticals, 436 Cambridge Science Park, Milton Road, Cambridge, CB4 0QA, UK.
Running title: ASTX660 is an antagonist of cIAP1/2 and XIAP
Key Words: XIAP, cIAP1, cIAP2, apoptosis, ASTX660, antagonist
Authors to whom correspondence should be addressed:
George A Ward, Astex Pharmaceuticals, 436 Cambridge Science Park, Milton Road, Cambridge, CB4 0QA,
UK. Tel: +44 1223 435068, Fax: +44 1223 226201, Email: [email protected]
Conflict of Interest Statement: GAW, EJL, JSA, CNJ, JFL, VM, JMM, SJR, TS, NTT, PAW, NEW, NGW, GC
are employees of Astex Pharmaceuticals
Total word count: 4902, Total Figures and Tables: 6
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

2
Abstract
Due to their roles in the evasion of apoptosis, Inhibitor of Apoptosis Proteins (IAPs) are considered
attractive targets for anti-cancer therapy. Antagonists of these proteins have the potential to switch pro-
survival signaling pathways in cancer cells towards cell death. Various SMAC-peptidomimetics with
inherent cIAP selectivity have been tested clinically and demonstrated minimal single agent efficacy.
ASTX660 is a potent, non-peptidomimetic, antagonist of cIAP1/2 and XIAP, discovered using fragment-
based drug design. The antagonism of XIAP and cIAP1 by ASTX660 was demonstrated on purified
proteins, cells and in vivo in xenograft models. The compound binds to the isolated BIR3 domains of
both XIAP and cIAP1 with nanomolar potencies. In cells and xenograft tissue direct antagonism of XIAP
was demonstrated by measuring its displacement from caspase-9 or SMAC. Compound-induced
proteasomal degradation of cIAP1 and 2, resulting in downstream effects of NIK stabilization and
activation of non-canonical NF-B signaling, demonstrated cIAP1/2 antagonism. Treatment with
ASTX660 led to TNF--dependent induction of apoptosis in various cancer cell lines in vitro, while dosing
in mice bearing breast and melanoma tumor xenografts inhibited tumor growth. ASTX660 is currently
being tested in a phase 1-2 clinical trial (NCT02503423) and we propose that its antagonism of cIAP1/2
and XIAP may offer improved efficacy over first-generation antagonists that are more cIAP1/2 selective.
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

3
Introduction
Evasion of apoptosis is one of the hallmarks of cancer (1) and can be achieved by overexpression of anti-
apoptotic proteins. Inhibitor of apoptosis proteins (IAPs), such as cellular IAP (cIAP) 1 and 2 and X-linked
IAP (XIAP), are key regulators of anti-apoptotic and pro-survival signaling pathways; XIAP directly inhibits
caspases, whilst cIAPs prevent the formation of pro-apoptotic signaling complexes. This leads to
suppression of apoptosis through both the extrinsic and intrinsic apoptosis pathways (2-4). Their
deregulation, through amplification, overexpression or loss of endogenous antagonists, occurs in various
cancers and is associated with tumor growth and poor prognosis, making them attractive targets for
anti-cancer therapy (5).
The IAPs are characterized by their baculovirus IAP repeat (BIR) domains which mediate protein:protein
interactions; some members of the family such as cIAP and XIAP also possess RING (Really Interesting
New Gene) zinc finger domains with E3 ubiquitin ligase activity (6,7). The anti-apoptotic activity of XIAP
is mediated by its direct binding to and inactivation of caspases 3, 7 and 9 via its BIR domains (8). IAP
antagonists such as the endogenous second mitochondria derived activator of caspases (SMAC), which is
released from mitochondria on induction of apoptosis, bind to the BIR domains of IAPs and can disrupt
interactions such as those between XIAP and caspase 9 (9). On binding to other IAPs (cIAP1 and cIAP2),
SMAC induces a conformational change, which activates their E3 ligase function leading to rapid
autoubiquitination and proteasomal degradation (10).
In response to TNF-α, cIAPs ubiquitinate RIP1 promoting the formation of complexes (e.g. complex I),
which activate survival signaling through the canonical NF-B pathway. Simultaneously, formation of
the death-inducing signaling complex (DISC), which drives apoptosis, is prevented. Antagonism and
subsequent degradation of the cIAP1/2 leads to the stabilization of NIK (NF-B-inducing kinase) which
activates the non-canonical NF-B pathway, resulting in the production of multiple cytokines including
TNF-α. Removal of the cIAP1/2 also allows the DISC to form, leading overall to a switch in TNF-α
signaling from pro-survival to pro-apoptotic (11-15). This loss of cIAP1/2 combined with release of the
XIAP-mediated block on caspases, which is essential for full activation of apoptosis, leads to a sustained
pro-apoptotic effect in the presence of TNF-α via the extrinsic apoptosis pathway. Tumors with
sufficient levels of TNF-α in their environment may, therefore, be particularly susceptible to IAP
antagonism (16). In addition, the antagonism of XIAP-mediated caspase inhibition promotes apoptosis
induced by stimulation of the intrinsic apoptosis pathway by agents such as chemotherapeutics or DNA
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

4
damaging agents (17). This suggests that cIAP1/cIAP2/XIAP antagonists can be used to promote
apoptosis through both the extrinsic and intrinsic pathways.
IAPs have been therapeutically targeted by antisense oligonucleotides and antagonist small molecules
(4). AEG35156, an antisense oligonucleotide targeted to XIAP, showed some evidence of clinical activity
(18) and sensitized cancer cells to chemotherapeutic agents and TRAIL receptor agonists in preclinical
models (3). The first generation of SMAC-mimetic IAP antagonists to enter the clinic, all containing
alanine moieties and inherently cIAP-selective, have shown some activity in preclinical models (19-22) ,
but thus far limited single agent efficacy in clinical trials (3,4,23-26).
We have previously reported the identification of lead compounds with activity against cIAP1 and XIAP
by fragment-based screening and structure-based drug design (27,28). Here we describe the discovery
and characterization of ASTX660, an antagonist of cIAP1/2 and XIAP, which is currently being tested in a
phase 1-2 clinical trial (NCT02503423). We hypothesize that such IAP antagonism may lead to improved
efficacy as a result of the more effective activation of apoptosis provided by blocking cIAP1/2 while
releasing the XIAP-block on caspases.
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

5
Materials and Methods
Materials
ASTX660 as the hydrochloride salt was synthesized using a chemical procedure similar to that used for
AT-IAP (27). The key step involved the coupling reaction between methyl-5-((R)-3-methyl-morpholin-4-
ylmethyl)-piperazine-1-carboxylic acid tert-butyl ester and 2-chloro-1-{6-[(4-fluorophenyl)methyl]-5-
(hydroxymethyl)-3,3-dimethyl-1H,2H,3H-pyrrolo[3,2-b]pyridin-1-yl}ethan-1-one (see Supplementary
Materials and Methods); purity was determined as greater than 95% by high-performance liquid
chromatography. All other reagents were purchased from Sigma (Poole, UK) unless otherwise stated.
BV-6 [(2S)-2-{[(2S)-1-[(2S)-2-cyclohexyl-2-[(2S)-2-(methylamino)propanamido]acetyl]pyrrolidin-2-
yl]formamido}-N-{6-[(2S)-2-{[(2S)-1-[(2S)-2-cyclohexyl-2-[(2S)-2-
(methylamino)propanamido]acetyl]pyrrolidin-2-yl]formamido}-3,3-diphenylpropanamido]hexyl}-3,3-
diphenylpropanamide] (14) was purchased from Selleckchem (Munich, Germany).
Protein Production and crystallography
A XIAP-BIR3 construct (amino acids 250-354) was expressed in E. coli, purified by affinity and size-
exclusion column chromatography and crystallized at approximately 10 mg/mL as described previously
(27). Crystals were soaked with 5 mM ASTX660 in 5% DMSO overnight at room temperature prior to
data collection. The crystals had cell dimensions of approximately 70 Å × 70 Å × 105 Å and belong to
space group P4122. The diffraction observed ranged from 1.7 to 3.0 Å.
Binding assays
Interaction between ASTX660 and the BIR3 domains of XIAP or cIAP1 was determined by measuring the
displacement of a fluorescent peptide tracer derived from SMAC (AbuRPFK(5&6FAM)-amide; Peptide
Synthetics Ltd, Fareham, Hampshire, UK ) by fluorescence polarization on a Pherastar plate reader (BMG
Labtech, Orttenburg, Germany). IC50 curves were generated using GraphPad Prism version 6 (La Jolla,
CA, USA) and fitted using the four parameter logistic curve fit.
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

6
Cell Lines
The human cell lines MDA-MB-231 and HEK293 were purchased from the European Collection of Cell
Cultures (ECACC) (Porton Down, UK); human melanoma cell lines, A375 and SK-MEL-28, were purchased
from American Type Culture Collection (ATCC) (Teddington, UK); and the diffuse large B-cell lymphoma
cell line, WSU-DLCL2, was purchased from DSMZ (Braunschweig, Germany). All were grown in DMEM
medium supplemented with 10% FBS and maintained at 37 oC in an atmosphere of 5% CO2 except WSU-
DLCL2 cells which were grown as above except in RPMI medium supplemented with 10% FBS. All cell
culture reagents were purchased from Invitrogen unless stated otherwise. These cells lines were not
passaged for more than 6 months (or 30 passages) after authentication by the cell bank (short tandem
repeat PCR), and were routinely screened for mycoplasma (MycoAlert, LONZA, Basel Switzerland).
Melanoma cell lines screened at ChemPartner (Shanghai, China) were purchased from ATCC, except
COLO679 (ECACC), GAK (Japanese Collection of Research Bioresources, Osaka, Japan), MMAC-SF (Riken
Cell Bank, Tsukuba, Japan) and NHDF (LONZA); and were mycoplasma screened, short tandem repeat
PCR verified, and not used beyond 10 passages.
Western blots
Cell lysate samples were resolved by SDS-PAGE and immunoblotted as described previously (27) with
antibodies specific for XIAP (AF8221), cIAP1 (AF8181) and cIAP2 (AF8171) from R&D Systems (Abingdon,
UK); and the following antibodies from Cell Signaling Technology (Hitchin, UK): cleaved caspase-3
(#9664), cleaved PARP (#9541), cleaved caspase-9 (#9505), phospho-p65 (S536) (#3033), phospho-IB
(Ser32) (#2859), total IB (#4814), NF-B1 (p105/p50) (#3035), NF-B2 (p100/p52) (#4882), SMAC
(#2954), NIK (4994) and -actin (#8457).
Immunoprecipitation with Anti-XIAP
Equivalent amounts of cell lysate were incubated overnight at 4 °C with protein A/G magnetic beads
(Pierce, Paisley, UK) coated with anti-XIAP polyclonal antibody (R&D Systems). The beads were washed
and boiled in SDS sample buffer containing DTT, before analysis of the eluted proteins by Western
blotting. Western blots of the same lysate before immunoprecipitation were used for comparison.
Antagonism of XIAP by ASTX660 was monitored by Western blotting for levels of SMAC
immunoprecipitated by XIAP.
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

7
Mesoscale Discovery Assays
An engineered HEK293 cell line (clonal isolate from stable transfection with full-length FLAG-tagged XIAP
expression construct (RC207627) and a full-length untagged caspase-9 construct (SC119362) (Origene,
Rockville, USA)) were used to detect XIAP binding to caspase-9. Cells were incubated with compound for
2 hours, lysed and lysates added to streptavidin Mesoscale Discovery (MSD) plates coated with
biotinylated anti-XIAP polyclonal antibody (R&D Systems), before washing and probing with anti-
caspase-9 or anti-XIAP antibody (Cell Signaling Technology) followed by the appropriate secondary
detection antibody.
A Mesoscale Discovery (MSD) plate-based assay was used to quantify levels of cIAP1 in MDA-MB-231
after two hour ASTX660 treatment, cells. Cells were incubated with compound for two hours, washed
and lysed. Lysates were applied to MSD plates as described previously (27).
Live cell imaging
Cells were imaged in real-time using the IncuCyte ZOOM live cell imager (Essen BioScience, Ann Arbor,
Michigan). Cells were incubated with compound in 0.1% (v/v) dimethyl sulfoxide (DMSO), with or
without neutralizing anti-TNF- antibody (R&D Systems) for 5 days and live images were taken every 3
hours using a 10× objective. IncuCyte software was used to calculate mean percent confluency from
four non-overlapping phase contrast images of each well.
Induction of apoptosis was measured over the first 24 hours by including the Essen BioScience
IncuCyte™ Caspase-3/7 Reagent at a final concentration of 2 µM in all the wells. Apoptotic cells were
identified by the appearance of green-labelled nuclei, and green fluorescence was measured in real-time
in the green FL1 channel of the IncuCyte ZOOM live cell microscope.
Apoptosis Cytometry Assays
After incubation of the cells with ASTX660 for the designated length of time, cells were harvested by
trypsinisation, spun down and 100 µl FACS buffer (PBS + 1% FBS) added. Cells were then added to a 96-
well plate and 100 µl of 2 X CellEvent reagent (Thermo, Paisley, UK) (4 µM in FACS buffer) was added.
The plate was incubated in the dark for 30 minutes before measuring fluorescent stained cells in a
Guava easyCyte HT cytometer (Millipore, Billerica, Massachusetts). Cleaved caspase-3 staining was
recorded in the FL1 channel, with unstained and DMSO control wells being used to set the gated stained
and unstained cell populations.
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

8
Cell Line Viability Screening
In-house cell viability assays were set up using alamarBlue reagent (Bio-Rad, Oxford, UK) as described
previously (27). A human melanoma cell line panel was analysed at ChemPartner (Shanghai, China) by
CellTiter-Glo luminescent cell proliferation assay (Promega, Madison, Wisconsin) after ASTX660
treatment for 72 hours in the presence or absence of 1 ng/mL TNF- (R&D Systems). Data were
normalised to 0.1% DMSO (v/v) control, and the drug response, measured as the area over the dose-
response curve (activity area), was determined for each cell line (29).
In vivo Studies
All mice were purchased from Envigo (Bicester, UK). The care and treatment of animals were in
accordance with the United Kingdom Coordinating Committee for Cancer Research guidelines and with
the United Kingdom Animals (Scientific Procedures) Act 1986 (30,31). The study protocols were
approved by the University of Cambridge Ethical Review Committee.
Initial pharmacokinetic studies were performed in male BALB/c wild type as described previously (27).
ASTX660 was either dissolved in saline and administered intravenously (IV) at 5 mg/kg in a dose volume
of 5 mL/kg or dissolved in water adjusted to pH5.5 with NaOH and administered by oral gavage (PO) at 5
to 30 mg/kg in 10 mL/kg. Blood samples were collected at various time points and plasma prepared by
centrifugation. Further pharmacokinetic and pharmacodynamic studies were performed using tumor-
bearing immunocompromised animals (see below). Tumors were excised at specific time points post
oral dose and immediately snap-frozen in liquid nitrogen before being stored at – 80 °C prior to analysis.
MDA-MB-231 xenografts were prepared by subcutaneously injecting 5x106 cells, suspended in 100 µl of
serum-free medium, into the right hind flank of male severe combined immunodeficient (SCID,
BALB/cJHan®Hsd-Prkdcscid) mice. A375 xenografts were prepared by subcutaneously injecting 5x106
cells, suspended in 100 µl of a 1:1 mixture of serum-free medium and Matrigel (approximately 10
mg/mL, Corning), into the right hind flank of male nude mice (BALB/cOlaHsd-Foxn1nu). Subcutaneous
xenograft tumors of HEK293 expressing FLAG-tagged human XIAP and caspase 9 were prepared as
previously described (27). Tumors were measured using digital calipers and volumes calculated by
applying the formula for ellipsoid.
For tumor growth inhibition studies, tumor-bearing animals were randomized into groups of 7 to 8 with
the average tumor volume of 100 mm3 (31 or 33 days after MDA-MB-231 cell injection and 19 days after
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

9
A375 injection). Mice were randomized and oral ASTX660 treatment started on Day 1. Control animals
received water. During the treatment period, tumors were measured at least twice a week and the
effect on body weight recorded daily where possible. Statistical analyses were performed using
GraphPad Prism version 6. The effects of treatments were compared using one-way ANOVA and two-
way ANOVA with Dunnett’s multiple comparisons test against vehicle control. Differences were deemed
statistically significant when P<0.05.
Analysis of Tumor Sample Pharmacodynamic Markers
Xenograft tumor lysates were prepared by grinding the frozen tissue to a fine powder with a
mortar/pestle under liquid nitrogen, and then adding ice-cold lysis buffer (1% Triton X-100, 150 mM
NaCl, 20 mM Tris·HCl pH 7.5, plus protease inhibitors (Roche, Burgess Hill, UK), 50 mM NaF and 1 mM
Na3V04), to the ground-up tumor powder. Samples were vortexed and left on ice for 30 min. Lysates
were cleared by centrifugation and samples of the supernatant removed for protein determination by
BCA assay (Pierce).
For Western blotting, equivalent amounts of protein lysate had SDS sample buffer and a final
concentration of 50 mM DTT added, before being boiled and analyzed by Western blotting as described
above.
For immunoprecipitation assay of tumor lysates, equivalent amounts of xenograft lysate were incubated
overnight at 4 °C with protein A/G magnetic beads (Pierce) coated with anti-XIAP polyclonal antibody
(R&D Systems) followed by western blot analysis. For the MSD assay, equivalent amounts of xenograft
lysate (200 µg/well) were incubated overnight at 4 °C in a streptavidin MSD plate coated with
biotinylated anti-XIAP polyclonal antibody (R&D Systems), before washing and probing with anti-SMAC,
anti-caspase-9 or anti-XIAP antibody (Cell Signaling Technology, Hitchin, UK) followed by the appropriate
secondary detection antibody.
Pharmacokinetic Analysis
Compound levels in plasma and tumor samples were measured and pharmacokinetic parameters
calculated as described previously with the exception of sample bioanalysis which was undertaken using
reverse-phase liquid chromatography-mass spectrometry (MS), using a Qtrap 4000® MS (AB Sciex, UK),
coupled to an Agilent 1200 HPLC system (Agilent, UK) or a Quattro Premier MS coupled to an Acquity
UPLC system (Waters, UK) (27).
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

10
Results
ASTX660 is a novel IAP antagonist that targets the BIR3 domain of cIAP1/2 and XIAP
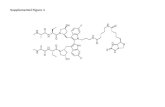
A fragment screen (28), subsequent structure-based drug design campaign (27) and further optimization
yielded ASTX660 (Figure 1A), a potent, non-peptidomimetic, orally-bioavailable, antagonist of cIAP1/2
and XIAP. ASTX660 potently inhibited the interactions between a SMAC-derived peptide and the BIR3
domains of XIAP (BIR3-XIAP) and cIAP1 (BIR3-cIAP1) with IC50 values less than 40 nM and 12 nM
respectively. The X-ray crystal structure of ASTX660 bound to BIR3-XIAP protein (PDB 5OQW) revealed
that this inhibitor binds to the surface of the protein by occupying the same 4 pockets (P1-P4) also
recognised by the N-terminal sequence of the endogenous ligand SMAC (Figure 1B) (32,33). The
piperazine ring occupies the P1 pocket, with the protonated nitrogen forming hydrogen bonds with the
side chain of Glu314 and the backbone carbonyl of Asp309. The methyl substituent is in van der Waals
contact with the side chain of Trp310, and hence efficiently fills a small lipophilic sub-pocket in P1. The
P2 pocket is occupied by the morpholine ring which stacks on the top of the central amide. The carbonyl
of the central amide forms a hydrogen bond with the backbone NH of Thr308. ASTX660 extends into the
P3 pocket with a 4-azaindoline bicycle which forms further van der Waals contacts with the side chain of
Trp323, the backbone carbonyl of Gly306 and the phenolic oxygen of the side chain of Tyr 324. Finally,
the benzylic substituent grows from C-6 of the azaindoline into P4 (Figure 1C).
ASTX660 potently antagonises XIAP in cells
Given the potent binding of ASTX660 to the isolated BIR3 domain of XIAP, we investigated the ability of
ASTX660 to antagonize the effects of XIAP in cells. Both caspases and SMAC bind to the BIR3 domain of
XIAP in cells and so should be displaced by a XIAP antagonist. We measured the displacement of
caspase-9 and SMAC in cells treated with ASTX660. To measure XIAP:caspase-9 binding we generated a
stably transfected HEK293 cell line in which full length XIAP (FLAG tagged) and caspase-9 are over-
expressed. This enabled us to observe the association of caspase-9 with XIAP. Two hours after addition
of ASTX660 to this engineered cell line, the association between XIAP and caspase 9 was potently
inhibited with an EC50 value of 2.8 nM (Figure 2A, Supplementary Table 1). To confirm the antagonism of
endogenous XIAP by ASTX660 we also investigated the displacement of SMAC from XIAP in A375,
melanoma cells. Cell lysates, treated overnight with a range of ASTX660 concentrations, were immuno-
precipitated with anti-XIAP. A clear reduction in SMAC levels immunoprecipitated with XIAP was
observed on treatment with concentrations of ASTX660 above 0.01 µM (Figure 2B). Furthermore,
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

11
exposure times as short as 5 minutes to 1 µM ASTX660 were sufficient to antagonise the interaction of
SMAC with XIAP in A375 cells (Figure 2C).
ASTX660 binds to and leads to the degradation of cIAP1/2 and stabilisation of NIK in cells
Binding of an IAP antagonist to the BIR3 domain of cIAP1/2 induces a conformational change which
activates the protein’s ubiquitin ligase activity from the C-terminal RING domain (34). This leads to rapid
auto-ubiquitination and proteasomal degradation of cIAP1/2 (14). In order to demonstrate the
antagonism of cIAPs by ASTX660, levels of cIAP1 were measured in the human breast cancer cell line,
MDA-MB-231, after treatment with ASTX660. Measurement of cIAP1 using an immunosorbent assay
MSD detection platform demonstrated that ASTX660 induced degradation of cIAP1, with an EC50 of 0.22
nM after two hours of treatment (Figure 3A, Supplementary Table 1). Further investigation
demonstrated a similar effect in A375 cells where cIAP1 levels dropped rapidly after treatment with 1
µM ASTX660 and remained significantly decreased for up to 48 hours after addition of compound
(Figure 3B). The human diffuse large B-cell lymphoma cell line, WSU-DLCL2, which has high basal cIAP2,
was used to demonstrate the effect of ASTX660 treatment on antagonism of both cIAP1 and cIAP2 over
a range of times and concentrations (Figure 3C). ASTX660 treatment of WSU-DLCL2 cells leads to
significant cIAP1 and cIAP2 degradation after 1 and 4 hours. Levels of cIAP2 were restored by 24 hours
possibly due to resistance previously observed with IAP antagonists after prolonged treatment in a high
NF-B signaling background(35). TNF--induced levels of cIAP2 are also antagonized in A375 and SK-
MEL-28 melanoma cell lines after ASTX660 treatment (see Figure 4B).
Degradation of cIAPs leads to the stabilization of NIK and activation of the non-canonical NF-B
pathway. The effects of ASTX660 treatment on NIK levels and NF-B signaling were also investigated.
On treatment of MDA-MB-231 cells with ASTX660, the rapid degradation of cIAP1 was accompanied by a
concomitant increase in the levels of NIK at 2 h which remained increased at 6 h (Figure 3D). The effect
of NIK stabilization on non-canonical NF-B signaling was further demonstrated by the depletion of NF-
B2 p100 and the increase in p52 after 24 and 48 h treatment with ASTX660. In addition, whilst
canonical NF-B signaling markers (phospho-p65 or NF-B1 (p105/p50)) remained largely unaltered,
levels of phospho-IB were rapidly increased post treatment and remained above basal levels up to 48
h after treatment (Figure 3D). Together these data suggest that ASTX660 binds to cIAP1 and 2 leading to
their degradation and downstream effects on NF-B signaling and apoptosis induction.
The mechanism by which ASTX660 induces apoptosis in cell lines is TNF--dependent
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

12
Effects of ASTX660 treatment on cell viabilityand apoptosis were investigated further in MDA-MB-231
cells. Overall viability of this cell line was reduced with an EC50 of 1.8 nM (Supplementary Table 1),
whilst cleavage of PARP and caspase-3, detected by Western blot 24 h after treatment with ASTX660,
indicated apoptosis was induced by ASTX660 treatment (Figure 3D). These effects were investigated
further by measuring cell viabilityand induction of cleaved capsase-3 in real time. Loss of viability was
observed soon after treatment with 0.1 µM ASTX660 (Figure 4A) and this was reversed on addition of a
neutralising anti-TNF-antibody, indicating that the effects of ASTX660 were TNF-α dependent and that
this cell line produces autocrine TNF- levels sufficient for this ASTX660-induced response. The loss of
viability was paralleled by an induction of apoptosis indicated by an increase in levels of cleaved
caspase-3, an effect which, again, was reversed by the addition of the neutralising anti-TNF-antibody
(Figure 4A and Supplementary Figure 1).
Induction of apoptosis was further studied in two melanoma cell lines, A375 and SK-MEL-28. Whilst
addition of 1 µM ASTX660 to these cell lines led to a degradation of both cIAP1 and cIAP2, apoptosis was
only observed in the presence of TNF- as demonstrated by an increase in the levels of cleaved PARP,
caspase-3 and caspase-9, again indicating the dependence on TNF-α (Figure 4B, Supplementary Figure
2). Activity of caspase-3, as a marker of apoptosis, was further investigated in these melanoma cell lines
by measuring the cleavage of a caspase-3 substrate using flow cytometry. Treatment with ASTX660 or
TNF- (1 ng/mL) alone showed no effect on caspase 3 activity, in either the A375 or SK-MEL-28 cells,
however when both treatments were combined the activity of caspase-3 was greatly increased at time
points up to 72 h indicating induction of apoptosis (Figure 4C).
Effects of ASTX660 treatment on cellular viability was also investigated in a wider panel of 33 melanoma
cell lines (Figure 4D). Addition of TNF-α increased or potentiated the inhibitory effect of ASTX660 for
the majority of cell lines, but not necessarily for all, suggesting that at least in these cell lines sufficient
levels of endogenous TNF-α are produced. Normal human dermal fibroblast (NHDF) cell viability was
not significantly altered by ASTX660 treatment (Figure 4D).
ASTX660 is orally bioavailable in mice and demonstrates prolonged antagonism of XIAP and cIAP1 in
vivo
The pharmacokinetic parameters of ASTX660 were evaluated following IV and PO administration to
mice. Following IV administration of ASTX660 at 5 mg/kg to male BALB/c mice, a moderate mean
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

13
clearance of 41 mL/min/kg was observed with a large volume of distribution of 2.7 L/kg. ASTX660 was
orally bioavailable (34%) following administration at 10 mg/kg to mice.
ASTX660 distributed into MDA-MB-231 (Figure 5A) tumor xenografts. ASTX660 was detectable in
tumors up to 168 h after a single oral dose of 20 mg/kg, and area under the curve (AUC)last was 129
µM∙h/mL. In contrast, plasma exposure achieved an AUClast 14.3 µM∙h/mL and concentrations were
below the limit of detection after 48h. A rapid reduction in cIAP1 levels coupled with antagonism of the
XIAP:SMAC interaction was detected from 30 min and for up to 3 days following a single dose in the
tumors in the same experiment (Figure 5B). Further experiments with Western blotting analyses
confirmed rapid cIAP1 degradation in MDA-MB-231 xenograft tumors after treatment and a
concomitant induction of apoptosis within one hour as demonstrated by an increase in cleaved PARP
and cleaved caspase-3 (Figure 5C). A decrease in association of XIAP and SMAC was also observed in
xenograft tumor tissue, again after one hour of treatment (Figure 5D), indicating antagonism of XIAP by
ASTX660. The antagonistic effects on XIAP were investigated further in a tumor model derived from
HEK293 cells genetically engineered to express XIAP and caspase-9 (Supplementary Figure 3). In this
model, a single dose of ASTX660 disrupted the interaction between XIAP and caspase-9 or SMAC for at
least 3 days. Tumor ASTX660 concentrations persisted over the duration of the study.
ASTX660 causes growth inhibition of MDA-MB-231 and A375 xenograft tumors in vivo.
The dose- and schedule-dependent anti-tumor effects of ASTX660 were investigated in the MDA-MB-
231 breast cancer xenograft model (Figures 6A and 6B, and Supplementary Figure 4A and 4B). Daily oral
administration of ASTX660 at 5, 10 and 20 mg/kg significantly inhibited tumor growth (P<0.05 from Day
15, 18 and Day 11, respectively) (Figure 6A). An intermittent schedule (cycles of 7 consecutive days of
dosing followed by 7 days of dose-holiday) at 20 mg/kg also achieved significant tumor growth inhibition
and its effects were equivalent to the continuous schedule in the same model (both P<0.05 vs vehicle
treatment from Day 8) (Figure 6B). The activity of ASTX660 was further investigated in an A375
melanoma xenograft model in nude mice (Figure 6C and Supplementary Figure 4C-i). Daily oral
administration of ASTX660 at 10 and 20 mg/kg caused significant tumor growth inhibition in an A375
model (both P<0.05 from Day 8), although in vitro this cell line was only sensitive to ASTX660 in the
presence of TNF- in vitro (Figure 4B and C).
In all three studies, the ASTX660 treatments were well-tolerated and no excessive bodyweight loss or
significant adverse effects was observed (Supplementary Figure 4A-i, B-i and C-i). It has been suggested
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

14
that antagonizing multiple IAPs may be lead to reduced tolerability in vivo (20,36). To investigate the
inflammatory effects of ASTX660, we tested the compound ex vivo on peripheral blood mononuclear
cells (PBMCs) from 2 healthy human donors. We observed no substantial evidence of increase in
secretion of pro-inflammatory cytokines (TNF-α, IL-1β, IL-2, IL-6, IL-8, IFNγ and MCP-1) in a multiplexed
cytokine assay (Meso Scale Discovery) after a 48-hour treatment with 10 µM of ASTX660 or with 10 µM
LCL161 (see Supplementary Table 2). This was in contrast to the bivalent antagonist, BV-6 (14)
(Genentech), which causes rapid cIAP1/2 depletion, potently antagonises XIAP (37), and induced
cytokine elevation in the same PBMC assays (Supplementary Table 2).
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

15
Discussion
Antagonism of IAPs is considered a promising therapeutic approach for activating apoptosis pathways in
cancer and a number of IAP antagonists have undergone evaluation in early clinical trials (38). However,
despite several observations of objective clinical response, single agent activity in the clinic has been
limited (39). The N-terminal sequence (AVPI) of the endogenous protein SMAC provided the starting
point for the first generation of peptidomimetic IAP antagonists. The AVPI peptide shows cIAP1
selectivity (~200 fold over XIAP) due to differential interactions formed by the alanine residue in the P1
pocket of the BIR3 domains of XIAP and cIAP1. We have shown that this intrinsic selectivity is retained in
the first generation of SMAC mimetics currently in the clinic (27). Given its function in inhibiting caspase
activation, it has been proposed that XIAP antagonism plays a key role in the activation of apoptosis
through both the intrinsic and extrinsic pathways (2,40). An IAP antagonist with increased XIAP potency
may lead to improved efficacy due to more effective activation of apoptosis provided by releasing the
XIAP-block on caspases [38, 45]. An increased therapeutic window may also be achieved between
efficacious dose and the onset of cytokine-induced toxicity that results from cIAP antagonism. Cytokine
release syndrome has been reported as the dose-limiting toxicity in a Phase I dose escalation study of
cIAP selective antagonist LCL161 (23) and such effects have also been described in detail in preclinical
toxicity studies with another cIAP1/2 antagonist GDC-0152 (41). A true antagonist of cIAP1/2 and XIAP
could better explore the clinical potential of IAP antagonism.
In order to develop such a compound we have applied our fragment-based drug discovery approach
(PyramidTM) to specifically identify non-alanine leads with a cIAP1-cIAP2-XIAP profile, avoiding the
intrinsic selectivity resulting from peptidomimetic approaches based on the AVPI tetrapeptide (27,28). A
subsequent lead optimization campaign focused on reducing off-target activities and improving
pharmacokinetic properties, resulting in ASTX660, a potent non-peptidic IAP antagonist, structurally
distinct from IAP antagonists previously reported. The resulting IAP antagonism of ASTX660 was
demonstrated both in vitro and in vivo. ASTX660 binds to both cIAP1 and XIAP with low-nM potency and
this inhibition translates to cells, where potent cIAP1/2 antagonism was demonstrated by their
degradation, which occurs on the induction of autoubiquitinylation upon antagonist binding; at the
same time antagonism of XIAP was measured by displacement of SMAC and casapse-9. Moreover, our
data also confirm here that ASTX660 has potent long-lasting antagonism of cIAP1 and XIAP in vivo
following oral administration accompanied by significant inhibition of xenograft tumor growth. In the
assays used here ASTX660 demonstrated potencies of 2.8 and 0.22 nM, respectively, for XIAP and cIAP1
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

16
antagonism (12.7-fold difference). In contrast, other clinical stage monovalent SMAC mimetics do not
achieve this level of potency for XIAP (range 10-35 nM) or this balance of antagonism (39-227 fold
difference) under the same conditions (see Supplementary Table 1), making ASTX660 a novel compound
for further investigating the effects of IAP antagonism.
There are several potential advantages to antagonising XIAP in addition to cIAP1/2. Aberrantly high
levels of XIAP have been linked to poor prognosis in a number of tumor types, including diffuse large B-
cell lymphoma, renal cell carcinoma, bladder cancer and colorectal cancer (42-45), whilst overexpression
of XIAP in response to chemotherapy and radiotherapy has been proposed to contribute to resistance to
these treatments (42,46,47). In addition, XIAP levels have also been implicated in modulating responses
to immunotherapy, with increased levels in Hodgkin’s lymphoma preventing cytotoxic lymphocyte-
mediated cytotoxicity by blocking granzyme B-induced apoptosis (48) and upregulation of XIAP in
inflammatory breast cancer driving resistance to antibody-dependent cell-mediated cytotoxicity (49),
further indicating potential for a molecule that potently antagonizes XIAP. Furthermore, the role of XIAP
in both extrinsic and intrinisc apoptosis suggests that a potent XIAP inhibitor, such as ASTX660, might
also be expected to show increased synergy with activators of both these apoptotic pathways such as
chemotherapeutics, radiotherapy, or TRAIL agonists compared with a more cIAP selective antagonist (2).
It has been suggested that certain bivalent compounds that potently antagonise cIAP and XIAP are not
well tolerated in vivo as they induce an excessive cytokine-mediated pro-inflammatory phenotype
(20,36). However, we have demonstrated that, unlike the bivalent compounds, ASTX660 does not elicit a
toxic cytokine signature, suggesting that the IAP antagonist properties of this compound are not a
disadvantage in this respect.
ASTX660 is currently being tested in a phase 1-2 study in subjects with advanced solid tumors and
lymphomas (NCT02503423). Further studies with this novel compound will allow the true clinical
potential of an IAP antagonist to be explored both as a single agent and in combination.
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

17
Acknowledgements
We warmly acknowledge scientific discussions with many Astex colleagues.
We also thank Anne Cleasby for critical discussions and crystallographic support.
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

18
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646-74 2. Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nature reviews
Drug discovery 2012;11:109-24 3. Dubrez L, Berthelet J, Glorian V. IAP proteins as targets for drug development in oncology.
OncoTargets and therapy 2013;9:1285-304 4. Bai L, Smith DC, Wang S. Small-molecule SMAC mimetics as new cancer therapeutics.
Pharmacology & therapeutics 2014;144:82-95 5. Hunter AM, LaCasse EC, Korneluk RG. The inhibitors of apoptosis (IAPs) as cancer targets.
Apoptosis : an international journal on programmed cell death 2007;12:1543-68 6. Deveraux QL, Reed JC. IAP family proteins--suppressors of apoptosis. Genes & development
1999;13:239-52 7. Vaux DL, Silke J. IAPs, RINGs and ubiquitylation. Nature reviews Molecular cell biology
2005;6:287-97 8. Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black
sheep of the family. EMBO Rep 2006;7:988-94 9. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-
dependent caspase activation by eliminating IAP inhibition. Cell 2000;102:33-42 10. Feltham R, Bettjeman B, Budhidarmo R, Mace PD, Shirley S, Condon SM, et al. Smac mimetics
activate the E3 ligase activity of cIAP1 protein by promoting RING domain dimerization. The Journal of biological chemistry 2011;286:17015-28
11. Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci U S A 2008;105:11778-83
12. Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Molecular cell 2008;30:689-700
13. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003;114:181-90
14. Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007;131:669-81
15. Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 2007;131:682-93
16. Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J, et al. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer cell 2007;12:445-56
17. Obexer P, Ausserlechner MJ. X-linked inhibitor of apoptosis protein - a critical death resistance regulator and therapeutic target for personalized cancer therapy. Frontiers in oncology 2014;4:197
18. Dean E, Jodrell D, Connolly K, Danson S, Jolivet J, Durkin J, et al. Phase I trial of AEG35156 administered as a 7-day and 3-day continuous intravenous infusion in patients with advanced refractory cancer. J Clin Oncol 2009;27:1660-6
19. Cai Q, Sun H, Peng Y, Lu J, Nikolovska-Coleska Z, McEachern D, et al. A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. Journal of medicinal chemistry 2011;54:2714-26
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

19
20. Condon SM, Mitsuuchi Y, Deng Y, LaPorte MG, Rippin SR, Haimowitz T, et al. Birinapant, a smac-mimetic with improved tolerability for the treatment of solid tumors and hematological malignancies. Journal of medicinal chemistry 2014;57:3666-77
21. Flygare JA, Beresini M, Budha N, Chan H, Chan IT, Cheeti S, et al. Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidate for the treatment of cancer (GDC-0152). Journal of medicinal chemistry 2012;55:4101-13
22. Hennessy EJ, Adam A, Aquila BM, Castriotta LM, Cook D, Hattersley M, et al. Discovery of a novel class of dimeric Smac mimetics as potent IAP antagonists resulting in a clinical candidate for the treatment of cancer (AZD5582). Journal of medicinal chemistry 2013;56:9897-919
23. Infante JR, Dees EC, Olszanski AJ, Dhuria SV, Sen S, Cameron S, et al. Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2014;32:3103-10
24. DiPersio JF, Erba HP, Larson RA, Luger SM, Tallman MS, Brill JM, et al. Oral Debio1143 (AT406), an antagonist of inhibitor of apoptosis proteins, combined with daunorubicin and cytarabine in patients with poor-risk acute myeloid leukemia--results of a phase I dose-escalation study. Clinical lymphoma, myeloma & leukemia 2015;15:443-9
25. Amaravadi RK, Schilder RJ, Martin LP, Levin M, Graham MA, Weng DE, et al. A Phase I Study of the SMAC-Mimetic Birinapant in Adults with Refractory Solid Tumors or Lymphoma. Molecular cancer therapeutics 2015;14:2569-75
26. Noonan AM, Bunch KP, Chen JQ, Herrmann MA, Lee JM, Kohn EC, et al. Pharmacodynamic markers and clinical results from the phase 2 study of the SMAC mimetic birinapant in women with relapsed platinum-resistant or -refractory epithelial ovarian cancer. Cancer 2016;122:588-97
27. Tamanini E, Buck IM, Chessari G, Chiarparin E, Day JEH, Frederickson M, et al. Discovery of a Potent Nonpeptidomimetic, Small-Molecule Antagonist of Cellular Inhibitor of Apoptosis Protein 1 (cIAP1) and X-Linked Inhibitor of Apoptosis Protein (XIAP). J Med Chem 2017;60:4611-25
28. Chessari G, Buck IM, Day JE, Day PJ, Iqbal A, Johnson CN, et al. Fragment-Based Drug Discovery Targeting Inhibitor of Apoptosis Proteins: Discovery of a Non-Alanine Lead Series with Dual Activity Against cIAP1 and XIAP. Journal of medicinal chemistry 2015;58:6574-88
29. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603-7
30. Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer 2010;102:1555-77
31. Hollands C. The Animals (scientific procedures) Act 1986. Lancet 1986;2:32-3 32. Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, et al. Structural basis for binding of
Smac/DIABLO to the XIAP BIR3 domain. Nature 2000;408:1004-8 33. Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, et al. Structural basis of IAP recognition by
Smac/DIABLO. Nature 2000;408:1008-12 34. Dueber EC, Schoeffler AJ, Lingel A, Elliott JM, Fedorova AV, Giannetti AM, et al. Antagonists
induce a conformational change in cIAP1 that promotes autoubiquitination. Science (New York, NY) 2011;334:376-80
35. Petersen SL, Peyton M, Minna JD, Wang X. Overcoming cancer cell resistance to Smac mimetic induced apoptosis by modulating cIAP-2 expression. Proceedings of the National Academy of Sciences of the United States of America 2010;107:11936-41
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

20
36. Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D'Cruz AA, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nature communications 2015;6:6282
37. Muller-Sienerth N, Dietz L, Holtz P, Kapp M, Grigoleit GU, Schmuck C, et al. SMAC mimetic BV6 induces cell death in monocytes and maturation of monocyte-derived dendritic cells. PloS one 2011;6:e21556
38. Derakhshan A, Chen Z, Van Waes C. Therapeutic Small Molecules Target Inhibitor of Apoptosis Proteins in Cancers with Deregulation of Extrinsic and Intrinsic Cell Death Pathways. Clin Cancer Res 2017;23:1379-87
39. Fulda S. Promises and Challenges of Smac Mimetics as Cancer Therapeutics. Clinical cancer research : an official journal of the American Association for Cancer Research 2015;21:5030-6
40. Ndubaku C, Varfolomeev E, Wang L, Zobel K, Lau K, Elliott LO, et al. Antagonism of c-IAP and XIAP proteins is required for efficient induction of cell death by small-molecule IAP antagonists. ACS chemical biology 2009;4:557-66
41. Erickson RI, Tarrant J, Cain G, Lewin-Koh SC, Dybdal N, Wong H, et al. Toxicity profile of small-molecule IAP antagonist GDC-0152 is linked to TNF-alpha pharmacology. Toxicol Sci 2013;131:247-58
42. Hussain AR, Uddin S, Ahmed M, Bu R, Ahmed SO, Abubaker J, et al. Prognostic significance of XIAP expression in DLBCL and effect of its inhibition on AKT signalling. The Journal of Pathology 2010;222:180-90
43. Ramp U, Krieg T, Caliskan E, Mahotka C, Ebert T, Willers R, et al. XIAP expression is an independent prognostic marker in clear-cell renal carcinomas. Human pathology 2004;35:1022-8
44. Li M, Song T, Yin ZF, Na YQ. XIAP as a prognostic marker of early recurrence of nonmuscular invasive bladder cancer. Chinese medical journal 2007;120:469-73
45. Xiang G, Wen X, Wang H, Chen K, Liu H. Expression of X-linked inhibitor of apoptosis protein in human colorectal cancer and its correlation with prognosis. Journal of surgical oncology 2009;100:708-12
46. Xiong Z, Fu Z, Shi J, Jiang X, Wan H. HtrA1 Down-regulation Induces Cisplatin Resistance in Colon Cancer by Increasing XIAP and Activating PI3K/Akt Pathway. Annals of clinical and laboratory science 2017;47:264-70
47. Holcik M, Yeh C, Korneluk RG, Chow T. Translational upregulation of X-linked inhibitor of apoptosis (XIAP) increases resistance to radiation induced cell death. Oncogene 2000;19:4174-7
48. Kashkar H SJ, Hombach A, Deggerich A, Yazdanpanah B, Utermöhlen O, Heimlich G, Abken H, Krönke M. XIAP targeting sensitizes Hodgkin lymphoma cells for cytolytic T-cell attack. Blood 2006;108:3434-40
49. Ravi R FE, Jain A, Pham V, Yoshimura K, Prouser T, Jalla S, Zhou X, Garrett-Mayer E, Kaufmann SH, Schulick RD, Pardoll DM, Bedi A. Resistance of cancers to immunologic cytotoxicity and adoptive immunotherapy via X-linked inhibitor of apoptosis protein expression and coexisting defects in mitochondrial death signaling. Cancer research 2006;108:1730-9
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

21
Figure Legends
Figure 1
ASTX660 is a novel antagonist of cIAP1/2 and XIAP. A, Chemical structure of non-peptidomimetic
cIAP1/2 and XIAP antagonist, ASTX660, derived by fragment-based drug discovery. B & C, X-ray crystal
structure of ASTX660 (in green) in complex with XIAP-BIR3 (PDB 5OQW). The Connolly surface of the
protein in B is colored by electrostatic potential (red = negative, blue = positive, grey = neutral).
Hydrogen bonds between ligand and protein in C are shown as dashed red lines.
Figure 2
ASTX660 treatment leads to potent antagonism of XIAP in cells. A, An engineered HEK293 cell line
overexpressing XIAP and caspase-9, was treated for 2 hours with indicated concentrations of ASTX660.
XIAP:caspase-9 binding was measured by immunoprecipitation of FLAG-tagged XIAP and quantitation of
XIAP-associated caspase-9 using an MSD plate-based assay. Results show the mean of duplicate values.
B, A375 cells were treated for 16 h with the indicated concentrations of ASTX660. Endogenous XIAP
antagonism was measured by Western blotting cell lysates for SMAC levels bound to XIAP following
immunoprecipitation with anti-XIAP (upper panel). Total XIAP and SMAC levels were determined by
Western blots of cell lysates before immunoprecipitation (lower panel). C, A375 cell lysates were treated
with 1 µM ASTX660 for the indicated times. XIAP antagonism was measured by Western blotting for
SMAC levels after immunoprecipitation with anti-XIAP (upper panel). Total XIAP and SMAC levels were
determined by Western blots of cell lysates before immunoprecipitation (lower panel).
Figure 3
Antagonism of cIAP1 by ASTX660 leads to its degradation in cancer cells. A, MDA-MB-231 cells were
treated with the indicated concentrations of ASTX660 for 2h, lysed and relative levels of cIAP1 measured
using a Meso Scale Discovery plate-based assay. Results show the mean of duplicate values. B, A375
cells were treated with 1 µM ASTX660 for the indicated times and cIAP1 or XIAP levels analyzed by SDS-
PAGE followed by immunoblotting. -actin was used as an internal control. C, WSU-DLCL2 cells were
treated with the indicated concentrations of ASTX660 for the indicated times. Levels of cIAP1 and cIAP2
were analysed by SDS-PAGE followed by immunoblotting. D, MDA-MB-231 cells were treated with the
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

22
indicated concentrations of ASTX660 for 2, 6, 24 and 48h, lysed and equal amounts of total protein were
then analysed by SDS-PAGE followed by immunoblotting with the indicated antibodies.
Figure 4
TNF- triggers ASTX660-induced apoptosis in cell lines. A, Viability (upper panel) and apoptosis (lower
panel) were measured in breast cancer MDA-MB-231 cells after treatment with 0.1 µM ASTX660 using
IncuCyte ZOOM live cell imaging. Anti-TNF- antibody at concentrations of 1 or 10 µg/mL was added to
neutralize TNF- in the cell supernatant in order to monitor effects on the cell survival. B, Melanoma
cell lines, A375 and SK-MEL-28, were treated with 1 µM ASTX660 plus or minus 1 ng/mL TNF- for 24 h.
Cells were then lysed, and equal amounts of total protein were analysed by SDS-PAGE followed by
immunoblotting with the indicated antibodies. C, A375 or SK-MEL-28 cells were treated with 1 µM
ASTX660 for 25 h in the presence or absence of 1 ng/mL TNF-. Cleaved caspase-3 activity was
measured by cytometry. D, Effect of 72 h ASTX660 treatment on the viability of 33 melanoma cell lines
(plus normal human dermal fibroblast (NHDF) cells (*), included as a control). Data generated by
CellTiterGlo in triplicate, in the presence or absence of 1 ng/mL TNF- were normalised to 0.1% DMSO
(v/v) control, and the drug response, measured as the area over the dose-response curve (activity area),
was determined for each cell line (29).
Figure 5
Orally administered ASTX660 treatment modulates pharmacodynamic markers in a MDA-MB-231
xenograft model. A, SCID mice bearing MDA-MB-231 xenograft were treated with a single 20 mg/kg
dose of ASTX660 and compound concentrations in plasma or tumor samples analysed. B, In the same
tumor samples, levels of cIAP1 and XIAP:SMAC association were determined by MSD assay. C, Mice
bearing MDA-MB-231 xenograft tumors received a single oral dose of ASTX660 at 30 mg/kg. Animals
were sacrificed at the indicated time points and protein levels in tumors were measured by
immunoblotting of whole cell lysates. D, In the same tumor lysates, XIAP:SMAC association was analyzed
by anti-XIAP immunoprecipitation. Each sample represents individual animals. MSD assay values
represent mean ± SEM from 3 animals.
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

23
Figure 6
Orally administered ASTX660 inhibits growth of MDA-MB-231 and A375 xenograft tumors. Each data
point represents mean ± SEM from 8 animals unless otherwise indicated. A, ASTX660 was orally
administered to SCID mice, bearing MDA-MB-231 xenografts, at 20, 10, and 5 mg/kg once daily for 25
days. Control animals received water. B, Comparison of the effects of varying ASTX660 administration
schedule on MDA-MB-231 tumors. One group of tumor-bearing animals was treated with ASTX660 for
28 consecutive days (q.d., N=7) and another was given 2 cycles of 7 consecutive days of dosing followed
by 7 days of dose-holiday (q.d. x 7/ no dose x 7). On dosing days, both treatment groups received
ASTX660 once daily at 20 mg/kg. The unconnected symbols represent the dosing events (red square:
q.d. and blue triangle: q.d. x 7/ no dose x 7). The control group received water as vehicle once daily for
25 consecutive days. C, A375-bearing nude mice were orally treated with 10 or 20 mg/kg of ASTX660 for
14 consecutive days.
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

Figure 1
P1
P2
P3
P4
Glu314
Asp309
Trp310
Trp323
Thr308
Tyr324
A B C
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

Figure 2
A B
C 0 5 10 15 20 25 30 40 50 60 120 240 Time (min)
XIAP
b-actin
SMAC
XIAP
SMAC
Cell Lysates
IP Anti-XIAP
0 5 10 15 20 25 30 40 50 60 120 240 Time (min)
IP Anti-XIAP
CellLysates
XIAP
SMAC
XIAP
SMAC
b-actin
0 0.001 0.01 0.1 1 10 [ASTX660], µM
0 0.001 0.01 0.1 1 10 [ASTX660], µM
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

A
C
Figure 3
B
D
0 5 10 15 20 25 30 40 50 60 120 240 Time (min)
cIAP1
XIAP
b-actin
cIAP2
cIAP1
b-actin
10 1 0.1 0.01 0.001 0 10 1 0.1 0.01 0.001 0 10 1 0.1 0.01 0.001 0 [ASTX660], µM
1 h 4 h 24 h
NIK
NF-kB2
p100
p52
cIAP1
cleaved PARP
cleaved caspase-3
phospho-IkBa[Ser32]
XIAP
b-actin
DMSO 0.001 0.01 0.1 1 10 DMSO 0.001 0.01 0.1 1 10 DMSO 0.001 0.01 0.1 1 10 DMSO 0.001 0.01 0.1 1 10 [ASTX660], µM2 h 6 h 24 h 48 h
phospho-p65[Ser536]
NF-kB1
p105
p50
total IkBa
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

A B
Figure 4
D
A375
DMSO: + - - - + - - -
TNF-a: - + - + - + - +
ASTX660: - - + + - - + +
SK-MEL-28
CleavedCaspase-9
CleavedCaspase-3
cIAP1
cIAP2
XIAP
Cleaved PARP
b-Actin
0
10
20
30
40
50
60
-4 -3 -2 -1 0 1 2
% C
ell
sC
leav
ed
Cas
pas
e-3
Po
siti
ve
Log 10 [ASTX660] (µM)
Control+ 1 ng/ml TNF-α
0
10
20
30
40
50
60
-4 -3 -2 -1 0 1 2
% C
ell
sC
leav
ed
Cas
pas
e-3
Po
siti
ve
Log 10 [ASTX660] (µM)
Control
+ 1 ng/ml TNF-α
C
0
1
2
3
4
5
6
7
8
A1
01
D
Hs2
94
T
A2
05
8
RP
MI-
79
51
SK-M
EL-2
8
A3
75
HT-
14
4
SK-M
EL-5
Mal
me
-3M
Hs6
95
T
Hs8
52
T
SH-4
Hs9
40
T
WM
-11
5
SK-M
EL-2
G-3
61
SK-M
EL-1
CO
LO8
29
GA
K
Me
Wo
SK-M
EL-3
1
Hs8
95
T
SK-M
EL-3
IGR
-1
Hs8
39
T
CH
L-1
WM
-26
6-4
C3
2
CO
LO6
79
HM
CB
Hs6
88
(A)T
MM
AC
-SF
SK-M
EL-2
4
NH
DF
Re
lati
ve A
nti
-Pro
life
rati
ve A
ctiv
ity
(Act
ivit
y A
rea)
ASTX660
ASTX660 + TNF-α
*
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

Figure 7: PD – MDA-MB-231 xenografts A
B D
C
Figure 5
1
10
100
1000
10000
0 24 48 72 96 120 144 168
Me
an A
STX
66
0 C
on
cen
trat
ion
(n
M)
Time post single dose (h)
Plasma
Tumor
0
10
20
30
40
50
60
70
80
90
100
110
0
10
20
30
40
50
60
70
80
90
100
110
0 24 48 72 96 120 144 168
cIA
P1
Le
vel
(%
ve
hic
le C
on
tro
l)
XIA
P:S
MA
C B
ind
ing
(%
Ve
hic
le C
on
tro
l)
Time (h)
XIAP:SMAC (IP)
cIAP1 Level
6 h 24 hVehicle Control
cIAP1
Cleaved PARP
Cleaved Caspase-3
1 h
XIAP
b-Actin
SMAC
SMAC
XIAP
6 h 24 hVehicle Control 1 h
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

A
Figure 6
B
0
100
200
300
400
500
600
0 7 14 21 28
Tum
or
volu
me
(m
m3)
Day
Vehicle Control
20 mg/kg ASTX660 q.d.
20 mg/kg ASTX660 q.d.x 7/no dose x 7
0
100
200
300
400
500
600
700
0 7 14 21 28
Tum
or
volu
me
(m
m3)
Day
Vehicle Control20 mg/kg ASTX66010 mg/kg ASTX6605 mg/kg ASTX660
0
100
200
300
400
500
600
700
800
0 7 14
Tum
or
volu
me
(m
m3)
Day
Vehicle Control
20 mg/kg ASTX660
10 mg/kg ASTX660
C
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848

Published OnlineFirst April 25, 2018.Mol Cancer Ther George A. Ward, Edward J Lewis, Jong Sook Ahn, et al. cancer cell lines and inhibits tumor growth
dependent apoptosis inαand XIAP, potently induces TNF-ASTX660, a novel non-peptidomimetic antagonist of cIAP1/2
Updated version
10.1158/1535-7163.MCT-17-0848doi:
Access the most recent version of this article at:
Material
Supplementary
http://mct.aacrjournals.org/content/suppl/2018/04/25/1535-7163.MCT-17-0848.DC1
Access the most recent supplemental material at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mct.aacrjournals.org/content/early/2018/04/25/1535-7163.MCT-17-0848To request permission to re-use all or part of this article, use this link
on March 29, 2020. © 2018 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 25, 2018; DOI: 10.1158/1535-7163.MCT-17-0848