Artículos destacados · 2014-02-07 · Una dieta rica en colesterol exacerba la fibrosis hepática...
68
Pancreatitis Paradigma de múltiples facetas Paradigma tripsina Tripsógeno Tripsina Autodigestión Factores etiológicos Tripsina NG-κG Ras Estrés ER ??? Inflamasomas en la inflamación y el cáncer intestinal Los fármacos antiinflamatorios no esteroides y las estatinas tienen efectos quimiopreventivos Eficacia del inhibidor de proteasa BI 201335, el inhibidor de polimerasa BI 207127 y la ribavirina Una dieta rica en colesterol exacerba la fibrosis hepática Artículos destacados www.gastrojournal.org Vl 1 Nú 2 M Ab il 2012 Volumen 1 Número 2 . Marzo-Abril 2012
Transcript of Artículos destacados · 2014-02-07 · Una dieta rica en colesterol exacerba la fibrosis hepática...
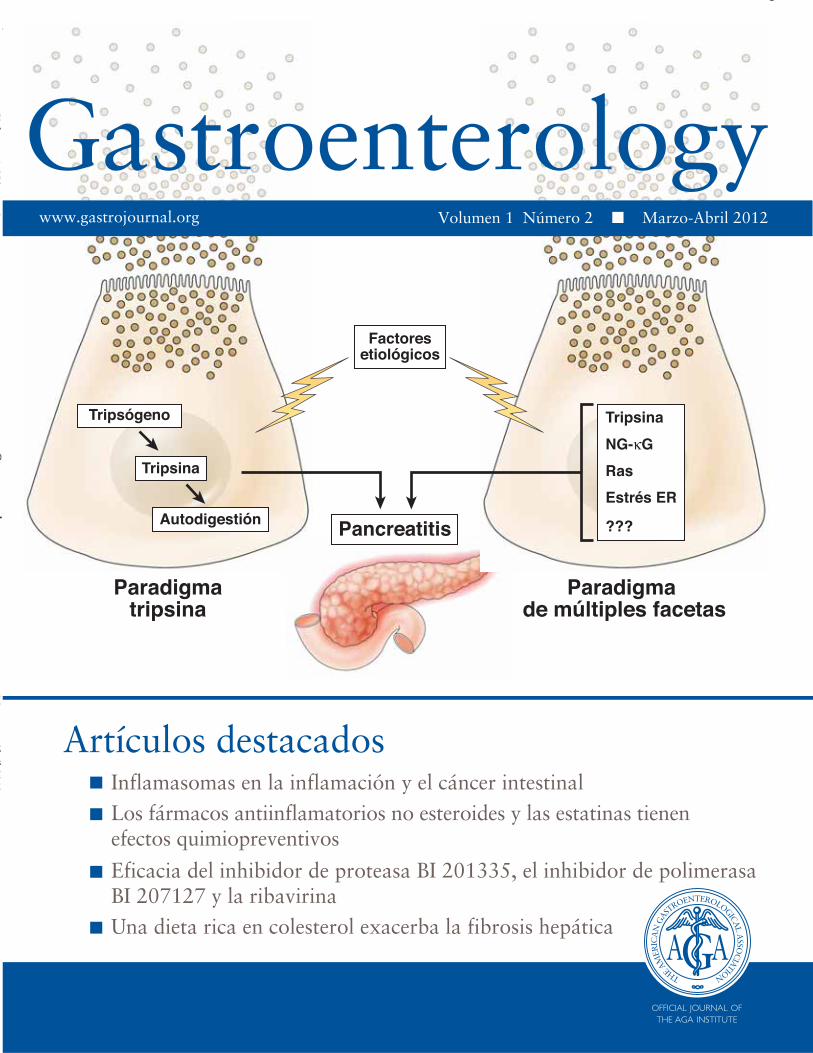
Pancreatitis
Paradigma de múltiples facetas
Paradigmatripsina
Tripsógeno
Tripsina
Autodigestión
Factoresetiológicos
Tripsina
NG-κG
Ras
Estrés ER
???
Inflamasomas en la inflamación y el cáncer intestinalLos fármacos antiinflamatorios no esteroides y las estatinas tienen efectos quimiopreventivos
Eficacia del inhibidor de proteasa BI 201335, el inhibidor de polimerasaBI 207127 y la ribavirinaUna dieta rica en colesterol exacerba la fibrosis hepática
Artículos destacados
www.gastrojournal.org
Vl
1N
ú2
MA
bil2012
Volumen 1 Número 2 . Marzo-Abril 2012
Portada Gastr_astra.pdf 1 3/16/12 11:10 AM


CO
NTEN
IDO
Gastroenterology
Edición MexicanaComité EditorialDr. Arturo Ballesteros AmozurrutiaDr. Enrique Wolpert Barraza (Coordinador)Dr. Francisco Javier Bosques PadillaDr. Luis Federico Uscanga DomínguezDr. José Ramón Nogueira de RojasDr. Juan Miguel Abdo Francis
Editor AsociadoDr. Óscar T. Teramoto Matsubara
Traducción Dr. Jorge Ramírez Peredo
Cuidado de la edición Lic. Cristina Segura Flores
Formación DCG. Marco A.M. Nava
Esta edición ha sido traducida, adaptada y producida en México
por Intersistemas, S. A. de C.V.
Política Editorial© COPYRIGHT 2012 por la American Gastroenterological Association. Reservados todos los derechos. Ninguna parte de esta publicación puede ser reproduci-da, almacenada en un sistema de recuperación o transmitida en ninguna forma o por ningún medio —electrónico, óptico, mecánico, fotocopiado o de cualquier clase— sin permiso escrito de la American Gastroenterological Association. Los artículos de Gastroenterology® en Español aparecieron publicados original-mente en inglés en Gastroenterology® por la American Gastroenterological Association (AGA), y son publicados en español por Intersistemas, S.A. de C.V.
Deslinde de responsabilidad: las afirmaciones y opiniones expresadas en cualquier artículo son del autor únicamente y no necesariamente de la revista, los editores, el Instituto AGA o de la editorial a menos que se especifique como tal. El Instituto AGA otorgó autorización al editor para producir esta publicación. El Instituto AGA no tuvo ningún papel en la selección de cuáles artículos serían resumidos y no es responsable de la integridad de los artículos. Se aconseja al lector referirse a la literatura médica apropiada, incluir el original que apareció en Gastroenterology®, y la información del producto proporcionada actualmente por el fabricante de cada medicamento que se va a administrar para verificar la dosis, el método y la duración de la administración y las contraindicaciones. Es responsabilidad del médico tratante o de otros profesionales de la salud, de acuerdo con la experiencia independiente y el conocimiento de su paciente, determinar las dosis de los medicamentos y el mejor tratamiento para el paciente. La publicación de un anuncio o de alguna otra mención del producto en la revista no debe tomarse como un aval del producto o de las afirmaciones del fabricante. Ni el Instituto AGA ni la editorial asumen ninguna responsabilidad por alguna lesión y/o daño a personas o propiedades que derive del uso relacionado con el material contenido en esta revista.
Ni la AGA ni Intersistemas, S.A. de C.V. se hacen responsables de cualquier inexactitud o error en el contenido de estos artículos, incluyendo cualquier imprecisión o error de traducción del idioma inglés al español. Además, ni la AGA ni Intersistemas, S.A. de C.V. respaldan el uso de, o garantizan (directa o indirectamente) la calidad o la eficacia de cualquier producto o servicio descrito en los anuncios u otro material de naturaleza comercial en Gastroenterology® en Español. El anunciante no tiene control sobre el contenido editorial en esta versión traducida. La AGA no respalda los anuncios adjuntos al contenido editorial.
Gastroenterology® (ISSN 0016-5085). Marca Registrada. Año 1 No. 2. Fecha de publicación: Marzo-Abril 2012. Revista bimestral, editada y publicada por Intersistemas, S.A. de C.V. Toda correspondencia deberá dirigirse a Aguiar y Seijas No. 75, C.P. 11000. Col. Lomas de Chapultepec, México, D.F. Tel. (5255) 5520-20-73; Fax (5255) 5540-37-64. Correo electrónico: [email protected]; Editor responsable: Pedro Vera Cervera, miembro de la Cámara Nacional de la Industria Editorial Mexicana. Número de Certificado de Reserva de derechos al uso exclusivo del título Gastroenterology®: En trámite, ante el Instituto Nacional del Derecho de Autor. Certificado de Licitud de Título y contenido: En trámite ante la Comisión Calificadora de Publicaciones y revistas ilustradas. Distribuidor exclusivo en México: Intersistemas, S.A. de C.V. Esta publicación consta de un tiro de 5 000 ejemplares e impresa en: Impresiones Editoriales FT ubicada en Calle 31 de julio de 1859, Col. Leyes de Reforma, México, D.F. INTERSISTEMAS, S.A. DE C.V. investiga sobre la seriedad de sus anunciantes, pero no se responsabiliza por la calidad o los beneficios de los productos o servicios de los mismos. Prohibida su reproducción parcial o total.
IMPRESA EN MÉXICO - PRINTED IN MEXICO.TODOS LOS DERECHOS RESERVADOS - ALL RIGHTS RESERVED.© Copyright 2012.
Volumen 1
No. 2
Marzo - abril 2012
EDITORIAL 4 Esquemas de tratamiento sin interferón
para la hepatitis C: ¿todavía no estamos ahí? Anna S. Lok
7 Los lípidos en la enfermedad hepática: una visión más allá de la esteatosis
Robert F. Schwabe y Jacquelyn J. Maher
REVISIONES DE GASTROENTEROLOGÍA Y HEPATOLOGÍA BÁSICAS Y CLÍNICAS11 Inflamasomas en la inflamación y el cáncer
intestinal Grace Y, Chen y Gabriel Núñez
CLÍNICA — TRACTO ALIMENTARIO25 Los fármacos antiinflamatorios no esteroi-
des y las estatinas tienen efectos quimiopre-ventivos en pacientes con esófago de Barrett
Florine Kastelein, Manon C. W. Spaander, Katharina Atha-rina Biermann, Ewout W. Steyerberg, Ernest J. Kuiprs, y Marco J. Bruno en nombre del Grupo de Estudio Probar
CLÍNICA — HÍGADO34 Eficacia del inhibidor de proteasa BI 201335,
el inhibidor de polimerasa BI 207127 y la ribavirina en pacientes con infección crónica por VHC
Stefan Zeumen, Tarik Asselah, Peter Angus, Jean-Pierre Zarski, Donique Larrey, Beat Müllhaupt, Ed Gane, Mar-cus Schuchmann, Ansgar Lohse, Stanislas Pol, Jean-Pierre Bronowicki, Stuart Roberts, Keikawus Arasteh, Fabien Zoulim, Markus Heim, Jerry O. Stern, George Kukolj, Ger-hard Nehmiz, Carla Haefner, y Wulf Otto Boecher
43 Una dieta rica en colesterol exacerba la fi-brosis hepática en ratones a través de la acu-mulación de colesterol libre en las células estelares hepáticas
Toshiaki Teratani, Kengo Tomita, Takahiro Suzuki, Tetsuya Os-hikawa, Hirokazu Yokoyama, Katsuyoshi Shimamura, Susumu Tominaga, Sadayuki Hiroi, Rie Irie, Yoshikiyo Okada, Chie Kuri-hara, Hirotoshi Ebinuma, Hidetsugu Saito, Ryota Hokari, Kazuo Sugiyama, Takanori Kanai,* Soichiro Miura, y Toshifumi Hibi
EVALUACIÓN63 Instructivo y cuestionario
preliminar gastro V1N2 1 3/14/12 11:26 AM

GastroenterologyEditor
M. Bishr Omary University of Michigan Medical School
Senior Associate Editors
John M. Carethers University of Michigan Medical School
Anna S. Lok University of Michigan Medical School
Chung Owyang University of Michigan Medical School
Associate Editors
Jonathan Braun University of California, Los Angeles
Douglas A. Corley Kaiser Permanente, Division of Research
William L. Hasler University of Michigan Medical School
David Lieberman Oregon Health and Science University
Malcolm J. Low University of Michigan Medical School
Jean-Michel Pawlotsky University of Paris–Est
Linda C. Samuelson University of Michigan Medical School
Bruce E. Sands Mount Sinai School of Medicine
Detlef Schuppan Johannes Gutenberg, University of Mainz
Diane M. Simeone University of Michigan Medical School
Jerrold R. Turner The University of Chicago
John A. Williams University of Michigan Medical School
Online Editor
John F. Kuemmerle Medical College of Virginia Campus
Virginia Commonwealth University
Special Section Editors
Covering the Cover
Anson W. Lowe Stanford University
Richard H. Moseley University of Michigan Medical School
Press Highlights
Grace L. Su University of Michigan Medical School
Mentoring, Education, and
Training Corner
John Del Valle University of Michigan Medical School
Clinical Challenges and Images
in GI
Grace Elta University of Michigan Medical School
Robert J. Fontana University of Michigan Medical School
Imaging and Advanced
Technology
Ralf Kiesslich Johannes Gutenberg, University of
Mainz
Thomas D. Wang University of Michigan Medical School
Selected Summaries
Philip S. SchoenfeldUniversity of Michigan Medical School
Print and Digital Media Reviews
Joel Rubenstein University of Michigan Medical School
Reviews in Basic and Clinical
Gastroenterology and Hepatology
Robert F. Schwabe Columbia University
John W. Wiley University of Michigan Medical School
Biostatistical Editors
Julie A. Douglas University of Michigan Medical School
Cathie Spino University of Michigan School of Public
Health
Advisory Committee
Juanita L. Merchant University of Michigan Medical School
Daniel K. Podolsky University of Texas Southwestern
Robert S. Sandler University of North Carolina
International Consultants
Minoti Apte Sydney, Australia
Ding-Shinn Chen Taipei, Taiwan
Toshifumi Hibi Tokyo, Japan
Jesús Prieto Pamplona, Spain
Jiaming Qian Beijing, China
Editors EmeritiAnil K. Rustgi 2006–2011
David A. Brenner 2001–2006
Daniel K. Podolsky 1996–2001
Nicholas F. LaRusso 1991–1996
Raj K. Goyal 1986–1991
Robert K. Ockner 1981–1986
John S. Fordtran 1977–1981
Robert M. Donaldson 1970–1977
Marvin H. Sleisenger 1965–1970
Morton I. Grossman 1959–1965
Abraham H. Aaron 1953–1959
Andrew C. Ivy 1950–1952
Walter C. Alvarez 1943–1950
Editorial Staff
Erin Dubnansky Senior Director of Scholarly Publishing
Christopher Lowe Assistant Managing Editor
Sarah Williamson Senior Medical Illustrator
Kristine Novak Science Editor
Lindsey Brounstein Publications and Graphics Manager
Laura Claus Senior Publications Coordinator
Jennifer Tarr Publications Coordinator
Linda Busha Editorial Assistant
Jillian Schweizter Editorial Assistant
Offices of the AGA Institute
President
C. Richard Boland Dallas, TX
President-Elect
Loren A. Laine Los Angeles, CA
Vice President
Anil K. Rustgi
Philadelphia, PA
Secretary/Treasurer
J. Sumner Bell, III
Virginia Beach, VA
Editorial Board
ChairAnil Rustgi Philadelphia, PA
Editorial Board
Maria T. Abreu Miami, FLDavid H. Adams Birmingham, United Kingdom Hana Algül Munich, GermanyShrikant Anant Kansas City, KSPaul Angulo Lexington, KYOlivier Barbier Québec, CanadaKim E. Barrett San Diego, CATerrence A. Barrett Chicago, ILRalf Bartenschlager Heidelberg, Germany Charles N. Bernstein Winnipeg, ManitobaRichard S. Blumberg Boston, MAJames L. Boyer New Haven, CT William R. Brugge Boston, MAMarkus W. Büchler Heidelberg, GermanyMichael Camilleri Rochester, MN Marcia Irene Canto Baltimore, MDElke Cario Essen, GermanyNaga Chalasani Indianapolis, INAndrew T. Chan Boston, MALin Chang Los Angeles, CATsutomu Chiba Kyoto, Japan Judy H. Cho New Haven, CTDaniel C. Chung Boston, MAPierre Clavien Zurich, SwitzerlandHans Clevers Utrecht, The NetherlandsNicholas O. Davidson St. Louis, MOLee A. DensonCincinnati, OH Mark Donowitz Baltimore, MDPradeep K. Dudeja Chicago, ILEmad M. El-Omar Aberdeen, ScotlandWael M. El-Rifai Nashville, TNMary K. Estes Houston, TXJames E. Everhart Bethesda, MDBrian G. Feagan London, OntarioEric R. Fearon Ann Arbor, MIScott L. Friedman New York, NYBin Gao Rockville, MDGuadalupe Garcia-Tsao New Haven, CTMichael D. Gershon New York, NYM. Eric Gershwin Davis, CAFayez Ghishan Tucson, AZRavinder K. Gill Chicago, IL
Jeffrey S. Glenn Los Angeles, CAAjay Goel Dallas, TXJames R. Goldenring Nashville, TNFrank J. Gonzalez Bethesda, MDFred Gorelick West Haven, CTThomas Gress Marburg, GermanyAnna S. Gukovskaya Los Angeles, CAAida Habtezion Stanford, CASteven-Huy Han Los Angeles, CAMatthias Hebrok San Francisco, CACourtney W. HouchenOklahoma City, OKJeanMarie Houghton Worcester, MAKenneth E. Hung Boston, MAKenichi Ikejima Tokyo, JapanJohn M. Inadomi Seattle, WABarbara Jung Chicago, ILPeter J. Kahrilas Chicago, ILFasiha Kanwal St. Louis, MOJohn Y. Kao Ann Arbor, MIMichael Karin San Diego, CAJonathan P. Katz Philadelphia, PAJonathan D. Kaunitz Los Angeles, CAKenneth W. Kinzler Baltimore, MDErnst J. Kuipers Rotterdam, The NetherlandsAndrew B. Leiter Worcester, MAFrederic Lemaigre Brussels, BelgiumWayne I. Lencer Boston, MAMarkus M. Lerch Greifswald, GermanyT. Jake Liang Bethesda, MDShelly C. Lu Los Angeles, CAPatrick Lynch Houston, TXFabio Marra Florence, ItalyBeth McConnell Atlanta, GAKenneth R. McQuaid San Francisco, CADidier Merlin Atlanta, GAJason C. Mills St. Louis, MOAtsushi Mizoguchi Boston, MASatdarshan (Paul) MongaPittsburgh, PAMarshall H. Montrose Cincinnati, OHTimothy R. Morgan Long Beach, CASteven F. Moss Providence, RIMark W. Musch Chicago, ILBruce Naliboff Los Angeles, CAGeoffrey C. Nguyen Toronto, OntarioMindie H. Nguyen Stanford, CA
Oliver G. Opitz Freiburg, GermanyRonald P.J. Oude Elferink Amsterdam, The NetherlandsStephen J. Pandol Los Angeles, CAJohn Pandolfino Chicago, ILTushar C. Patel Jacksonville, FLRichard M. Peek Nashville, TNScott E. Plevy Chapel Hill, NCStephen J. Polyak Seattle, WAFiona Powrie Oxford, United KingdomDouglas K. Rex Indianapolis, INCharles M. Rice New York, NYDouglas J. Robertson White River Junction, VTK. Lenhard Rudolph Ulm, GermanyHamid M. Said Long Beach, CAPedro J. Salas Miami, FL Ashok Saluja Minneapolis, MNSushil K. Sarna Galveston, TXGünter Schneider München, GermanyThomas Seufferlein Halle (Saale), GermanyYatrik Shah Ann Arbor, MIStuart Sherman Indianapolis, INRamesh A. Shivdasani Boston, MANoah F. Shroyer Cincinnati, OHEric Sibley Stanford, CAJens T. Siveke Munich, GermanyScott Snapper Boston, MALudvig Sollid Oslo, NorwayRhonda F. Souza Dallas, TXBen Z. Stanger Philadelphia, PAPavel Strnad Ulm, GermanyJan F. Tack Leuven, BelgiumRobert Thimme Freiburg, GermanyDavid Thomas Baltimore, MDNikolai A. Timchenko Houston, TXMichael Trauner Graz, AustriaChristian Trautwein Aachen, GermanyHide Tsukamoto Los Angeles, CAFernando S. Velayos San Francisco, CAArnold Wald Madison, WIDavid C. Whitcomb Pittsburgh, PAAllan W. Wolkoff Bronx, NYHoward J. Worman New York, NY Nicholas A. Wright London, United Kingdom
preliminar gastro V1N2 2 3/14/12 11:26 AM

Editorial
GASTROENTEROLOGY 2011;141:1963–1967
El tratamiento combinado con interferón pegilado (IFN-PEG) y ribavirina (RBV) es el estándar del tratamiento
de la hepatitis C crónica (HCC) durante más de una década. Las tasas de respuesta virológica sostenida (RVS) varían de 40 a 45% en los pacientes con infección con el genotipo 1 a 75 a 80% en los pacientes con el genotipo 2 o 3.1 Sin embargo, el tratamiento con IFN-PEG y RBV se asocia con muchos efec-tos secundarios. En el registro de estudios que incluyeron pacientes altamente seleccionados, 13 a 15% de los pacien-tes interrumpieron el tratamiento de forma prematura, y 25 a 42% requirieron disminución de la dosis debido a eventos adversos o anormalidades de laboratorio.1-3 Debido a la to-lerabilidad deficiente, muchos pacientes con HCC prefieren no continuar con el tratamiento, o no se les ofreció el tra-tamiento. El IFN y la RBV también están contraindicados en muchos trastornos, como enfermedades autoinmunes y alteraciones psiquiátricas graves/no controladas.1 Por lo tanto, existe una necesidad urgente de tratamiento eficaz y mejor tolerado para la HCC.
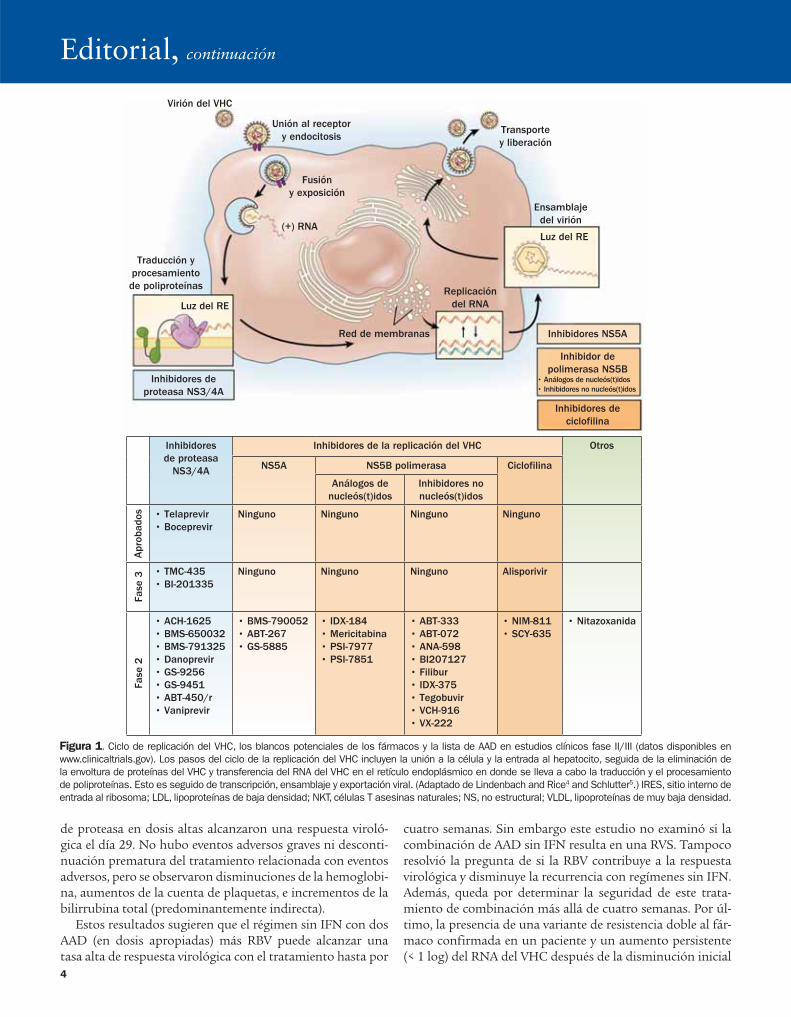
Los avances en la comprensión del ciclo de vida del virus de la hepatitis C (VHC) llevaron al desarrollo de muchos agentes antivirales de acción directa (AAD) prometedores en la última década (Figura 1).4,5 Dos de estos AAD —telaprevir y bocepre-vir— inhibidores de la serina proteasa NS3/4A fueron aproba-dos para el tratamiento del VHC en Estados Unidos en mayo de 2011. Aunque la aprobación del boceprevir y el telaprevir re-presenta un gran avance en el tratamiento de la HCC con tasas de RVS del 67 y 73%, respectivamente, en los pacientes con el genotipo 1 vírgenes al tratamiento, ambos fármacos requieren el uso concomitante de IFN-PEG y RBV para alcanzar una RVS y evitar el resurgimiento viral por resistencia a los fármacos.6,7 Además, ambos fármacos deben administrarse cada 8 horas y se asocian con eventos adversos adicionales.
Con el desarrollo de los AAD dirigidos a múltiples blancos en el ciclo de vida del VHC (Figura 1), la pregunta obvia es, “¿estamos listos para los regímenes de tratamiento sin IFN?” Los estudios clínicos del telaprevir y el boceprevir en mo-noterapia mostraron una disminución rápida de los niveles plasmáticos del RNA del VHC desde el primer día, seguida de resurgimiento virológico desde el día 3.8,9 El VHC circula como cuasiespecie, una mezcla de virus con secuencias virales heterogéneas. Se ha calculado que las variantes preexistentes de resistencia a fármacos con una, dos, tres e incluso cuatro mutaciones pueden estar presentes en la mayoría de los pa-cientes infectados con VHC, y son responsables del desarrollo de resistencia a los fármacos con la exposición a los AAD. El surgimiento de variantes resistentes a los fármacos clínicamen-te relevantes depende de varios factores, como la potencia del fármaco, la barrera genética a la resistencia y la adaptación de la replicación de las variantes de resistencia.10,11 Con base en
experimentos en modelos se sugiere que un régimen sin IFN que pueda superar la presencia de variantes con mutaciones de resistencia a cuatro fármacos requiere una combinación de ≥ 3 AAD con una baja barrera genética a la resistencia, a saber, AAD que seleccionen variantes resistentes de aminoá-cidos únicos.12 Cada uno de estos fármacos debe tener activi-dad antiviral potente, poseer perfil de resistencia sin sobre-posición, interacción medicamentosa limitada o manejable, y mínimos eventos adversos. Además, estos fármacos deben estar en estadios similares de desarrollo clínico para que pue-dan ser probados en combinación.
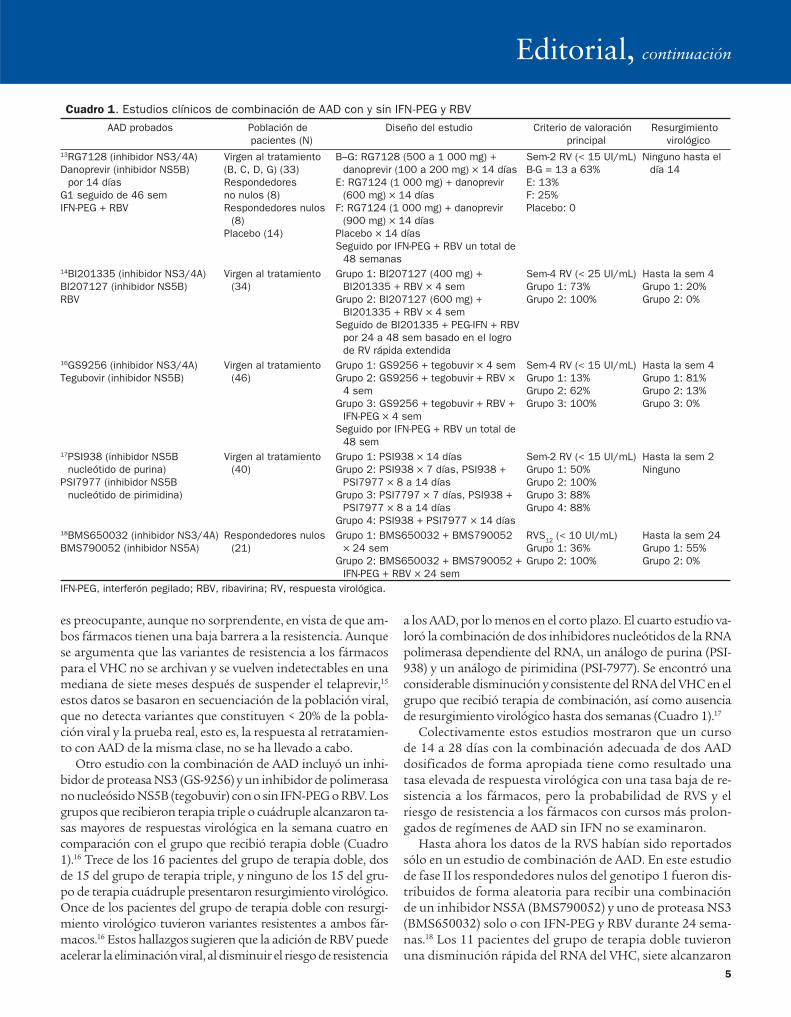
El primer estudio de combinación de AAD, el estudio INFORM-1, incluyó la combinación de un inhibidor de poli-merasa NS5B (RG7128) y un inhibidor NS3/4A (danoprevir). Este estudio incluyó sujetos vírgenes al tratamiento así como pacientes que habían sido tratados.13 En el día 14, 13 a 63% de los pacientes vírgenes al tratamiento y 25% de respondedores nulos tenían RNA del VHC indetectable (Cuadro 1). Ningu-no de los pacientes presentó resurgimiento virológico en los 14 días del régimen sin IFN, lo que sugirió que la adición de RG7128, que tiene una alta barrera a la resistencia, pudo ha-ber evitado la emergencia de resistencia al danoprevir.
Estos resultados prometedores han alentado otros es-tudios de combinación de AAD. El diseño y los resultados preliminares de estos estudios se resumen en el Cuadro 1. Todos los estudios reportados hasta ahora sólo incluyeron pa-cientes con infección con el genotipo 1. Los resultados de un estudio de fase Ib de la combinación de un inhibidor de proteasa NS3/4A BI201335, un inhibidor de polimerasa NS5B BI207127 y RBV fueron publicados en un número de GASTROENTEROLOGY.14 En este estudio los autores distribuye-ron de forma aleatoria a 34 pacientes con HCC vírgenes al tratamiento para recibir 400 o 600 mg TID de BI207127, 120 mg una vez al día de BI201335, y RBV de acuerdo con el peso durante cuatro semanas. Todos los pacientes se cambia-ron a terapia triple (BI201335 + IFN PEG + RBV) del día 29 hasta la semana 24 o 48 según la respuesta virológica rápida ampliada. El criterio de valoración principal fue la respuesta vi-rológica el día 29. Los cinco pacientes con el genotipo 1b y sólo seis de los 10 con genotipo 1a del grupo que recibió el inhibidor de proteasa en dosis bajas alcanzaron una respuesta virológica el día 29 (Cuadro 1). Un paciente con el genotipo 1a tuvo resur-gimiento virológico el día 22, con variantes resistentes a ambos fármacos (R155K en NS3 y P495L en NS5B), y otro paciente tuvo un aumento del RNA del VHC de 0.7 log10 UI/mL del na-dir, pero no pudieron determinarse las secuencias por el bajo nivel del RNA del VHC. Ambos pacientes tuvieron una dis-minución del RNA del VHC < 100 UI/mL después de 10 días de BI201335, IFN-PEG y RBV. Todos los pacientes (ocho con genotipo 1a y ocho con genotipo 1b) del grupo del inhibidor
Esquemas de tratamiento sin interferón para la hepatitis C: ¿todavía no estamos ahí?
preliminar gastro V1N2 3 3/14/12 11:26 AM
Editorial, continuación
4
de proteasa en dosis altas alcanzaron una respuesta viroló-gica el día 29. No hubo eventos adversos graves ni desconti-nuación prematura del tratamiento relacionada con eventos adversos, pero se observaron disminuciones de la hemoglobi-na, aumentos de la cuenta de plaquetas, e incrementos de la bilirrubina total (predominantemente indirecta).
Estos resultados sugieren que el régimen sin IFN con dos AAD (en dosis apropiadas) más RBV puede alcanzar una tasa alta de respuesta virológica con el tratamiento hasta por
cuatro semanas. Sin embargo este estudio no examinó si la combinación de AAD sin IFN resulta en una RVS. Tampoco resolvió la pregunta de si la RBV contribuye a la respuesta virológica y disminuye la recurrencia con regímenes sin IFN. Además, queda por determinar la seguridad de este trata-miento de combinación más allá de cuatro semanas. Por úl-timo, la presencia de una variante de resistencia doble al fár-maco confirmada en un paciente y un aumento persistente (< 1 log) del RNA del VHC después de la disminución inicial
Figura 1. Ciclo de replicación del VHC, los blancos potenciales de los fármacos y la lista de AAD en estudios clínicos fase II/III (datos disponibles en
www.clinicaltrials.gov). Los pasos del ciclo de la replicación del VHC incluyen la unión a la célula y la entrada al hepatocito, seguida de la eliminación de
la envoltura de proteínas del VHC y transferencia del RNA del VHC en el retículo endoplásmico en donde se lleva a cabo la traducción y el procesamiento
de poliproteínas. Esto es seguido de transcripción, ensamblaje y exportación viral. (Adaptado de Lindenbach and Rice4 and Schlutter5.) IRES, sitio interno de
entrada al ribosoma; LDL, lipoproteínas de baja densidad; NKT, células T asesinas naturales; NS, no estructural; VLDL, lipoproteínas de muy baja densidad.
Inhibidores
de proteasa
NS3/4A
Inhibidores de la replicación del VHC Otros
NS5A NS5B polimerasa Ciclofilina
Análogos de
nucleós(t)idos
Inhibidores no
nucleós(t)idos
• Telaprevir
• Boceprevir
Ninguno Ninguno Ninguno Ninguno
• TMC-435
• BI-201335
Ninguno Ninguno Ninguno Alisporivir
• ACH-1625
• BMS-650032
• BMS-791325
• Danoprevir
• GS-9256
• GS-9451
• ABT-450/r
• Vaniprevir
• BMS-790052
• ABT-267
• GS-5885
• IDX-184
• Mericitabina
• PSI-7977
• PSI-7851
• ABT-333
• ABT-072
• ANA-598
• BI207127
• Filibur
• IDX-375
• Tegobuvir
• VCH-916
• VX-222
• NIM-811
• SCY-635
• Nitazoxanida
Apro
bados
Fase
3Fase
2
Virión del VHC
Inhibidores NS5A
Unión al receptor
y endocitosisTransporte
y liberación
Fusión
y exposición
(+) RNA
Ensamblaje
del virión
Traducción y
procesamiento
de poliproteínas
Inhibidores de
proteasa NS3/4A
Red de membranas
Replicación
del RNA
Luz del RE
Luz del RE
Inhibidores de
ciclofilina
Inhibidor de
polimerasa NS5B• Análogos de nucleós(t)idos
• Inhibidores no nucleós(t)idos
preliminar gastro V1N2 4 3/14/12 11:26 AM
Editorial, continuación
5
es preocupante, aunque no sorprendente, en vista de que am-bos fármacos tienen una baja barrera a la resistencia. Aunque se argumenta que las variantes de resistencia a los fármacos para el VHC no se archivan y se vuelven indetectables en una mediana de siete meses después de suspender el telaprevir,15 estos datos se basaron en secuenciación de la población viral, que no detecta variantes que constituyen < 20% de la pobla-ción viral y la prueba real, esto es, la respuesta al retratamien-to con AAD de la misma clase, no se ha llevado a cabo.
Otro estudio con la combinación de AAD incluyó un inhi-bidor de proteasa NS3 (GS-9256) y un inhibidor de polimerasa no nucleósido NS5B (tegobuvir) con o sin IFN-PEG o RBV. Los grupos que recibieron terapia triple o cuádruple alcanzaron ta-sas mayores de respuestas virológica en la semana cuatro en comparación con el grupo que recibió terapia doble (Cuadro 1).16 Trece de los 16 pacientes del grupo de terapia doble, dos de 15 del grupo de terapia triple, y ninguno de los 15 del gru-po de terapia cuádruple presentaron resurgimiento virológico. Once de los pacientes del grupo de terapia doble con resurgi-miento virológico tuvieron variantes resistentes a ambos fár-macos.16 Estos hallazgos sugieren que la adición de RBV puede acelerar la eliminación viral, al disminuir el riesgo de resistencia
a los AAD, por lo menos en el corto plazo. El cuarto estudio va-loró la combinación de dos inhibidores nucleótidos de la RNA polimerasa dependiente del RNA, un análogo de purina (PSI-938) y un análogo de pirimidina (PSI-7977). Se encontró una considerable disminución y consistente del RNA del VHC en el grupo que recibió terapia de combinación, así como ausencia de resurgimiento virológico hasta dos semanas (Cuadro 1).17
Colectivamente estos estudios mostraron que un curso de 14 a 28 días con la combinación adecuada de dos AAD dosificados de forma apropiada tiene como resultado una tasa elevada de respuesta virológica con una tasa baja de re-sistencia a los fármacos, pero la probabilidad de RVS y el riesgo de resistencia a los fármacos con cursos más prolon-gados de regímenes de AAD sin IFN no se examinaron.
Hasta ahora los datos de la RVS habían sido reportados sólo en un estudio de combinación de AAD. En este estudio de fase II los respondedores nulos del genotipo 1 fueron dis-tribuidos de forma aleatoria para recibir una combinación de un inhibidor NS5A (BMS790052) y uno de proteasa NS3 (BMS650032) solo o con IFN-PEG y RBV durante 24 sema-nas.18 Los 11 pacientes del grupo de terapia doble tuvieron una disminución rápida del RNA del VHC, siete alcanzaron
Cuadro 1. Estudios clínicos de combinación de AAD con y sin IFN-PEG y RBV
AAD probados Población de
pacientes (N)
Diseño del estudio Criterio de valoración
principal
Resurgimiento
virológico
13RG7128 (inhibidor NS3/4A)
Danoprevir (inhibidor NS5B)
por 14 días
G1 seguido de 46 sem
IFN-PEG + RBV
Virgen al tratamiento
(B, C, D, G) (33)
Respondedores
no nulos (8)
Respondedores nulos
(8)
Placebo (14)
B–G: RG7128 (500 a 1 000 mg) +
danoprevir (100 a 200 mg) × 14 días
E: RG7124 (1 000 mg) + danoprevir
(600 mg) × 14 días
F: RG7124 (1 000 mg) + danoprevir
(900 mg) × 14 días
Placebo × 14 días
Seguido por IFN-PEG + RBV un total de
48 semanas
Sem-2 RV (< 15 UI/mL)
B-G = 13 a 63%
E: 13%
F: 25%
Placebo: 0
Ninguno hasta el
día 14
14BI201335 (inhibidor NS3/4A)
BI207127 (inhibidor NS5B)
RBV
Virgen al tratamiento
(34)
Grupo 1: BI207127 (400 mg) +
BI201335 + RBV × 4 sem
Grupo 2: BI207127 (600 mg) +
BI201335 + RBV × 4 sem
Seguido de BI201335 + PEG-IFN + RBV
por 24 a 48 sem basado en el logro
de RV rápida extendida
Sem-4 RV (< 25 UI/mL)
Grupo 1: 73%
Grupo 2: 100%
Hasta la sem 4
Grupo 1: 20%
Grupo 2: 0%
16GS9256 (inhibidor NS3/4A)
Tegubovir (inhibidor NS5B)
Virgen al tratamiento
(46)
Grupo 1: GS9256 + tegobuvir × 4 sem
Grupo 2: GS9256 + tegobuvir + RBV ×
4 sem
Grupo 3: GS9256 + tegobuvir + RBV +
IFN-PEG × 4 sem
Seguido por IFN-PEG + RBV un total de
48 sem
Sem-4 RV (< 15 UI/mL)
Grupo 1: 13%
Grupo 2: 62%
Grupo 3: 100%
Hasta la sem 4
Grupo 1: 81%
Grupo 2: 13%
Grupo 3: 0%
17PSI938 (inhibidor NS5B
nucleótido de purina)
PSI7977 (inhibidor NS5B
nucleótido de pirimidina)
Virgen al tratamiento
(40)
Grupo 1: PSI938 × 14 días
Grupo 2: PSI938 × 7 días, PSI938 +
PSI7977 × 8 a 14 días
Grupo 3: PSI7797 × 7 días, PSI938 +
PSI7977 × 8 a 14 días
Grupo 4: PSI938 + PSI7977 × 14 días
Sem-2 RV (< 15 UI/mL)
Grupo 1: 50%
Grupo 2: 100%
Grupo 3: 88%
Grupo 4: 88%
Hasta la sem 2
Ninguno
18BMS650032 (inhibidor NS3/4A)
BMS790052 (inhibidor NS5A)
Respondedores nulos
(21)
Grupo 1: BMS650032 + BMS790052
× 24 sem
Grupo 2: BMS650032 + BMS790052 +
IFN-PEG + RBV × 24 sem
RVS12
(< 10 UI/mL)
Grupo 1: 36%
Grupo 2: 100%
Hasta la sem 24
Grupo 1: 55%
Grupo 2: 0%
IFN-PEG, interferón pegilado; RBV, ribavirina; RV, respuesta virológica.
preliminar gastro V1N2 5 3/14/12 11:26 AM

Editorial, continuación
6
un nivel indetectable del RNA del VHC; sin embargo seis presentaron resurgimiento virológico y tuvieron variantes resistentes a ambos AAD seleccionados (Cuadro 1). Aunque la mayoría de estos pacientes respondió al tratamiento de rescate con la adición de IFN-PEG y RBV, no es claro si ten-drán una RVS. Cuatro de los 11 pacientes alcanzaron una RVS.12 Las respuestas fueron alentadoras en el grupo de terapia cuádruple, donde los 10 pacientes alcanzaron una RVS.12 Estos datos muestran que la adición de dos AAD al IFN-PEG y la RBV tiene como resultado una tasa mayor de RVS en comparación con el AAD en no respondedores al IFN-PEG y la RBV.19 Más importante aún es que proporcio-nó la prueba del concepto de que se puede alcanzar un RVS con la combinación de AAD únicamente.
Similar al estudio de Zeuzem y colaboradores,14 todos los pacientes con resurgimiento virológico en el estudio de Lok y colaboradores tuvieron infección con el gentoipo 1a.18 Los ge-notipos Ia y Ib del VHC pueden diferir en su susceptibilidad a los AAD. Además, se requiere un número mayor de cambios de nucleótidos para crear una variante de resistencia al inhibidor de proteasa clínicamente significativa para el genotipo 1b (ma-yor barrera a la resistencia) que para el genotipo 1a del VHC.10,11 Por ejemplo, se requieren dos cambios de nucleótidos para ge-nerar la mutación de resistencia R155K para cultivos 1b (CGG → AAG), mientras que sólo se requiere el cambio de un nucleó-tido para los cultivos 1a (AGG → AAG).19 Estos datos indican que pueden requerirse diferentes estrategias para el genotipo 1a y 1b de la infección en la era de la combinación de AAD.
Ocho años después del primer estudio clínico de AAD,20 el desarrollo de tratamientos de acción directa del VHC ahora se mueve a un paso rápido con muchos productos que mues-tran resultados prometedores. Los regímenes sin IFN ya no son un sueño, sino una realidad que va a estar disponible en la clínica en los próximos cinco años. Es posible que algu-nos de estos regímenes también sean sin RBV. Estas serán buenas noticias para los pacientes que desean ser tratados pero que tienen que diferir el tratamiento debido a contra-indicaciones del uso de IFN-PEG o RBV, o la preocupación respecto a su capacidad para tolerar estos medicamentos. No obstante se debe tener cuidado en seleccionar cuáles AAD combinar, así como las dosis y la duración apropiadas del tratamiento para cada genotipo y subgenotipo del VHC para prevenir la resistencia a múltiples fármacos.
PRATIMA SHARMAANNA S. LOKDivisión de GastroenterologíaUniversidad de MichiganAnn Arbor, Michigan
Referencias1. Ghany MG, Strader DB, Thomas DL, et al. Diagnosis, manage-
ment, and treatment of hepatitis C: an update. Hepatology 2009; 49:1335–1374.
2. Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 2002;347:975–982.
3. Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 2001;358:958–965.
4. Lindenbach BD, Rice CM. Unravelling hepatitis C virus replication from genome to function. Nature 2005;436:933–938.
5. Schlutter J. Therapeutics: new drugs hit the target. Nature 474: S5–S7.
6. Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 364:2405–2416.
7. Poordad F, McCone J Jr, Bacon BR, et al. Boceprevir for untreated chro-nic HCV genotype 1 infection. N Engl J Med 2011;364:1195–1206.
8. Reesink HW, Zeuzem S, Weegink CJ, et al. Rapid decline of viral RNA in hepatitis C patients treated with VX-950: a phase Ib, placebo con-trolled, randomized study. Gastroenterology 2006; 131:997–1002.
9. Susser S, Welsch C, Wang Y, et al. Characterization of resistance to the protease inhibitor boceprevir in hepatitis C virus-infected patients. Hepatology 2009;50:1709–1718.
10. Pawlotsky JM. Treatment failure and resistance with direct-acting an-tiviral drugs against hepatitis C virus. Hepatology; 53:1742–1751.
11. Sarrazin C, Zeuzem S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 2010; 138:447–462.
12. Rong L, Dahari H, Ribeiro RM, et al. Rapid emergence of protease inhibitor resistance in hepatitis C virus. Sci Transl Med 2:30ra32.
13. Gane EJ, Roberts SK, Stedman CA, et al. Oral combination the-rapy with a nucleoside polymerase inhibitor (RG7128) and dano-previr for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 2010;376:1467–1475.
14. Zuezem S, Asselah T, Angus P, et al. Efficacy of the protease in-hibitor BI 201335, polymerase inhibitor BI 207127, and ribavirin in patients with chronic HCV infection. Gastroenterology 2011; 141:2047–2055.
15. Sullivan JC, De Meyer S, Bartels DJ, et al. Evolution of treatmen-temergent resistant variants in Telaprevir phase 3 clinical trials. J Hepatol 2011;54:s4.
16. Zeuzem S, Buggisch P, Agarwal K, et al. Dual, triple and quadruple combination treatment with a protease inhibitor (GS-9256) and a polymerase inhibitor (GS-9190) alone and in combination with ri-bavirin (RBV) or PEGIFN/RBV for up to 28 days in treatment naïve genotype 1 HCV subjects. Hepatology 2010;52:LB-1, 400A.
17. Lawitz E, Rodriguez-Torres M, et al. Once daily dual-nucleotide combination of PSI-938 and PSI-7977 provides 94% HCV RNA < LOD at day 14: first purine/pyrimidine clinical combination data (the NUCLEAR study). J Hepatol 2011;54:S543.
18. Lok AS, Gardiner DF, Lawitz E, et al. Quadruple therapy with BMS-790052, BMS-650032 and Peg-IFN/RBV for 24 weeks results in 100% SVR12 in HCV genotype 1 null responders. J Hepatol 2011;54:s536.
19. Zeuzem S, Andreone P, Pol S, et al. Telaprevir for retreatment of HCV infection. N Engl J Med;364:2417–2428.
20. Lamarre D, Anderson PC, Bailey M, et al. An NS3 protease inhibi-tor with antiviral effects in humans infected with hepatitis C virus. Nature 2003;426:186–189.
Solicitud de reimpresosDirigir la solicitud de reimpresos a: Anna S. Lok, MD, Alice Lohrman An-
drews Research Professor in Hepatology, University of Michigan Health
System, 1500 E Medical Center Drive, 3912, Taubman Center, SPC 5362,
Ann Arbor, Michigan 48109. Correo electrónico: [email protected];
fax: (734) 936-7392.
Conflicto de interésLa Dra. Lok recibe subvenciones para investigación de Bristol-Myers
Squibb, GlaxoSmithKline, Gilead, Roche, y Merck, y ha estado en el
Panel de Monitorización de Datos y Seguridad de Abbott, Bristol-Myers
Squibb, GlaxoSmithKline, Gilead, and Roche.
RecursosLa Dra. Sharma tiene subvención KO8 DK-088946 de los National Institutes of Health (NIH).
preliminar gastro V1N2 6 3/14/12 11:26 AM

GASTROENTEROLOGY 2012;142:8–11
7
Los lípidos en la enfermedad hepática: una visión más allá de la esteatosis
El hígado es un sitio clave para la generación de energía y para el metabolismo de los lípidos. La acumulación exce-
siva de grasa en el hígado predispone al desarrollo subsecuen-te de fibrosis, cirrosis y cáncer hepatocelular.1 La esteatosis es una comorbilidad importante que potencía el daño hepático y la progresión de enfermedades de otras etiologías, como son al-gunas afecciones por tóxicos, virales y colestásicas. Se descono-ce el grado en el cual los lípidos y el metabolismo de los lípidos influyen en las enfermedades hepáticas cuando la esteatosis está ausente. Existen indicios de que la acumulación de lípidos per se no es un factor clave para la lesión de órganos. De hecho las gotas de lípidos son organelos altamente especializados que permiten a las células almacenar y liberar lípidos en forma regulada,2 y se argumenta que la división de los lípidos en las gotas previene el daño.3-5 En cambio, el flujo de lí pidos dentro y fuera de estas gotas, permite la conversión de moléculas inertes en mediadores tóxicos potenciales, se coloca como una hipó-tesis del mecanismo para el daño hepático mediado por los lí-pidos. Un segundo punto que se pasa por alto es que los lípidos afectan otras poblaciones celulares en el hígado, además de los hepatocitos. Las células estelares hepáticas (CEH), antes llama-das “lipocitos” hepáticos, contienen grandes cantidades de lípi-dos almacenados en forma de gotas. Es concebible que las CEH y otras poblaciones de células no parenquimatosas puedan ser blancos relevantes de los lípidos en las enfermedades hepáticas. Una tercera consideración importante está en relación con los tipos de lípidos que inducen el daño y la enfermedad hepática. Aunque el consumo global de calorías y grasas es una deter-minante clave del desarrollo de obesidad y esteatosis hepática, lípidos específicos de la alimentación como el colesterol o las grasas trans son más relevantes para el desarrollo de la en-fermedad, incluidos daño hepático, inflamación y fibrosis.6,7 En este número de GASTROENTEROLOGY dos estudios investigan el papel de los lípidos en el desarrollo del daño hepático y la fibrosis.8,9 Aunque estos estudios investigan diferentes aspec-tos, y utilizan distintos modelos, cuestionan nuestra com-prensión actual del papel y los sitios blancos de los lípidos en la enfermedad hepática, y sugieren un papel más amplio de los lípidos en los procesos de la enfermedad hepática de lo que antes se pensaba.
Teratani y colaboradores, investigaron los efectos de una dieta con alto contenido de colesterol sobre la fibro-sis hepática inducida por la ligadura del conducto biliar (LCB) y el tetracloruro de carbono, dos modelos comunes de lesión biliar y daño hepático tóxico.8 El resultado prin-cipal de sus estudios radica en el hallazgo de que una dieta con alto contenido de colesterol no tiene un impacto sobre el daño de los hepatocitos valorado por las determinacio-nes de alanina aminotransferasa y la tinción TUNEL, pero afecta gravemente las CEH y la fibrosis hepática. Por otra parte, los autores excluyeron la contribución de las células de Kupffer, porque notaron efectos de la dieta rica en coles-
terol favorece la fibrosis en ratones depletados de células de Kupffer. En cambio los autores observaron niveles ma-yores de colesterol en las CEH de los ratones que recibieron una dieta rica en colesterol, y un aumento en la regula-ción positiva de los genes profibrogénicos en respuesta al factor transformador del crecimiento (TGF)-β. Aunque los autores no notaron efecto de las dietas ricas en colesterol en la expresión de los receptores TGF-β, hallaron una expresión aumentada de los receptores de lipopolisacáridos semejantes a Toll 4 (TLR4), con un aumento subsecuente de las señales del TGF-β y regulación a la baja de Bambi, un seudorrecep-tor inhibidor del TGF altamente expresado en las CEH y que afecta de modo negativo las señales del TGF-β en estas célu-las.10 Los autores confirmaron estos hallazgos en un segun-do modelo genético en el cual los niveles intracelulares de colesterol aumentaron debido a la ausencia de la proteína NPC1. Es interesante notar que en ambos modelos el coles-terol no aumentó los niveles del mRNA TLR4, lo que llevó a los autores a sugerir que el colesterol previene la degrada-ción de la proteína TLR4. Lo novedoso de este estudio es que demuestra los efectos del colesterol sobre las CEH y la activación fibrogénica (Figura 1).
Debido a que las gotas de lípidos en las CEH contienen alrededor de 20% de colesterol o ésteres del colesterol,11 pa-rece lógico que el colesterol de la dieta aumente estos niveles y afecte la biología de las CEH. Sin embargo el estudio deja abierta la pregunta de si otras células en el hígado se afectan
Figura 1. Activación aumentada de las CEH por el colesterol de la dieta.
El colesterol aumentado en la dieta regula a la alta los niveles de la proteína
TLR4 en las CEH, que a su vez regulan a la baja el seudorreceptor Bambi del
TGF-β inhibidor. El aumento resultante de las señales del TGF-β favorece el
aumento de la activación de las CEH y la fibrosis hepática.
Dieta rica en
colesterol
↑TLR4
TGFβRI
TGFβ
↓Bambi
Células
estelares
hepáticas
Fibrosis
hepática
HígadoEl colesterol elevado aumentó los TLR4
en las CEH
El TLR4 aumenta las señales del
TGFβ en las CEH
Activación aumentada de las CEH por El TGFβ
Activación aumentada de las CEH por El TGFβ
preliminar gastro V1N2 7 3/14/12 11:26 AM

8 LOS LÍPIDOS EN LA ENFERMEDAD HEPÁTICA GASTROENTEROLOGY • Volumen 1 • Núm. 2
por el colesterol de la dieta. Se sabe que los hepatocitos tienen capacidad para captar grandes cantidades de colesterol del plasma y eliminar el exceso de colesterol a través de la ex-creción en la bilis en un proceso conocido como transporte inverso del colesterol.12 De manera similar los macrófagos se afectan por una dieta rica en colesterol.12 Es importante destacar que estudios previos han demostrado que una die-ta rica en colesterol sensibiliza a los ratones para desarrollar esteatohepatitis y muerte hepatocelular mediadas por el fac-tor de necrosis tumoral y el Fas. Aunque Teratani y colabo-radores no observaron influencia del colesterol de la dieta sobre la apoptosis o la lesión necrótica después de la LCB o la inyección de tetraclouro de carbono, parece poco proba-ble que el colesterol de la dieta se dirija de forma selectiva a las CEH sin ejercer efectos en otras poblaciones de células hepáticas. Se podría vaticinar que el organelo blanco y los efectos predominantes del colesterol dependen de la enfer-medad subyacente, y que el colesterol puede ser redirigido hacia las CEH cuando la excreción biliar está bloqueada o la captación de los hepatocitos está alterada.
Aunque el estudio de Teratani y colaboradores muestra por primera vez la forma en que los lípidos alteran las res-puestas específicamente en las CEH, las implicaciones de estos resultados para la enfermedad hepática en los pacien-tes con colesterol elevado no son claras. Es concebible que el colesterol de la dieta amplifique la respuesta fibrogénica en las CEH según lo sugerido por Teratani y colaboradores, y que afecte también el daño hepático y la inflamación como un “segundo impacto” de los hepatocitos en otras enferme-dades, como lo sugirieron Mari y colaboradores.6 Es impor-tante destacar que ambos estudios utilizaron una dieta con elevadas cantidades de colesterol que se traduce en 1 000 a 2 000 mg/día, mientras que el promedio del consumo hu-mano es de sólo 300 mg/día, lo que hace difícil extrapolar-los a la clínica. Estudios previos con los datos de la National Health and Nutrition Examination Survey relacionan el con-sumo mayor de colesterol con la muerte por enfermedad hepática, pero no analizaron la enfermedad hepática sub-yacente.13 Por lo tanto, se requieren estudios bien diseñados en grupos específicos de pacientes, como es en la hepatitis C y el hígado graso no alcohólico, para valorar el impacto del consumo de colesterol en diferentes enfermedades hepáticas y el desarrollo de fibrosis. Aunque quedan muchas pregun-tas por contestar respecto al colesterol de la alimentación y la enfermedad hepática, una lección importante del estudio de Teratani y colaboradores es que necesitamos enfocarnos en los efectos de los lípidos en las poblaciones celulares dife-rentes a los hepatocitos, como las CEH.
En un segundo artículo publicado en este número de GASTROENTEROLOGY, Moustafa y colaboradores9 descubrie-ron una característica única de los lípidos hepáticos en un modelo de ratón con enfermedad hepática colestásica cau-sada por deleción genética del casete de unión al ATP de la subfamilia B miembro 4 (Abcb4–/–). Abcb4 codifica una lipasa de fosfolípidos que favorece la secreción biliar de fos-folípidos y protege al epitelio biliar de los efectos dañinos
de los ácidos biliares. Los ratones deficientes de Abcb4 de-sarrollan daño hepatocelular, colestasis y fibrosis hepáti-ca;14 las mutaciones Abcb4 también ocurren en humanos, y son responsables del síndrome conocido como colestasis intrahepática familiar progresiva tipo 3 (CIFP3).15
Aunque los ratones Abcb4–/– desarrollan muchas altera-ciones bioquímicas e histológicas que se ven en los humanos con colestasis, también tienen algunas alteraciones únicas en el metabolismo de los lípidos. En específico, los ratones Abcb4–/– tienen hipocolesterolemia y niveles bajos de trigli-céridos hepáticos, mientras que los humanos con colestasis por lo regular tienen hipercolesterolemia. A pesar de esta discrepancia, existe una posibilidad de que las anormali-dades de los lípidos en los ratones Abcb4–/– son clínicamente relevantes porque los polimorfismos de ABCB4 en los hu-manos llevan a disminución de los niveles circulantes de colesterol.16 Existe la curiosidad de saber si las alteraciones relacionadas con los lípidos en los ratones Abcb4-/- se relacio-nan de manera causal con la enfermedad hepática, Moustafa y colaboradores compararon la expresión del gen hepático en ratones noqueados con controles del tipo silvestre. Ellos encontraron que los genes que afectan el metabolismo de los lípidos estaban influenciados por la deficiencia de Abcb4. También determinaron que la expresión hepática del gen en ratones Abcb4–/– antes y después del tratamiento con el ácido 24-NorUrsodesoxicólico (NorUDCA), una variante de la cadena lateral del ácido ursodesoxicólico que mejora el daño hepático colestásico. Una vez más, los genes que afectan el metabolismo de los lípidos fueron de los más influenciados por el tratamiento con NorUDCA.
Al examinar a los ratones Abcb4–/– tratados con Nor-UDCA se observó que el fármaco revirtió tanto la hipo-colesterolemia como la hipofosfolipidemia y normalizó el contenido hepático de triglicéridos, y se revirtieron así las anormalidades causadas por el gen noqueado. Además existió un incremento en la incorporación de colesterol y fosfolípidos en las lipoproteínas y una disminución de la actividad de la hidrolasa de triglicéridos. Este último ha-llazgo fue particularmente interesante porque sugirió que la enfermedad hepática en los ratones Abcb4–/– se debe al aumento de la hidrólisis de triglicéridos en el hígado.
Los ácidos grasos liberados de los triglicéridos son in-ductores del receptor activador de la proliferación del pe-roxisoma (PPAR)-α. Moustafa y colaboradores postulan que el PPAR-α contribuía al exceso de lipólisis de trigli-céridos en los ratones Abcb4–/–, basados en la evidencia de que varias enzimas involucradas en el catabolismo de los triglicéridos son inducibles por PPAR-α.17 Cruzaron enton-ces ratones Abcb4–/– con ratones PPAR-α–/–, lo que resultó en una notable protección contra la colestasis y el daño hepá-tico. Los ratones doblemente noqueados tuvieron niveles de lípidos séricos y niveles de triglicéridos hepáticos eleva-dos que los ratones Abcb4–/– así como una menor expresión hepática de los genes lipolíticos. Por el contrario, cuando los ratones Abcb4–/– se trataron con el agonista PPAR-α fe-nofibrato el daño hepático se agravó.
preliminar gastro V1N2 8 3/14/12 11:26 AM

LOS LÍPIDOS EN LA ENFERMEDAD HEPÁTICA 9GASTROENTEROLOGY • Volumen 1 • Núm. 2
Después de haber demostrado en dos escenarios experi-mentales que la mejoría del daño hepático colestásico se re-lacionó con disminución de la hidrólisis de triglicéridos, los autores examinaron luego un tercer método para desplazar el equilibrio hepático lejos de la hidrólisis de triglicéridos me-diante una dieta alta en grasas. Igual que con el tratamiento con NorUDCA y la deficiencia de PPAR-α, la dieta rica en gra-sa mejoró la colestasis, la inflamación hepática y la fibrosis en los ratones Abcb4–/–, y la mejoría coincidió con la disminu-ción significativa de la actividad de la fosfolipasa hepática y de la lipasa de triglicéridos (Figura 2).
Estos resultados son sorprendentes porque implican que la homeostasis hepática de los lípidos es una determi-nante importante del resultado en una enfermedad hepá-tica sin esteatosis hepática. Son únicos porque demuestran que la enfermedad hepática colestásica, por lo menos en los ratones Abcb4–/–, mejora al aumentar, en lugar de dismi-nuir, los niveles de triglicéridos hepáticos. ¿Significa esto que hay algún nivel óptimo de triglicéridos hepáticos que pueda llevar a enfermedad si el aporte se desplaza excesi-vamente hacia arriba o hacia abajo? ¡Probablemente no! Más bien la enfermedad hepática puede estar en función de la actividad metabólica de los lípidos hepáticos. Estu-dios previos muestran que el exceso de ácidos grasos en el hígado es nocivo si se impide que se incorporen a los trigli-céridos.3 El estudio de Moustafa y colaboradores ilustra el punto contrario —que el daño hepático ocurre también si las gotitas de lípidos no son capaces de mantenerse en sus triglicéridos. Tomados en conjunto, estos hallazgos incri-minan a los ácidos grasos como compuestos hepatotóxicos y refuerzan la noción de que la síntesis de triglicéridos en los hepatocitos es un mecanismo citoprotector.4
Aunque Moustafa y colaboradores evitaron de manera eficaz el daño hepatocelular en los ratones Abcb4–/– con va-rios tratamientos que disminuyen la hidrólisis de los trigli-céridos, no se sabe por qué estas manipulaciones también mejoraron la colestasis. No hubo evidencia de que la de-ficiencia de PPAR-α o la alimentación con alto contenido
de grasa mejorara el f lujo de bilis, aunque esto ocurre con NorUDCA. El efecto benéfico de la deficiencia de PPAR-α sobre el daño hepático en los ratones Abcb4–/– es también curioso en vista de otros reportes de que la colestasis experi-mental debida a la LCB, así como el hígado graso experimen-tal, mejoran con agonistas PPAR-α.18-19 Estos resultados son difícilies de reconciliar, pero están acordes con los efectos complejos y algunas veces paradójicos de la manipulación del PPAR-α en otros órganos.5,20 Hasta que los hallazgos puedan colocarse en su perspectiva apropiada, es prematu-ro considerar la supresión de PPAR-α como tratamiento de la colestasis en los humanos.
Por último, aunque los ratones Abcb4–/– se utilizan de forma generalizada como un modelo de enfermedad he-pática colestásica en los seres humanos, no es seguro que las anormalidades relacionadas con los lípidos en estos ratones puedan generalizarse a los humanos —incluso a humanos con CIFP3. Un paso siguiente importante será determinar si los pacientes con CIFP3 tienen la misma re-ducción de triglicéridos hepáticos y aumento de la hidróli-sis de triglicéridos que los ratones Abcb4–/–. Si es así, puede llevarnos a un nuevo enfoque de esta enfermedad, y cierta-mente se estimulará una mayor investigación de las anor-malidades relacionadas con los lípidos en otros trastornos colestásicos crónicos.
ROBERT F. SCHWABEDepartamento de MedicinaInstituto de Nutrición HumanaUniversidad de ColumbiaNueva York, Nueva York
JACQUELYN J. MAHERDepartamento de Medicina y Centro de HígadoUniversidad de California, San FranciscoSan Francisco, California
Referencias1. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old
questions and new insights. Science 2011;332:1519–1523.
2. Thiele C, Spandl J. Cell biology of lipid droplets. Curr Opin Cell
Biol 2008;20:378–385.
3. Yamaguchi K, Yang L, McCall S, et al. Inhibiting triglyceride
synthesis improves hepatic steatosis but exacerbates liver da-
mage and fibrosis in obese mice with nonalcoholic steatohepa-
titis. Hepatology 2007;45:1366–1374.
4. Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogene-
sisof nonalcoholic steatohepatitis: the central role of nontrigly-
ceridefatty acid metabolites. Hepatology 2010;52:774–788.
5. Son NH, Yu S, Tuinei J, et al. PPARgamma-induced cardiolipotoxicity
in mice is ameliorated by PPARalpha deficiency despite increases
in fatty acid oxidation. J Clin Invest 2010;120:3443–3454.
6. Mari M, Caballero F, Colell A, et al. Mitochondrial free choleste-
rol loading sensitizes toTNF- and Fas-mediated steatohepatitis.
Cell Metab 2006;4:185–198.
7. Kohli R, Kirby M, Xanthakos SA, et al. High-fructose, medium
chaintrans fat diet induces liver fibrosis and elevates plasma
coenzyme Q9 in a novel murine model of obesity and nonalcoho-
lic steatohepatitis.Hepatology 2010;52:934–944.
8. Teratani T, Tomita K, Suzuki T, et al. A high-cholesterol diet exa-
cerbates liver fibrosis in mice via accumulation of free cholesterol
in hepatic stellate cells. Gastroenterology 2011;142:152–165.
Figura 2. La hidrólisis de los triglicéridos en la gotas de los lípidos hepáticos
libera ácidos grasos tóxicos, que pueden activar un asa de retroalimenta-
ción positiva nociva al activar PPAR-α y estimular la lipasa de los triglicéri-
dos. El NorUDCA, la inhibición de PPAR-α y la dieta rica en grasas pueden
interrumpir el ciclo a evitar una mayor hidrólisis y desplazar el equilibrio del
hepatocito hacia la síntesis y almacenamiento de triglicéridos.
NorUDCA
PPAR–/–
HFD
↑ PPARα
↑ Lipólisis
Gotita
de lípido
Abcb4–/–
LipólisisHepatotoxicidad
por colestasis (?)
↑ Ácidos grasos
preliminar gastro V1N2 9 3/14/12 11:26 AM

10 LOS LÍPIDOS EN LA ENFERMEDAD HEPÁTICA GASTROENTEROLOGY • Volumen 1 • Núm. 2
9. Moustafa T, Fickert P, Magnes C, et al. Alterations in lipid me-
tabolism mediate inflammation, fibrosis, and proliferation in a
mousemodel of chronic cholestatic liver injury. Gastroenterology
2011;142;140–151.
10. Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-
beta signaling and hepatic fibrosis. Nat Med 2007;13:1324–
1332.
11. Blaner WS, O’Byrne SM, Wongsiriroj N, et al. Hepatic stellate
cell lipid droplets: a specialized lipid droplet for retinoid storage.
Biochim Biophys Acta 2009;1791:467–1473.
12. Tabas I. Consequences of cellular cholesterol accumulation:
basic concepts and physiological implications. J Clin Invest
2002;110:905–911..
13. Ioannou GN, Morrow OB, Connole ML, et al. Association bet-
ween dietary nutrient composition and the incidence of cirrho-
sis or liver cancer in the United States population. Hepatology
2009;50:175–184.
14. Smit JJ, Schinkel AH, Oude Elferink RP, et al. Homozygous dis-
ruptionof the murine mdr2 P-glycoprotein gene leads to a com-
plete absence of phospholipid from bile and to liver disease.
Cell 1993; 75:451–462.
15. Davit-Spraul A, Gonzales E, Baussan C, et al. The spectrum of li-
ver diseases related to ABCB4 gene mutations: pathophysiology
and clinical aspects. Semin Liver Dis 2010;30:134–146.
16. Acalovschi M, Tirziu S, Chiorean E, et al. Common variants of
ABCB4 and ABCB11 and plasma lipid levels: a study in sib pairs
with gallstones, and controls. Lipids2009;44:521–526.
17. Rakhshandehroo M, Sanderson LM, Matilainen M, et al. Com-
prehensive analysis of PPARalpha-dependent regulation of
hepatic lipid metabolism by expression profiling. PPAR Res
2007;2007:26839.
18. Cindoruk M, Kerem M, Karakan T, et al. Peroxisome proliferator-
sactivated alpha agonist treatment ameliorates hepatic damage
in rats with obstructive jaundice: an experimental study. BMC
Gastroenterol 2007;7:44.
19. Ip E, Farrell G, Hall P, et al. Administration of the potent PPA-
Ralpha agonist, Wy-14,643, reverses nutritional fibrosis and
steatohepatitis in mice. Hepatology 2004;39:1286–1296.
20. Haemmerle G, Moustafa T, Woelkart G, et al. ATGL-mediated
fat catabolism regulates cardiac mitochondrial function via PPA-
Ralpha and PGC-1. Nat Med 2011;17:1076–1085.
Solicitud de reimpresosDirigir la solicitud de reimpresos a: Robert F.Schwabe, MD, Russ Berrie
Pavililion, Room 415, Columbia University, 1150 St. Nicholas Avenue, New
York, New York 10032. Correo electrónico: [email protected]
Conflictos de interésLos autores declaran no tener conflictos.
preliminar gastro V1N2 10 3/14/12 11:26 AM

GASTROENTEROLOGY 2011;141:1986–1999
11
REVISIONES DE GASTROENTEROLOGÍA Y HEPATOLOGÍA BÁSICAS Y CLÍNICAS
Robert F. Schwabe y John W. Wiley, Editores de Sección
Los inflamasomas son complejos de multiproteínas mediadoras de la activación de la caspasa-1, que favorecen la secreción de las citocinas proinflamatorias ineterleucina-1β e interleucina-18 y la piroptosis, una forma de muerte celular inducida por los fagocitos patógenos bacterianos. Los miembros de la familia de los receptores semejantes a Nod (incluidos NLrp1, Nlrp3 y Nlrc4), el sensor de DNA Aim2, la proteína adaptadora semejante a speck asociada con la apoptosis (ASC) y la pro-caspasa-1 son componentes importantes de los inflamasomas. La estimulación con moléculas específicas microbianas y endógenas lleva al ensamblaje del inflamasoma y a la activación de la caspasa-1. Se cree que los inflamasomas son mediadores de la defensa del huésped contra patógenos microbianos y de la homeostasis de los tejidos intestinales, y su desregulación podría contribuir a las enfermedades inflamatorias y al cáncer intestinal. Mejorar nuestra comprensión de las vías de señales del inflamasoma podría propocionar información de la patogenia de muchos trastornos gastrointestinales y el desarrollo tanto de blancos terapéuticos como de enfoques para tratar afecciones como las enfermedades inflamatorias intestinales y los cánceres gastrointestinales.
Palabras clave: inmune; regulación; CRC; EII; microbiota.
Dentro del tracto gastrointestinal (GI) los receptores de la inmunidad innata funcionan como un mecanismo
inmediato de defensa contra los patógenos invasores. En 2004 Rakoff-Nahoum y colaboradores descubrieron que los receptores de reconocimiento de patrones (PRR, Pattern Recognition Receptors), que funcionan como sensores de las bacterias comensales, mantienen la homeostasis intestinal y la resistencia al daño.1 Esto no fue una sorpresa porque los PRR envían las señales a través de las vías inflamatorias que incluyen factores como el factor nuclear κB, la protein-cinasa activada por mitógenos, y la caspasa-1. Los PRR están involucrados no sólo en el control de la infección y la co-lonización bacteriana, sino también en la regulación de la función de barrera epitelial intestinal, la reparación epitelial y la homeostasis inmune.1-5 Por lo tanto, en el tracto GI los
defectos de la función de los PRR podrían estar involucra-dos en la patogenia de trastornos como la colitis infecciosa, las enfermedades inflamatorias intestinales (EII) y el cáncer.Hay por lo menos cuatro clases de PRR involucrados en el re-conocimiento de patógenos. Estos son los receptores seme-jantes a Toll (TLR), los receptores semejantes a Nod (NLR), los receptores semejantes a RIG-I y los receptores de tipo C de la lectina. Por lo regular estos PRR funcionan como sensores de motivos estructurales conservados o de patrones molecu-lares asociados con los patógenos (PAMP, Pathogen-associated molecular patterns) de los microbios, como los lipopolisacá-ridos y los peptidoglucanos que se encuentran en la pared celular de las bacterias. Los PRR son capaces de reconocer los patrones moleculares vinculados con el daño endógeno derivado del huésped (DAMP, Damage-associated molecular patterns), que se suelen generar durante el daño celular o ti-sular.6 En contraste con los TLR, que están localizados en la superficie extracelular de las células y en los endosomas, los NLR están localizados dentro del citoplasma y reconocen a los PAMP y los DAMP. Los NLR se definen por una estruc-tura tripartita: 1) un dominio N-terminal de reclutamiento de la caspasa (CARD), un dominio de la pirina (PYD), un dominio transactivador acídico, o repeticiones del inhibi-dor del baculovirus que son mediadores de las interacciones proteína-proteína; 2) un dominio de oligomerización cen-tral de la unión a nucleótidos (NOD), mediador de la autoo-ligomerización importante durante la activación; y 3) repe-ticiones ricas en leucina en el C-terminal que determinan la especificidad del ligando.7
**División de Hematología y Oncología, Departamento de Medicina Interna, y Centro Integral de Cáncer, y ‡Departamento de Patología y Centro Integral de Cáncer,
Universidad de Michigan, Ann Arbor, Michigan
GRACE Y, CHEN* y GABRIEL NÚÑEZ‡
Inflamasomas en la inflamación y el cáncer intestinal
Abreviaturas utilizadas en este artículo: AOM, azoximetano; ASC, pro-teína adaptadora semejante a speck asociada con la apoptosis; CAC, cáncer de colon asociado con colitis; CARD; dominio de reclutamiento
de la caspasa; DAMP, patrón molecular asociado con el daño; SSD, sulfato sódico dextrán; IL, interleucina; MDP, muramil dipéptido; NLR,
receptor semejante a Nod; NOD, dominio de oligomerización de la unión a nucleótidos; PAMP, patrón molecular asociado con patógenos; PRR,
receptor de reconocimiento de patrones; PYD, dominio de la pirina; P2X7R, receptor purinérgico P2X7; ROS, especies reactivas de oxígeno; T3SS, sistema de secreción de tipo III; T4SS, sistema de secreción de
tipo IV; TLR, receptor semejante a Toll.© 2011 by the AGA Institute
doi:10.1053/j.gastro.2011.10.002
01Inflamasomas 11 3/14/12 11:28 AM
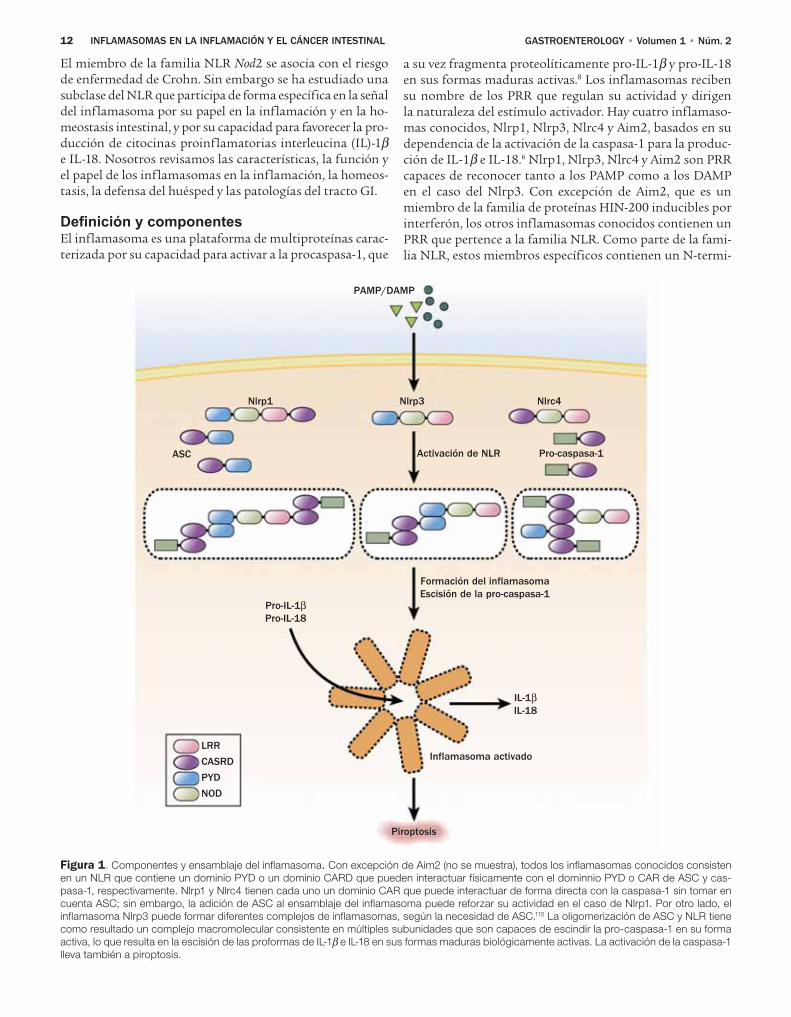
12 INFLAMASOMAS EN LA INFLAMACIÓN Y EL CÁNCER INTESTINAL GASTROENTEROLOGY • Volumen 1 • Núm. 2
Figura 1. Componentes y ensamblaje del inflamasoma. Con excepción de Aim2 (no se muestra), todos los inflamasomas conocidos consisten
en un NLR que contiene un dominio PYD o un dominio CARD que pueden interactuar físicamente con el dominnio PYD o CAR de ASC y cas-
pasa-1, respectivamente. Nlrp1 y Nlrc4 tienen cada uno un dominio CAR que puede interactuar de forma directa con la caspasa-1 sin tomar en
cuenta ASC; sin embargo, la adición de ASC al ensamblaje del inflamasoma puede reforzar su actividad en el caso de Nlrp1. Por otro lado, el
inflamasoma Nlrp3 puede formar diferentes complejos de inflamasomas, según la necesidad de ASC.115 La oligomerización de ASC y NLR tiene
como resultado un complejo macromolecular consistente en múltiples subunidades que son capaces de escindir la pro-caspasa-1 en su forma
activa, lo que resulta en la escisión de las proformas de IL-1β e IL-18 en sus formas maduras biológicamente activas. La activación de la caspasa-1
lleva también a piroptosis.
El miembro de la familia NLR Nod2 se asocia con el riesgo de enfermedad de Crohn. Sin embargo se ha estudiado una subclase del NLR que participa de forma específica en la señal del inflamasoma por su papel en la inflamación y en la ho-meostasis intestinal, y por su capacidad para favorecer la pro-ducción de citocinas proinflamatorias interleucina (IL)-1β e IL-18. Nosotros revisamos las características, la función y el papel de los inflamasomas en la inflamación, la homeos-tasis, la defensa del huésped y las patologías del tracto GI.
Definición y componentesEl inflamasoma es una plataforma de multiproteínas carac-terizada por su capacidad para activar a la procaspasa-1, que
a su vez fragmenta proteolíticamente pro-IL-1β y pro-IL-18 en sus formas maduras activas.8 Los inflamasomas reciben su nombre de los PRR que regulan su actividad y dirigen la naturaleza del estímulo activador. Hay cuatro inflamaso-mas conocidos, Nlrp1, Nlrp3, Nlrc4 y Aim2, basados en su dependencia de la activación de la caspasa-1 para la produc-ción de IL-1β e IL-18.6 Nlrp1, Nlrp3, Nlrc4 y Aim2 son PRR capaces de reconocer tanto a los PAMP como a los DAMP en el caso del Nlrp3. Con excepción de Aim2, que es un miembro de la familia de proteínas HIN-200 inducibles por interferón, los otros inflamasomas conocidos contienen un PRR que pertence a la familia NLR. Como parte de la fami-lia NLR, estos miembros específicos contienen un N-termi-
Activación de NLRASC Pro-caspasa-1
Formación del inflamasoma
Escisión de la pro-caspasa-1
LRR
CASRD
PYD
NOD
Inflamasoma activado
Piroptosis
Nlrc4Nlrp3
PAMP/DAMP
Nlrp1
IL-1βIL-18
Pro-IL-1βPro-IL-18
01Inflamasomas 12 3/14/12 11:28 AM

INFLAMASOMAS EN LA INFLAMACIÓN Y EL CÁNCER INTESTINAL 13GASTROENTEROLOGY • Volumen 1 • Núm. 2
nal PYD o un dominio CARD que puede interactuar con el dominio PYD de la proteína adaptadora semejante a speck asociada con la apoptosis (ASC) y un dominio CARD de la caspasa-1, respectivamente. ASC contiene en forma similar un dominio CARD además de su dominio PYD que puede interactuar con el domino CARD de la caspasa-1 durante la activación. Nlrc4, que no contiene un dominio PYD N-ter-minal como los otros, tiene en su lugar un dominio CARD N-terminal que en teoría puede reclutar de manera directa a la caspasa-1 a través de la interacción CARD-CARD. Ba-sados en estudios genéticos, bioquímicos y de resonancia magnética nuclear, el modelo prevalente del ensamblaje del inflamasoma y de la activación de la caspasa-1 es el siguien-te8-10 (Figura 1). La activación de los PRR por PAMP y DAMP resulta en la oligomerización a través del dominio NOD del PRR. De manera subsecuente, a través de interacciones pro-teína-proteína CARD-CARD y PYD-PYD se ensambla un complejo grande macromolecular semejante al apoptosoma involucrado en la activación de la caspasa-9 vía Apaf-1 de la apoptosis,11 que funciona como el andamiaje para el reclu-tamiento de la procaspasa-1 y la autofragmentación en cas-pasa-1 activa. La activación de la caspasa-1 conduce a la pro-ducción de IL-1β e IL-18 activas y el procesamiento de otros sustratos como la procaspasa-7.12 Además la activación de la caspasa-1 es mediadora de la piroptosis, una forma es-pecífica de muerte celular temprana inducida por patóge-nos intracelulares que favorece la lisis celular y la liberación de contenidos inflamatorios intracelulares para estimular
vías adicionales de señales inflamatorias.13,14 Por lo tanto, los eventos celulares que requieren producción de IL-1β e IL-18 o piroptosis por lo regular implican la activación del inflamasoma. No obstante hay algunas excepciones conoci-das en las cuales la fragmentación de pro-IL-1β y pro- IL-18 puede ocurrir de manera independiente al inflamasoma.15,16
Activadores del inflamasomaEstímulos microbianos y no microbianos pueden inducir la activación de los inflamasomas. Es importante notar que la activación de cada inflamasoma NLR o Aim2 es inducida por moléculas microbianas específicas o endógenas (Cuadro 1). Aunque la base molecular de la especificidad no se com-prende bien, la activación específica de cada inflamasoma es determinada en gran parte por que las moléculas activadoras particulares son reconocidas por NLR individuales o Aim2. A diferencia de Aim2, que se une de forma directa al DNA cito-sólico de doble cadena, no hay todavía evidencia concluyente de que los NLR involucrados en el inflamasoma interactúen directamente con moléculas microbianas o endógenas ac-tivadoras. Por lo tanto, es posible que el reconocimiento de las moléculas por los NLR del inflamasoma sea indirecto y mediado a través de factores intermedios, como se ha pro-puesto para otros miembros de la familia NLR de mamífe-ros o sus homólogos de las plantas.17,18 En el texto siguiente discutimos los estímulos específicos y los mecanismos que activan a los inflamasomas que tienen más relevancia en la inflamación intestinal, incluidos Nlrc4, Nlrp1 y Nlrp3.
Cuadro 1. Reconocimiento de patógenos por diferentes inflamasomas
Patógenos Activador microbiano Inflamasoma Referencias
Bacterianos
Staphylococcus aureus Hemolisinas Nlrp3 Munoz-Planillo et al, 2010
Vibrio cholerae HlyA y MARTXVc
Nlrp3 Toma et al, 2010
Streptococcus pyogenes Etreptolisina O Nlrp3 Harder et al, 2010
Chlamydia pneumonia Desconocido Nlrp3 He et al, 2010
Neisseria gonorrhea Desconocido Nlrp3 Duncan et al, 2009
Mycobacterium tuberculosis Desconocido Nlrp3, Nlrc4 Koo et al, 2008; Master et al, 2008
Listeria monocytogenes LLO, flagelina, DNA bacteriano Nlrp3, Nlrc4, Aim2 Mariathasan, 2006; Warren et al, 2008; Sauer et al, 2010;
Wu et al, 2010; Tsuchiya K, 2010; Meixenberger K, 2010
Salmonella typhimurium Flagelina, PrgJ Nlrc4, Nlrp3 Franchi et al, 2006; Miao et al, 2006; Miao et al, 2010;
Broz et al, 2011
Shigella flexneri MxiI? Nlrc4; Nlrp3 Susuki et al, 2007; Miao et al, 2010; Willingham SB, 2007
Pseudomonas aeruginosa Flagelina Nlrc4 Franchi et al, 2007; Galle et al, 2007
Legionella pneumophila Flagelina Amer et al; Lightfield KL, 2008; Zamboni DS, 2006
Bacillus anthracis Toxina letal Nlrp1b Nour et al, 2009, Boyden ED, 2006; Terra JK, 2010
Francisella tularensis DNA bacteriano Aim2 Fernandes-Alnemri et al, 2010; Rathinam et al, 2010;
Jones et al, 2010
Hongos
Candida albicans Desconocido Nlrp3 Gross et al, 2009; Hise et al, Joly S, 2009
Aspergillus fumigatus Desconocido Nlrp3 Said-Sadier et al, 2009
Virales
Sendai virus Desconocido Nlrp3 Kanneganti et al, 2006
Influenza A M2 Viral, RNA viral? Nlrp3 Thomas et al, 2009; Allen et al, 2009; Ichinohe et al, 2009
Adenovirus Desconocido Nlrp3 Muruve et al, 2008
Varicella-zoster Desconocido Nlrp3 Nour et al, 2011
Citomegalovirus DNA viral de doble cadena Aim2 Rathinam et al, 2010;
Virus de la vacuna DNA viral de doble cadena Aim2 Hornung et al, 2009; Rathinam et al, 2010
01Inflamasomas 13 3/14/12 11:28 AM

14 INFLAMASOMAS EN LA INFLAMACIÓN Y EL CÁNCER INTESTINAL GASTROENTEROLOGY • Volumen 1 • Núm. 2
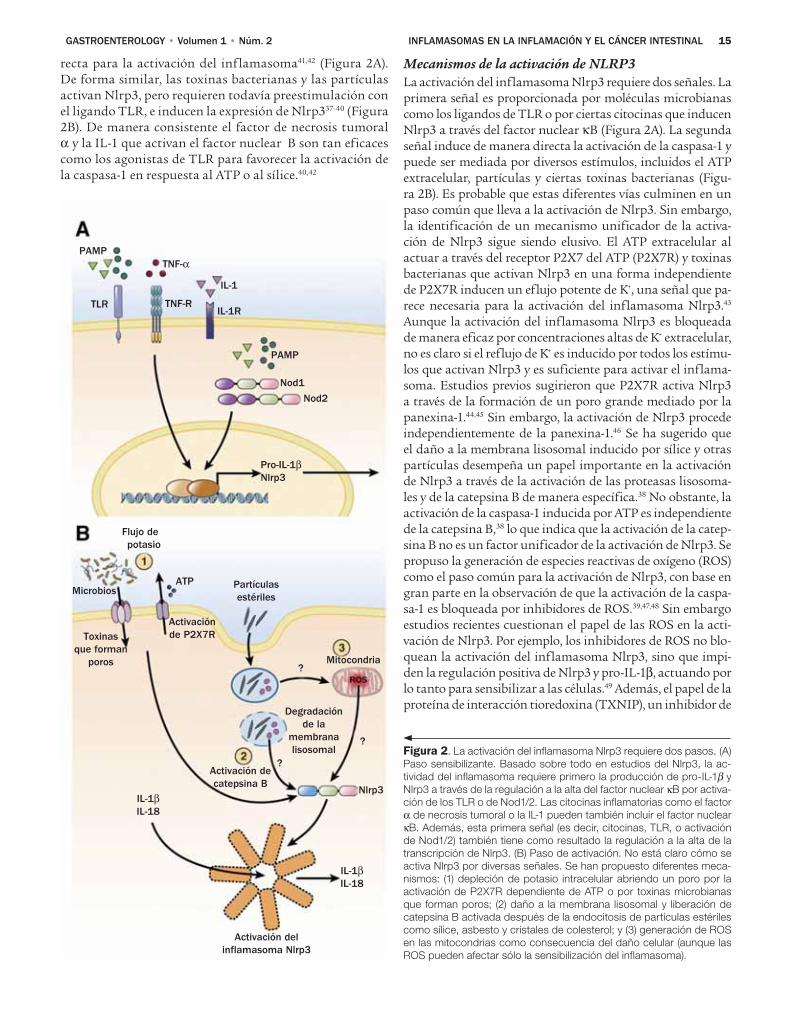
NLRC4Diversas bacterias gramnegativas, incluidas Legionella pneumophila, Pseudomonas aeruginosa, y los patógenos en-téricos Salmonella typhimurium y Shigella f lexneri, inducen la activación de la caspasa-1 vía el inf lamasoma Nlrc4. La activación del inf lamasoma Nlrc4 requiere un sistema de secreción tipo III intacto (T3SS) para S. typhimurium, S. f lexneri, y P. aureginosa o sistema de secreción tipo IV (T4SS) para L. pneumophila.19-22 En la infección estos siste-mas de secreción forman poros en las membranas celula-res del huésped y son mediadores de la translocación de un amplio conjunto de factores de virulencia (proteínas efectoras) en el citosol de la célula, que es crucial para la colonización de patógenos y la patología.23 Los análisis de mutantes de S. typhimurium, L. pneumophila y P. aeruginosa revelaron que la f lagelina, un componente del aparato f lagelar necesario para lo motilidad bacteriana, es indis-pensable para la activación de la caspasa-1 vía Nlrc4.19-21 Debido a que la liberación o expresión de la f lagelina es suficiente para inducir la activación del inf lamasoma Nlrc4, pequeñas cantidades de f lagelina que se fugan a través de T3SS o T4SS en el citosol del huésped parecen ser la señal para la activación de Nlrc4 durante la infección bacteriana. La activación de la caspasa-1 inducida por f la-gelina es independiente de TLR5, otro PRR que reconoce este componente del f lagelo.19 Por lo tanto, la f lagelina es reconocida por dos diferentes sensores: TLR5 reconoce a la f lagelina extracelular, mientras que Nlrc4 rconoce a la f la-gelina del citosol. Además de la f lagelina, hay evidencia de que PrgJ, un componente interno conservado del T3SS de la Salmonella, puede activar a Nlrc4.24 Es importante notar que Shigella no expresa f lagelina pero activa fuertemente a la caspasa-1 vía Nlrc4.24 Aunque no se ha identificado la molécula específica producida por Shigella que activa Nlrc4, es posible que su homóloga PrgJ conservada esté involucra-da porque se requiere la expresión de un T3SS funcional para la activación de la caspasa-1.
Hay evidencia de que la activación de Nlrc4 requiere la presencia de Naip5, otro miembro de la familia NLR. Naip5 parece reconocer la porción C-terminal de la flagelina de L. pneumophila y es necesaria para la activación del inflama-soma Nlrc4 en respuesta a esta bacteria.25 Se ha propuesto que Naip5 se relaciona físicamente con Nlrc4 para formar un inflamasoma complejo en respuesta a L. pneumophila. En contraste, la activación de la caspasa-1 inducida por S. typhi-murium y P. aeruginosa, que es también activada por la fla-gelina citosólica, es en gran parte independiente de Naip5.25 Debido a que L. pneumophila que expresa flagelina de la Sal-monella activa Nlrc4 en una forma dependiente de Naip5,26 el requisito diferencial de Naip5 no se debe a diferencias en la molécula de la flagelina entre las dos especies bacterianas. En fecha reciente se encontró que la flagelina se asocia de forma directa con Naip5, mientras que Naip2 se une a las proteínas del TTSS.27,28 Estos resultados indican que la activación del inflamasoma Nlrc4 es mediada de manera indirecta a través de la interacción de moléculas microbianas específicas con proteínas Naip diferentes.
NLRP1El inflamasoma humano NLRP1 fue el primer complejo de proteínas activador de la caspasa-1 en ser identificado.8 Además, el NLRP1 fue el único inflamasoma reconstituido in vitro con los componentes purificados, que mostró que el NLRP1 oligomeriza con la caspasa-1 en presencia del mu-ramil dipéptido (MDP).9 Mediante el uso de este sistema in vitro se propuso que la caspasa-1 es activada a través de un mecanismo de dos pasos en el cual el MDP microbiano in-duce un cambio conformacional del NLRP1, que a su vez le permite unirse al nucleótido y oligomerizarse, llevando a la activación de la caspasa-1.9 Sin embargo no hay evidencia di-recta de que MDP se una a NLRP1, por lo que el mecanismo que induce la oligomerización de NLRP1 no es claro. Nota-blemente, la molécula adaptadora ASC aumentó pero no fue necesaria para la activación in vitro de la caspasa-1 mediada por NLRP1,9 compatible con la observación de que ASC no es esencial para la activación de la caspasa-1 mediada por Nlrp1b en los macrófagos de los ratones.29 Por lo tanto, la proteína adaptadora ASC es necesaria para la activación de la caspasa-1 en la mayoría, pero no en todos los inflamaso-mas. A diferencia de los humanos, que tienen un solo gen NLRP1, los ratones tienen tres parálogos Nlrp1: Nlrp1a, b, y c. El inflamasoma Nlrp1b es activado por una toxina letal, una toxina bipartida secretada por Bacillus anthracis compuesta de antígenos protectores, una subunidad que forma poros que libera un factor letal, una metaloproteasa, en el citosol de las células infectadas.29 La observación de que los ratones con un alelo de susceptibilidad Nlrp1b son más sensibles a la toxina letal30,31 señala un papel importante del inflamaso-ma Nlrp1b en la defensa del huésped. Hay evidencia de que la activación de la caspasa-1, en respuesta a la infección con B anthracis, beneficia al huésped.32,33 Sin embargo no se com-prende bien el mecanismo por el cual el factor letal induce la activación del inflamasoma Nlrp1b.
NLRP3A diferencia de Nlrc4, que es activado sobre todo por la f la-gelina citosólica, se ha reportado que un conjunto grande de estímulos microbianos y no microbianos activan el in-flamasoma Nlrp3 en los macrófagos. Estos incluyen varios agonistas de los TLR y el agonista de Nod2 MDP en pre-sencia de trifosfato de adenosina (ATP) extracelular.34-36 Además, el Nrlp3 es acivado por ciertas toxinas bacteria-nas y partículas, incluidos cristales de uratos, asbestos, β-amiloide e hidróxido de aluminio en fagocitos preesti-mulados con ligandos microbianos como los lipopolisacá-ridos.37-40 La capacidad de múltiples PAMP para activar al inflamasoma Nlrp3 es desconcertante porque la mayoría de estas moléculas, incluso los ligandos TLR, no están re-lacionadas de manera estructural. Hallazgos recientes su-gieren que la mayoría, si no es que todos los agonistas de los TLR así como MDP, no actúan como activadores di-rectos de Nlrp3.41,42 Mas bien estos estímulos microbianos favorecen la activación del inflamasoma de forma indirec-ta a través de la inducción de Nlrp3 vía señales del factor nuclear κB, mientras que el ATP proporciona la señal di-
01Inflamasomas 14 3/14/12 11:28 AM
INFLAMASOMAS EN LA INFLAMACIÓN Y EL CÁNCER INTESTINAL 15GASTROENTEROLOGY • Volumen 1 • Núm. 2
recta para la activación del inflamasoma41,42 (Figura 2A). De forma similar, las toxinas bacterianas y las partículas activan Nlrp3, pero requieren todavía preestimulación con el ligando TLR, e inducen la expresión de Nlrp337-40 (Figura 2B). De manera consistente el factor de necrosis tumoral α y la IL-1 que activan el factor nuclear B son tan eficaces como los agonistas de TLR para favorecer la activación de la caspasa-1 en respuesta al ATP o al sílice.40,42
Mecanismos de la activación de NLRP3La activación del inflamasoma Nlrp3 requiere dos señales. La primera señal es proporcionada por moléculas microbianas como los ligandos de TLR o por ciertas citocinas que inducen Nlrp3 a través del factor nuclear κB (Figura 2A). La segunda señal induce de manera directa la activación de la caspasa-1 y puede ser mediada por diversos estímulos, incluidos el ATP extracelular, partículas y ciertas toxinas bacterianas (Figu-ra 2B). Es probable que estas diferentes vías culminen en un paso común que lleva a la activación de Nlrp3. Sin embargo, la identificación de un mecanismo unificador de la activa-ción de Nlrp3 sigue siendo elusivo. El ATP extracelular al actuar a través del receptor P2X7 del ATP (P2X7R) y toxinas bacterianas que activan Nlrp3 en una forma independiente de P2X7R inducen un eflujo potente de K+, una señal que pa-rece necesaria para la activación del inflamasoma Nlrp3.43 Aunque la activación del inflamasoma Nlrp3 es bloqueada de manera eficaz por concentraciones altas de K+ extracelular, no es claro si el reflujo de K+ es inducido por todos los estímu-los que activan Nlrp3 y es suficiente para activar el inflama-soma. Estudios previos sugirieron que P2X7R activa Nlrp3 a través de la formación de un poro grande mediado por la panexina-1.44,45 Sin embargo, la activación de Nlrp3 procede independientemente de la panexina-1.46 Se ha sugerido que el daño a la membrana lisosomal inducido por sílice y otras partículas desempeña un papel importante en la activación de Nlrp3 a través de la activación de las proteasas lisosoma-les y de la catepsina B de manera específica.38 No obstante, la activación de la caspasa-1 inducida por ATP es independiente de la catepsina B,38 lo que indica que la activación de la catep-sina B no es un factor unificador de la activación de Nlrp3. Se propuso la generación de especies reactivas de oxígeno (ROS) como el paso común para la activación de Nlrp3, con base en gran parte en la observación de que la activación de la caspa-sa-1 es bloqueada por inhibidores de ROS.39,47,48 Sin embargo estudios recientes cuestionan el papel de las ROS en la acti-vación de Nlrp3. Por ejemplo, los inhibidores de ROS no blo-quean la activación del inflamasoma Nlrp3, sino que impi-den la regulación positiva de Nlrp3 y pro-IL-1β, actuando por lo tanto para sensibilizar a las células.49 Además, el papel de la proteína de interacción tioredoxina (TXNIP), un inhibidor de
Figura 2. La activación del inflamasoma Nlrp3 requiere dos pasos. (A)
Paso sensibilizante. Basado sobre todo en estudios del Nlrp3, la ac-
tividad del inflamasoma requiere primero la producción de pro-IL-1β y
Nlrp3 a través de la regulación a la alta del factor nuclear κB por activa-
ción de los TLR o de Nod1/2. Las citocinas inflamatorias como el factor
α de necrosis tumoral o la IL-1 pueden también incluir el factor nuclear
κB. Además, esta primera señal (es decir, citocinas, TLR, o activación
de Nod1/2) también tiene como resultado la regulación a la alta de la
transcripción de Nlrp3. (B) Paso de activación. No está claro cómo se
activa Nlrp3 por diversas señales. Se han propuesto diferentes meca-
nismos: (1) depleción de potasio intracelular abriendo un poro por la
activación de P2X7R dependiente de ATP o por toxinas microbianas
que forman poros; (2) daño a la membrana lisosomal y liberación de
catepsina B activada después de la endocitosis de partículas estériles
como sílice, asbesto y cristales de colesterol; y (3) generación de ROS
en las mitocondrias como consecuencia del daño celular (aunque las
ROS pueden afectar sólo la sensibilización del inflamasoma).
PAMP
PAMP
Nod1
Nod2
TLR
TNF-α
Pro-IL-1βNlrp3
Flujo de
potasio
Microbios
Toxinas
que forman
poros
Activación
de P2X7R
Partículas
estériles
Degradación
de la
membrana
lisosomal
Mitocondria
Nlrp3
Activación de
catepsina B
Activación del
inflamasoma Nlrp3
IL-1βIL-18
IL-1βIL-18
ATP
TNF-RIL-1R
IL-1
?
?
?
01Inflamasomas 15 3/14/12 11:28 AM

16 INFLAMASOMAS EN LA INFLAMACIÓN Y EL CÁNCER INTESTINAL GASTROENTEROLOGY • Volumen 1 • Núm. 2
la tioredoxina antioxidante, para la activación de Nlrp3 no ha sido reproducido.50 Por lo tanto, el papel de ROS para activar el inflamasoma Nlrp3 es dudoso. Se requieren más estudios para comprender el mecanismo del inflamasoma Nlrp3.
NLRP6 como un posible regulador de la inflamaciónNlrp6 es un NLR mal caracterizado todavía que contiene un dominio N-terminal PYD, un dominio central NOD y repeti-ciones C-terminales ricas en leucina. Aunque la naturaleza de la señal que activa Nlrp6 no se conoce, estudios previos basa-dos en la sobreexpresión de NLRP6 sugieren su papel como participante del inflamasoma.51 De manera específica, la coexpresión de NLRP6 con ASC resultó en la producción coo-perativa de IL-1β en células COS-71 dependientes de la cas-pasa-1 y requirieron la presencia del dominio PYD.51 Además, los estudios de inmunofluorescencia mostraron localización del NLRP6 con ASC con un patrón moteado característico dentro del citoplasma.51 Es posible que el dominio PYN del NLRP6 interactúe de forma directa con el dominio PYD de ASC para permitir la activación de la caspasa-1, aunque nun-ca se ha demostrado una interacción directa con ASC o con la caspasa-1. Más recientemente, ratones deficientes en Nlrp6 mostraron niveles alterados del estado estable de la produc-ción de IL-18 en el suero y en los cultivos de explantes de colon en comparación con ratones de tipo silvestre,52 compatible con un papel de señal en la inflamación, aunque no hemos observado diferencias en los niveles basales de IL-18 entre ho-mogenados de tejidos del colon de Nlrp6–/– y de tipo silvestre.53
Hasta hace poco tiempo se desconocía la función del Nlrp6. El análisis de la expresión de Nlrp6 a nivel del RNA mensajero mostró que se expresa en gran medida en el intes-tino y significativamente menos en las células hematopoyéti-cas.51,53-55 Sin embargo, en las células hematopoyéticas Nlrp6 se expresa de forma diferencial; se detectaron mayores niveles de expresión en los granulocitos y linfocitos en comparación con los macrófagos y células dendríticas.51 Otros han repor-tado niveles elevados de expresión en los miofibroblastos, que tienen un papel importante en la reparación del epitelio intestinal,55,56 aunque no se sabe si el Nlrp6 funciona den-tro de este grupo de células. Han habido varios reportes que confirman un papel del Nlrp6 para favorecer la homeostasis intestinal (ver texto siguiente). Sin embargo queda por deter-minar si el Nlrp6 en realidad funciona como parte de un in-flamasoma in vivo; necesitamos una mayor comprensión de las señales del Nlrp6 y si la producción de IL-1β e IL-18 vía Nlrp6 es dependiente de ASC y de la caspasa-1.
Defensa del huéspedLa diarrea infecciosa aguda es una causa importante de morbilidad y mortalidad a nivel mundial, en especial en países en desarrollo y niños. Patógenos como Staphylococcus aureus, Escherichia coli enteropatógena y Vibrio cholerae pro-ducen toxinas que causan diarrea, y otros como Salmonella, Campylobacter, Shigella y rotavirus son invasores y causan inflamación intestinal grave. Los inflamasomas tienen un papel en la defensa del huésped contra las bacterias ente-ropatógenas. Por ejemplo, la activación del inflamasoma Nlrc4 funciona como una estrategia de defensa del huésped
contra las infecciones. En el caso de S. typhimurium y S. f lex-neri, la activación de la caspasa-1 dependiente de Nlrc4 se acompaña de secreción de IL-1β e induce la piroptosis.19,57 Es sorprendente notar que aunque se requiere Nlrc4 y la molécula adaptadora ASC para la activación de la caspasa-1 y la secreción de IL-1β, Nlrc4, pero no ASC, se requiere para la inducción de piroptosis.21,22 Hay evidencia de que el in-flamasoma es importante en la defensa del huésped contra la infección por Salmonella in vivo. Por ejemplo, los ratones nulos de caspasa-1 son susceptibles a la infección oral, que se asocia con aumento de la carga bacteriana en el hígado y ganglios linfáticos mesentéricos.58,59 La caspasa-1 tiene un papel en la fase sistémica de la infección, porque los rato-nes deficientes en caspasa-1 fueron más susceptibles que los ratones de tipo silvestre a la infección por Salmonella cuando se inyectaron de forma intraperitoneal.59 Además, se ha propuesto a la IL-18 como responsable de la defensa del huésped contra la Salmonella.59 Sin embargo los ratones deficientes en Nlrc4 fueron susceptibles de igual forma que los ratones de tipo silvestre;58 esto podría ocurrir debido a que Salmonella regula negativamente la expresión de la fla-gelina durante la fase sistémica de la infección.60 De manera consistente Nlrc4 tiene un papel importante en la defensa del huésped contra las bacterias cuando la flagelina es ex-presada por Salmonella durante la fase sistémica de la infec-ción.61 Salmonella codifica dos T3SS, SPI-1 favorece la inva-sión de las células epiteliales intestinales, y SPI-2 promueve la replicación en los macrófagos.62 En las cepas deficientes en SPI-1 de Salmonella, Nlrc4 actúa redundantemente con Nlrp3 para inducir la activación de la caspasa-1.63 Sin em-bargo no se conoce el mecanismo por el cual la activación de SPI-2 activa el inflamasoma Nlrp3.
La infección de los macrófagos por varios patóge-nos bacterianos, incluida Salmonella, induce la piroptosis durante la activación de la caspasa-1.64 Sorprendentemen-te, mientras que se requiere Nlrc4 y la proteína adaptadora ASC para la activación de la caspasa-1 y la secreción de IL-1β, Nlcr4, pero no ASC, es crucial para la inducción de la pirop-tosis.21,22,65 Aunque el mecanismo exacto no se comprende bien, hay evidencia de que la secreción de IL-1β y la pirop-tosis son mediadas por distintas vías de señales de la caspa-sa-1 que pueden ser separadas por la involucración de ASC.65 En la fase intestinal la Salmonella expresa SPI-1 y flagelina, lo que lleva a la activación de la caspasa-1 vía Nlrc4, mien-tras que en la fase sistémica la Salmonella no expresa SPI-1 ni flagelina. Sin embargo, la expresión forzada de flagelina durante la fase sistémica de la infección reveló un papel im-portante de la piroptosis dependiente de Nlrc4 para favore-cer la eliminación bacteriana. Específicamente, la piroptosis inducida por Nlrc4 favoreció la liberación de bacterias intra-celulares de las células fagocíticas, llevando a la captación y muerte del patógeno por los neutrófilos.61 Este proceso ocu-rrió independiente de la IL-1β y la IL-18, y explica por qué no se requiere la proteína adaptadora ASC para regular la susceptibilidad a la infección por Salmonella.58,61
Varios microorganismos patógenos, incluidos ciertos virus, hongos y bacterias, inducen la activación del infla-
01Inflamasomas 16 3/14/12 11:28 AM

INFLAMASOMAS EN LA INFLAMACIÓN Y EL CÁNCER INTESTINAL 17GASTROENTEROLOGY • Volumen 1 • Núm. 2
masoma Nlrp3. Entre las bacterias patógenas, S. aureus, Streptococcus pyogenes y V. cholerae activan a la caspasa-1 vía Nlrp3.66-68 De forma específica las toxinas producidas por las bacterias patógenas que forman poros o dañan a las membranas son importantes en la activación inducida del inflamasoma Nlrp3.66-68 A diferencia del ATP extracapsular, la activación del Nlrp3 inducida por la infección bacteria-na es independiente de P2X7R.66-68 Nlrp3 también regula la producción de IL-1β en respuesta a diversos virus como in-fluenza A, hongos como Candida albicans, y parásitos, inclui-do Plasmodium.69,70 Sin embargo se requieren estudios para determinar si el Nlrp3 regula la defensa del huésped contra virus o helmintos que causan de manera específica trastor-nos del intestino.
Reparación tisular y homeostasis GI IL-1β vs. IL-18La enfermedad inflamatoria intestinal (EII) comprende un grupo de trastornos crónicos que afectan a cerca de 1.4 millones de individuos en Estados Unidos y a 2.2 millones en Europa.71 La EII incluye a la colitis ulcerosa (CU) y la en-fermedad de Crohn (EC); en estos trastornos, el intestino se inflama de forma crónica, lo que lleva a la presentación clínica común de dolor abdominal, diarrea sanguinolenta y pérdida de peso. La CU y la EC se distinguen por criterios clínicos, endoscópicos e histológicos. En la CU la inflama-ción está restringida en la capa mucosa del colon, por lo re-gular en el recto, aunque puede extenderse continuamente hasta abarcar otras áreas del colon. En la EC la inflama-ción es transmural y puede ocurrir de una forma no con-tigua (lesiones salteadas) que involucran cualquier parte del tracto GI, con más frecuencia el íleon distal. Debido a la naturaleza transmural de la afección, pueden ocurrir complicaciones como estenosis intestinales y formación de fístulas que raras veces se asocian con la CU. La patogenia de la EII no es clara, pero los estudios indican tanto una contribución amnbiental como genética.72,73 El modelo prevalente del desarrollo de la EII implica una respuesta inmune aberrante a las bacterias comensales y/o un des-equilibrio en la estructura de la microbiota GI.74
El interés en el papel de los inflamasomas en la patogenia de la EII surgió de las observaciones de que los polimorfis-mos de los genes que codifican la IL-18 y las proteínas acce-sorias de IL-18 se asocian con aumento de la susceptibilidad a la EC.75,76 Además, los polimorfismos de componentes es-pecíficos del inflamasoma, en particular Nlrp3, que altera la producción de IL-1β por los monocitos estimulados por lipopolisacáridos, tuvieron susceptibilidad aumentada a la EC,77 aunque esta observación no fue reproducible en dife-rentes publicaciones.78 No obstante estos estudios de enlace genético indican un papel de la señal del inflamasoma y de la producción de IL-1β e IL-18 en el desarrollo de la EII.
Hay varios modelos de EII en ratones se utilizan para estudiar los mecanismos patogénicos de la EII, pero el que se emplea con más frecuencia para investigar el papel del inflamasoma es la inducción de colitis por sulfato sódico dextrán (SSD).73 El SSD produce daño epitelial directo, lo
que causa muerte de células epiteliales y aumento de la per-meabilidad intestinal, y resulta en ulceración y erosión de la mucosa, infiltración de células inflamatorias, así como regulación positiva de las citocinas proinflamatorias.79-81 Las lesiones por lo regular sólo afectan a la mucosa princi-palmente en el colon y el recto como ocurre en la CU, pero pueden desarrollarse de forma discontinua. El modelo de SSD ha sido criticado por representar lo que ocurre en el daño epitelial agudo, que el daño de la enfermedad mediada por células T; la colitis inducida por SSD afecta también a animales deficientes de células T, aunque la células T con-tribuyen a la gravedad de la enfermedad.82-85 Sin embargo el modelo de SSD ha sido útil para identificar factores impor-tantes de la reparación epitelial intestinal y la función de ba-rrera, así como las respuestas inmunes innatas involucradas en el desarrollo y control de la inflamación entérica.
Debido a que la EII se asocia con una regulación positi-va en la inducción de citocinas proinflamatorias, incluidas IL-1β e IL-18,86,87 una regulación negativa de estas citocinas proinflamatorias podría relacionarse con disminución en la gravedad de la enfermedad. Los ratones Balb/c deficientes en caspasa-1 fueron menos susceptibles a la colitis inducida por SSD, al tener mejores puntuaciones clínicas e histoló-gicas en comparación con los ratones de tipo silvestre.88 De manera consistente los ratones transgénicos que sobreex-presan IL-18 tuvieron aumento de la colitis89 e inhibición química de la enfermedad reducida de IL-18 o caspasa-1.90,91
No obstante, estudios recientes muestran un papel regu-lador negativo de la señal del inflamasoma en el desarrollo de la colitis. De forma específica, los ratones B6 deficientes tanto en ASC como en caspasa-1 tuvieron mayor suscep-tiblidad al SSD, con proliferación y restitución epitelial alterada, aumento de la permeabilidad intestinal, y mayor translocación de las bacterias comensales a la mucosa co-lónica y a los ganglios linfáticos mesentéricos.92 Aunque la deficiencia de caspasa-1 y ASC se asoció con disminución de la producción de IL-1β e IL-18, el mecanismo de la gravedad en la inflamación en ratones deficientes en caspsasa-1 se vinculó con menor producción de IL-18; la administración de IL-18 recombinante fue suficiente para rescatar a los ra-tones B6 deficientes en caspasa-1.92 Se propuso que la fuente de IL-18 son las células epiteliales, basados en los cultivos in vitro de células intestinales enriquecidas con células epi-teliales versus de lámina propia de ratones que recibieron SSD.92 La alteración de la producción de IL-18 en los ratones deficientes en caspasa-1 durante la colitis inducida por SSD y la capacidad para revertir este efecto con la administración de IL-18 se reprodujeron por un grupo diferente, apoyando un modelo en el cual la producción de IL-18 dependiente de caspasa-1, tal vez por el epitelio intestinal, es importante para la reparación y regeneración epitelial en la colitis inducida por SSD.93 (Figura 3). Sin embargo el epitelio podría no ser la úni-ca fuente de IL-18; hay evidencia de que la IL-18 se produce por células epiteliales y de la lámina propia en la EII.94
La IL-18 tradicionalmente se considera proinflamatoria, con importancia para inducir respuestas Th1; de manera específica interferón gamma, y por lo tanto el mecanismo
01Inflamasomas 17 3/14/12 11:28 AM
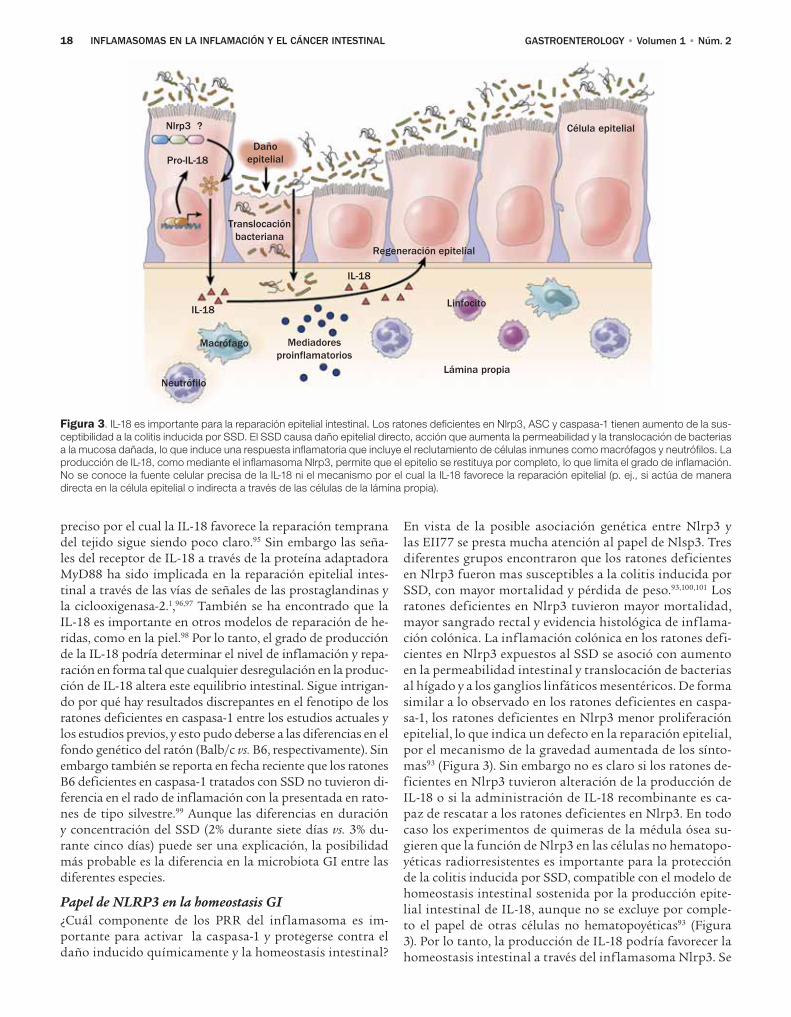
18 INFLAMASOMAS EN LA INFLAMACIÓN Y EL CÁNCER INTESTINAL GASTROENTEROLOGY • Volumen 1 • Núm. 2
preciso por el cual la IL-18 favorece la reparación temprana del tejido sigue siendo poco claro.95 Sin embargo las seña-les del receptor de IL-18 a través de la proteína adaptadora MyD88 ha sido implicada en la reparación epitelial intes-tinal a través de las vías de señales de las prostaglandinas y la ciclooxigenasa-2.1,96,97 También se ha encontrado que la IL-18 es importante en otros modelos de reparación de he-ridas, como en la piel.98 Por lo tanto, el grado de producción de la IL-18 podría determinar el nivel de inflamación y repa-ración en forma tal que cualquier desregulación en la produc-ción de IL-18 altera este equilibrio intestinal. Sigue intrigan-do por qué hay resultados discrepantes en el fenotipo de los ratones deficientes en caspasa-1 entre los estudios actuales y los estudios previos, y esto pudo deberse a las diferencias en el fondo genético del ratón (Balb/c vs. B6, respectivamente). Sin embargo también se reporta en fecha reciente que los ratones B6 deficientes en caspasa-1 tratados con SSD no tuvieron di-ferencia en el rado de inflamación con la presentada en rato-nes de tipo silvestre.99 Aunque las diferencias en duración y concentración del SSD (2% durante siete días vs. 3% du-rante cinco días) puede ser una explicación, la posibilidad más probable es la diferencia en la microbiota GI entre las diferentes especies.
Papel de NLRP3 en la homeostasis GI¿Cuál componente de los PRR del inflamasoma es im-portante para activar la caspasa-1 y protegerse contra el daño inducido químicamente y la homeostasis intestinal?
En vista de la posible asociación genética entre Nlrp3 y las EII77 se presta mucha atención al papel de Nlsp3. Tres diferentes grupos encontraron que los ratones deficientes en Nlrp3 fueron mas susceptibles a la colitis inducida por SSD, con mayor mortalidad y pérdida de peso.93,100,101 Los ratones deficientes en Nlrp3 tuvieron mayor mortalidad, mayor sangrado rectal y evidencia histológica de inflama-ción colónica. La inflamación colónica en los ratones defi-cientes en Nlrp3 expuestos al SSD se asoció con aumento en la permeabilidad intestinal y translocación de bacterias al hígado y a los ganglios linfáticos mesentéricos. De forma similar a lo observado en los ratones deficientes en caspa-sa-1, los ratones deficientes en Nlrp3 menor proliferación epitelial, lo que indica un defecto en la reparación epitelial, por el mecanismo de la gravedad aumentada de los sínto-mas93 (Figura 3). Sin embargo no es claro si los ratones de-ficientes en Nlrp3 tuvieron alteración de la producción de IL-18 o si la administración de IL-18 recombinante es ca-paz de rescatar a los ratones deficientes en Nlrp3. En todo caso los experimentos de quimeras de la médula ósea su-gieren que la función de Nlrp3 en las células no hematopo-yéticas radiorresistentes es importante para la protección de la colitis inducida por SSD, compatible con el modelo de homeostasis intestinal sostenida por la producción epite-lial intestinal de IL-18, aunque no se excluye por comple-to el papel de otras células no hematopoyéticas93 (Figura 3). Por lo tanto, la producción de IL-18 podría favorecer la homeostasis intestinal a través del inflamasoma Nlrp3. Se
Figura 3. IL-18 es importante para la reparación epitelial intestinal. Los ratones deficientes en Nlrp3, ASC y caspasa-1 tienen aumento de la sus-
ceptibilidad a la colitis inducida por SSD. El SSD causa daño epitelial directo, acción que aumenta la permeabilidad y la translocación de bacterias
a la mucosa dañada, lo que induce una respuesta inflamatoria que incluye el reclutamiento de células inmunes como macrófagos y neutrófilos. La
producción de IL-18, como mediante el inflamasoma Nlrp3, permite que el epitelio se restituya por completo, lo que limita el grado de inflamación.
No se conoce la fuente celular precisa de la IL-18 ni el mecanismo por el cual la IL-18 favorece la reparación epitelial (p. ej., si actúa de manera
directa en la célula epitelial o indirecta a través de las células de la lámina propia).
Nlrp3 ?
Pro-IL-18
IL-18
IL-18
Macrófago
Linfocito
Neutrófilo
Célula epitelial
Regeneración epitelial
Daño
epitelial
Translocación
bacteriana
Mediadores
proinflamatorios
Lámina propia
01Inflamasomas 18 3/14/12 11:28 AM
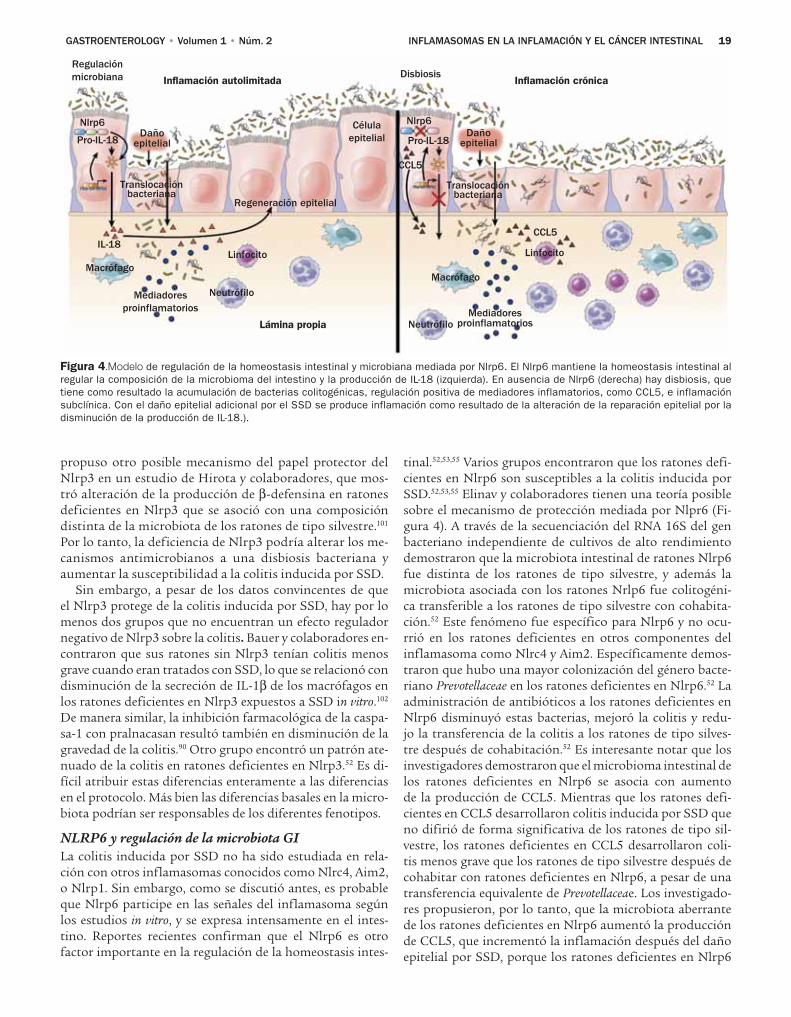
INFLAMASOMAS EN LA INFLAMACIÓN Y EL CÁNCER INTESTINAL 19GASTROENTEROLOGY • Volumen 1 • Núm. 2
propuso otro posible mecanismo del papel protector del Nlrp3 en un estudio de Hirota y colaboradores, que mos-tró alteración de la producción de β-defensina en ratones deficientes en Nlrp3 que se asoció con una composición distinta de la microbiota de los ratones de tipo silvestre.101 Por lo tanto, la deficiencia de Nlrp3 podría alterar los me-canismos antimicrobianos a una disbiosis bacteriana y aumentar la susceptibilidad a la colitis inducida por SSD.
Sin embargo, a pesar de los datos convincentes de que el Nlrp3 protege de la colitis inducida por SSD, hay por lo menos dos grupos que no encuentran un efecto regulador negativo de Nlrp3 sobre la colitis. Bauer y colaboradores en-contraron que sus ratones sin Nlrp3 tenían colitis menos grave cuando eran tratados con SSD, lo que se relacionó con disminución de la secreción de IL-1β de los macrófagos en los ratones deficientes en Nlrp3 expuestos a SSD in vitro.102
De manera similar, la inhibición farmacológica de la caspa-sa-1 con pralnacasan resultó también en disminución de la gravedad de la colitis.90 Otro grupo encontró un patrón ate-nuado de la colitis en ratones deficientes en Nlrp3.52 Es di-fícil atribuir estas diferencias enteramente a las diferencias en el protocolo. Más bien las diferencias basales en la micro-biota podrían ser responsables de los diferentes fenotipos.
NLRP6 y regulación de la microbiota GILa colitis inducida por SSD no ha sido estudiada en rela-ción con otros inflamasomas conocidos como Nlrc4, Aim2, o Nlrp1. Sin embargo, como se discutió antes, es probable que Nlrp6 participe en las señales del inflamasoma según los estudios in vitro, y se expresa intensamente en el intes-tino. Reportes recientes confirman que el Nlrp6 es otro factor importante en la regulación de la homeostasis intes-
tinal.52,53,55 Varios grupos encontraron que los ratones defi-cientes en Nlrp6 son susceptibles a la colitis inducida por SSD.52,53,55 Elinav y colaboradores tienen una teoría posible sobre el mecanismo de protección mediada por Nlpr6 (Fi-gura 4). A través de la secuenciación del RNA 16S del gen bacteriano independiente de cultivos de alto rendimiento demostraron que la microbiota intestinal de ratones Nlrp6 fue distinta de los ratones de tipo silvestre, y además la microbiota asociada con los ratones Nrlp6 fue colitogéni-ca transferible a los ratones de tipo silvestre con cohabita-ción.52 Este fenómeno fue específico para Nlrp6 y no ocu-rrió en los ratones deficientes en otros componentes del inflamasoma como Nlrc4 y Aim2. Específicamente demos-traron que hubo una mayor colonización del género bacte-riano Prevotellaceae en los ratones deficientes en Nlrp6.52 La administración de antibióticos a los ratones deficientes en Nlrp6 disminuyó estas bacterias, mejoró la colitis y redu-jo la transferencia de la colitis a los ratones de tipo silves-tre después de cohabitación.52 Es interesante notar que los investigadores demostraron que el microbioma intestinal delos ratones deficientes en Nlrp6 se asocia con aumento de la producción de CCL5. Mientras que los ratones defi-cientes en CCL5 desarrollaron colitis inducida por SSD que no difirió de forma significativa de los ratones de tipo sil-vestre, los ratones deficientes en CCL5 desarrollaron coli-tis menos grave que los ratones de tipo silvestre después de cohabitar con ratones deficientes en Nlrp6, a pesar de una transferencia equivalente de Prevotellaceae. Los investigado-res propusieron, por lo tanto, que la microbiota aberrante de los ratones deficientes en Nlrp6 aumentó la producción de CCL5, que incrementó la inflamación después del daño epitelial por SSD, porque los ratones deficientes en Nlrp6
Figura 4.Modelo de regulación de la homeostasis intestinal y microbiana mediada por Nlrp6. El Nlrp6 mantiene la homeostasis intestinal al
regular la composición de la microbioma del intestino y la producción de IL-18 (izquierda). En ausencia de Nlrp6 (derecha) hay disbiosis, que
tiene como resultado la acumulación de bacterias colitogénicas, regulación positiva de mediadores inflamatorios, como CCL5, e inflamación
subclínica. Con el daño epitelial adicional por el SSD se produce inflamación como resultado de la alteración de la reparación epitelial por la
disminución de la producción de IL-18.).
Mediadores
proinflamatorios MediadoresproinflamatoriosLámina propia
Inflamación autolimitada Inflamación crónica
Regulación
microbiana Disbiosis
Nlrp6 Nlrp6
Pro-IL-18 Pro-IL-18
IL-18CCL5
CCL5
Macrófago
Regeneración epitelial
Célula
epitelial
Neutrófilo
Neutrófilo
Linfocito Linfocito
Daño epitelial
Translocación bacteriana
Translocación bacteriana
Daño epitelial
Macrófago
01Inflamasomas 19 3/14/12 11:28 AM
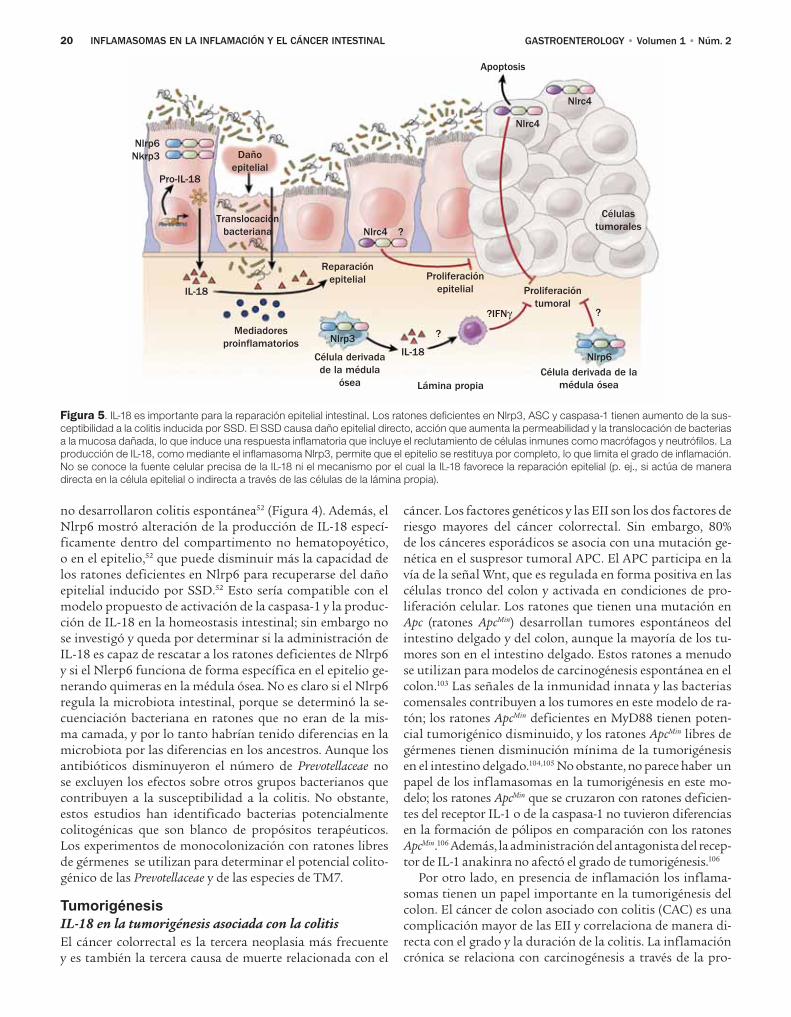
20 INFLAMASOMAS EN LA INFLAMACIÓN Y EL CÁNCER INTESTINAL GASTROENTEROLOGY • Volumen 1 • Núm. 2
no desarrollaron colitis espontánea52 (Figura 4). Además, el Nlrp6 mostró alteración de la producción de IL-18 especí-ficamente dentro del compartimento no hematopoyético, o en el epitelio,52 que puede disminuir más la capacidad de los ratones deficientes en Nlrp6 para recuperarse del daño epitelial inducido por SSD.52 Esto sería compatible con el modelo propuesto de activación de la caspasa-1 y la produc-ción de IL-18 en la homeostasis intestinal; sin embargo no se investigó y queda por determinar si la administración de IL-18 es capaz de rescatar a los ratones deficientes de Nlrp6 y si el Nlerp6 funciona de forma específica en el epitelio ge-nerando quimeras en la médula ósea. No es claro si el Nlrp6 regula la microbiota intestinal, porque se determinó la se-cuenciación bacteriana en ratones que no eran de la mis-ma camada, y por lo tanto habrían tenido diferencias en la microbiota por las diferencias en los ancestros. Aunque los antibióticos disminuyeron el número de Prevotellaceae no se excluyen los efectos sobre otros grupos bacterianos que contribuyen a la susceptibilidad a la colitis. No obstante, estos estudios han identificado bacterias potencialmente colitogénicas que son blanco de propósitos terapéuticos. Los experimentos de monocolonización con ratones libres de gérmenes se utilizan para determinar el potencial colito-génico de las Prevotellaceae y de las especies de TM7.
TumorigénesisIL-18 en la tumorigénesis asociada con la colitisEl cáncer colorrectal es la tercera neoplasia más frecuente y es también la tercera causa de muerte relacionada con el
cáncer. Los factores genéticos y las EII son los dos factores deriesgo mayores del cáncer colorrectal. Sin embargo, 80% de los cánceres esporádicos se asocia con una mutación ge-nética en el suspresor tumoral APC. El APC participa en la vía de la señal Wnt, que es regulada en forma positiva en las células tronco del colon y activada en condiciones de pro-liferación celular. Los ratones que tienen una mutación en Apc (ratones ApcMin) desarrollan tumores espontáneos del intestino delgado y del colon, aunque la mayoría de los tu-mores son en el intestino delgado. Estos ratones a menudo se utilizan para modelos de carcinogénesis espontánea en el colon.103 Las señales de la inmunidad innata y las bacterias comensales contribuyen a los tumores en este modelo de ra-tón; los ratones ApcMin deficientes en MyD88 tienen poten-cial tumorigénico disminuido, y los ratones ApcMin libres de gérmenes tienen disminución mínima de la tumorigénesis en el intestino delgado.104,105 No obstante, no parece haber un papel de los inflamasomas en la tumorigénesis en este mo-delo; los ratones ApcMin que se cruzaron con ratones deficien-tes del receptor IL-1 o de la caspasa-1 no tuvieron diferencias en la formación de pólipos en comparación con los ratones ApcMin.106 Además, la administración del antagonista del recep-tor de IL-1 anakinra no afectó el grado de tumorigénesis.106
Por otro lado, en presencia de inflamación los inflama-somas tienen un papel importante en la tumorigénesis del colon. El cáncer de colon asociado con colitis (CAC) es una complicación mayor de las EII y correlaciona de manera di-recta con el grado y la duración de la colitis. La inflamación crónica se relaciona con carcinogénesis a través de la pro-
Figura 5. IL-18 es importante para la reparación epitelial intestinal. Los ratones deficientes en Nlrp3, ASC y caspasa-1 tienen aumento de la sus-
ceptibilidad a la colitis inducida por SSD. El SSD causa daño epitelial directo, acción que aumenta la permeabilidad y la translocación de bacterias
a la mucosa dañada, lo que induce una respuesta inflamatoria que incluye el reclutamiento de células inmunes como macrófagos y neutrófilos. La
producción de IL-18, como mediante el inflamasoma Nlrp3, permite que el epitelio se restituya por completo, lo que limita el grado de inflamación.
No se conoce la fuente celular precisa de la IL-18 ni el mecanismo por el cual la IL-18 favorece la reparación epitelial (p. ej., si actúa de manera
directa en la célula epitelial o indirecta a través de las células de la lámina propia).
Apoptosis
Nlrp6
Nkrp3
Pro-IL-18
Daño
epitelial
Translocación
bacteriana
Mediadores
proinflamatorios
Reparación
epitelial Proliferación
epitelial
Células
tumorales
Nlrc4
Nlrc4
Nlrp3
Nlrp6
Nlrc4
?IFNγ
Proliferación
tumoral
Célula derivada
de la médula
óseaCélula derivada de la
médula ósea
IL-18
IL-18
?
?
?
Lámina propia
01Inflamasomas 20 3/14/12 11:28 AM

INFLAMASOMAS EN LA INFLAMACIÓN Y EL CÁNCER INTESTINAL 21GASTROENTEROLOGY • Volumen 1 • Núm. 2
ducción de radicales libres de oxígeno que dañan al DNA, mediadores proinflamatorios que favorecen la superviven-cia celular, y aumento de la proliferación y angiogénesis, así como la remodelación tisular que produce un microam-biente que favorece la tumorigénesis.107 Los ratones azoxi-metano (AOM)/SSD son un modelo para el CAC; el carci-nógeno azoximetano se utilliza para introducir mutaciones genómicas mediante metilación, seguidas de rondas repe-tidas de SSD para inducir un patrón de colitis crónica.108 Después de tres rondas de SSD los ratones desarrollan ade-nomas macroscópicamente visibles; con el tiempo la pro-porción de adenocarcinomas aumenta, lo que indica que este modelo recapitula eventos implicados en la progresión de los adenomas premalignos a los adenocarcinomas en los humanos.109 Sin embargo este modelo es criticado porque no incluye características importantes del CAC; las muta-ciones APC se desarrollan de forma temprana en el modelo AOM/SSD, pero tardía y con menos frecuencia en el CAC,110 y la predominancia de pólipos adenomatosos en los ratones AOM/SSD difiere de la lesiones planas displásicas observa-das en pacientes con EII.111 No obstante este modelo es útil para identificar factores y vías de señales importantes para modular la inflamación y la posterior carcinogénesis.
En vista de la asociación entre la inflamación y el cáncer colorrectal no sorprende que los inflamasomas, que protegen al intestino de la inflamación excesiva después del daño quí-micamente inducido, tengan un papel en la susceptibilidad al CAC (Figura 5). En el modelo AOM/SSD, aunque los ratones deficientes en IL-1R desarrollan un número similar de tumo-res que los ratones de tipo silvestre, los ratones deficientes en IL-18 e IL-18R tienen un número aumentado de tumores, lo que indica un papel del inflamasoma en las señales de su-presión tumoral en el contexto de la inflamación crónica.112 Los ratones deficientes en MyD88, con disminución en las señales de IL-18, tuvieron aumento de tumores en el modelo AOM/SSD, asociados con niveles disminuidos de varios fac-tores de reparación del DNA y expresión aumentada de genes mitogénicos y angiogénicos.112 Las alteraciones de los meca-nismos de reparación del DNA podrían, por lo tanto, aumen-tar la tumorigénesis en los ratones deficientes en MyD88, corriente abajo de las señales del receptor de IL-18.
NLRP3 en el CACCon un aumento de IL-18, se reporta que el Nlrp3 regula en forma negativa la tumorigénesis asociada con la colitis100,113 por mecanismos desconocidos. Zaki y colaboradores encon-traron que los ratones deficientes en caspasa-1 tuvieron más tumores, en forma similar a los ratones deficientes en Nlrp3, lo cual se asoció con disminución de la producción de IL-18 y activación de STAT1.106 La administración de IL-18 resta-bleció los niveles de fosforilación de STAT1 de los ratones de tipo silvestre, por lo que los investigadores propusieron que las señales de la IL-18 son importantes para la inducción de interferón gamma y la vigilancia inmune.113 Sin embargo se aclaró si la normalización de las señales de STAT1 con la ad-ministración de IL-18 resultaron en menos tumores. Allen y colaboradores notaron mayor susceptibilidad de los ratones
deficientes en Nlrp3 y consistentemente deficientes en ASC a los tumores asociados con la colitis con un defecto en la pro-ducción de IL-18 durante la inducción de la inflamación en el modelo AOM/SSD.100 En forma interesante, los experimen-tos de quimeras de la médula ósea muestran que el Nlrp3 es importante en el compartimento hematopoyético más que en el compartimento epitelial, lo que sugiere un papel del infla-masoma en la supresión del tumor independiente de la pro-ducción de IL-18 por el epitelio intestinal. En contraste con la inflamación, la tumorigénesis podría requerir la función del inflamasoma en el compartimento de células hematopoyéti-cas más que en el compartimento epitelial. NLRP6 suprime la tumorigénesis y la inflamación intestinal
En forma similar al Nlrp3, el Nlrp6 (particularmente en el compartimento hematopoyético) protege del desarrollo de tumores asociados con la colitis.53 Esto es interesante porque el Nlrp6 se expresa más en el epitelio que en las cé-lulas hematopoyéticas, aunque es posible que en forma se-mejante al Nlpr3 la expresión del Nlrp6 sea inducida por la activación.53 La tumorigénesis aumentada se relacionó con aumento en las respuestas inflamatorias con produc-ción deficiente de IL-18 en el colon después del tratamiento con AOM/SSD.53 No es clara la forma en que esto aumenta el potencial de tumores, en vista de la predominancia de la producción de IL-18 por el epitelio intestinal, más que por las células derivadas de la lámina propia del colon o de la médula ósea.53 El análisis de la expresión de genes de tumo-res en los ratones Nlrp6 y de tipo silvestre reveló diferencias en las moléculas de señales importantes para la prolifera-ción y transformación epitelial, como en las vías Notch y Wnt, por lo que la desregulación de esas vías en los ratones deficientes en Nlrp6 podrían contribuir a la tumorigéne-sis.55 Se requieren estudios para investigar la relación entre la producción de IL-18, la función del Nlrp6 en las células hematopoyéticas, y otras vías de señales carcinogénicas po-tencialmente reguladas por el Nlrp6.
NLRC4 regula la tumorigénesis independientemente de la inflamaciónHa habido reportes controvertidos de que el NLrc4 tam-bién suprime la formación tumoral; un estudio no en-contró un papel del Nlrc4100 en el desarrollo de tumores asociados con la colitis, y otro mostró un papel regulador negativo del Nlrc4.99 Hu y colaboradores mostraron que los ratones deficientes en Nlrc4 desarrollaron más tumores en el modelo AOM/SSD, sin relación con la inflamación, y la gravedad de la colitis en estos ratones no fue diferente de la observada en los ratones de tipo silvestre.99 Más bien la deficiencia de Nlrc4 aumentó la proliferación epitelial y disminuyó la apoptosis en tumores avanzados.99 Aunque los mecanismos de este efecto no son claros, los investiga-dores utilizaron experimentos de quimeras de la médula ósea para mostrar que la función del Nlrc4 en el compar-timento de células no hematopoyéticas es importante para la supresión tumoral, y proponen que las señales del Nlrc4 dentro del epitelio regulan la apoptosis y la proliferación de los tumores.99,114 Sin embargo se necesitó la función de
01Inflamasomas 21 3/14/12 11:28 AM

22 INFLAMASOMAS EN LA INFLAMACIÓN Y EL CÁNCER INTESTINAL GASTROENTEROLOGY • Volumen 1 • Núm. 2
la caspasa-1 en el compartimento hematopoyético y epi-telial,99 tal vez debido a la influencia de otros componen-tes del inflamasoma que funcionan en el compartimento hematopoyético para la supresión tumoral (p. ej., Nlrp3 o Nlrp6). Es interesante notar que Allen y colaboradores no encontraron diferencias en el potencial tumoral entre los ratones Nlrc4 y de tipo silvestre con el modelo AOM/SSD,100 lo que indica que las diferencias en la microbiota intestinal en diferentes instalaciones podrían afectar los resultados de la deficiencia de Nlrc4.
ConclusiónLos inflamasomas, como los PRR, son importantes en la de-fensa de organismos patógenos que pueden invadir el tracto GI. Sin embargo los inflamasomas mantienen la integridad del epitelio intestinal y favorecen la reparación, lo que hace que sean factores importantes en la patogenia de las EII y del cáncer. Estudios recientes también han encontrado que los in-flamasomas regulan el microbioma GI y afectan, por lo tanto, la susceptibilidad a enfermedades más allá del tracto GI, in-cluidas la obesidad y la diabetes. Al aprender más respecto al repertorio de las señales derivadas de los microbios y del hués-ped reconocidas por los inflamasomas, las estrategias para mo-dular la actividad de los inflamasomas podrían utilizarse para desarrollar tratamientos para los trastornos GI y no GI.
Referencias1. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, et al. Recogni-
tion of commensal microflora by toll-like receptors is required for
intestinal homeostasis. Cell 2004;118:229–241.
2. Chen GY, Shaw MH, Redondo G, et al. The innate immune re-
ceptor Nod1 protects the intestine from inflammation-induced
tumorigenesis. Cancer Res 2008;68:10060–10067.
3. Bouskra D, Brezillon C, Berard M, et al. Lymphoid tissue genesis
induced by commensals through NOD1 regulates intestinal ho-
meostasis. Nature 2008;456:507–510.
4. Fukata M, Chen A, Vamadevan AS, et al. Toll-like receptor-4 pro-
motes the development of colitis-associated colorectal tumors.
Gastroenterology 2007;133:1869–1881.
5. Saleh M, Trinchieri G. Innate immune mechanisms of colitis and
colitis-associated colorectal cancer. Nat Rev Immunol 2011;11:
9–20.
6. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to
damage. Nat Rev Immunol 2010;10:826–837.
7. Chen G, Shaw MH, Kim YG, et al. NOD-like receptors: role in
innate immunity and inflammatory disease. Annu Rev Pathol
2009;4:365–398.
8. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular
platform triggering activation of inflammatory caspases and pro-
cessing of proIL-beta. Mol Cell 2002;10:417–426.
9. Faustin B, Lartigue L, Bruey JM, et al. Reconstituted NALP1 in-
flammasome reveals two-step mechanism of caspase-1 activa-
tion. Mol Cell 2007;25:713–724.
10. de Alba E. Structure and interdomain dynamics of apoptosi-
sassociated speck-like protein containing a CARD (ASC). J Biol
Chem 2009;284:32932–32941.
11. Bratton SB, Salvesen GS. Regulation of the Apaf-1-caspase-9
apoptosome. J Cell Sci 2010;123:3209–3214.
12. Lamkanfi M, Kanneganti TD, Van Damme P, et al. Targeted peptide-
centric proteomics reveals caspase-7 as a substrate of the caspa-
se-1 inflammasomes. Mol Cell Proteomics 2008;7:2350–2363.
13. Fernandes-Alnemri T, Wu J, Yu JW, et al. The pyroptosome: a
supramolecular assembly of ASC dimers mediating inflam-
matory cell death via caspase-1 activation. Cell Death Differ
2007;14:1590–1604.
14. Fink SL, Cookson BT. Pyroptosis and host cell death responses
during Salmonella infection. Cell Microbiol 2007;9:2562–2570.
15. Mayer-Barber KD, Barber DL, Shenderov K, et al. Caspase-1 in-
dependent IL-1beta production is critical for host resistance to
mycobacterium tuberculosis and does not require TLR signaling
in vivo. J Immunol 2010;184:3326–3330.
16. van de Veerdonk FL, Netea MG, Dinarello CA, et al. Inflammaso-
me activation and IL-1beta and IL-18 processing during infec-
tion. Trends Immunol 2011;32:110–116.
17. DeYoung BJ, Innes RW. Plant NBS-LRR proteins in pathogen sen-
sing and host defense. Nat Immunol 2006;7:1243–1249.
18. Franchi L, Warner N, Viani K, et al. Function of Nod-like recep-
tors in microbial recognition and host defense. Immunol Rev
2009;227:106–128.
19. Franchi L, Amer A, Body-Malapel M, et al. Cytosolic flagellin requi-
res Ipaf for activation of caspase-1 and interleukin 1beta in sal-
monella-infected macrophages. Nat Immunol 2006;7:576–582.
20. Amer A, Franchi L, Kanneganti TD, et al. Regulation of Legionella
phagosome maturation and infection through flagellin and host
Ipaf. J Biol Chem 2006;281:35217–35223.
21. Franchi L, Stoolman J, Kanneganti TD, et al. Critical role for Ipaf
in Pseudomonas aeruginosa-induced caspase-1 activation. Eur
J Immunol 2007;37:3030–3039.
22. Suzuki T, Franchi L, Toma C, et al. Differential regulation of cas-
pase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in
Shigella-infected macrophages. PLoS Pathog 2007;3:e111.
23. Galan JE, Wolf-Watz H. Protein delivery into eukaryotic cells by
type III secretion machines. Nature 2006;444:567–573.
24. Miao EA, Mao DP, Yudkovsky N, et al. Innate immune detection
of the type III secretion apparatus through the NLRC4 inflam-
masome. Proc Natl Acad Sci U S A 2010;107:3076–3080.
25. Lightfield KL, Persson J, Brubaker SW, et al. Critical function
for Naip5 in inflammasome activation by a conserved carboxy-
terminal domain of flagellin. Nat Immunol 2008;9:1171–1178.
26. Lightfield KL, Persson J, Trinidad NJ, et al. Differential requi-
rements for NAIP5 in activation of the NLRC4 inflammasome.
Infect Immun 2011;79:1606–1614.
27. Zhao Y, Yang J, Shi J, et al. The NLRC4 inflammasome receptors
for bacterial flagellin and type III secretion apparatus. Nature
2011;477:596–600.
28. Kofoed EM, Vance RE. Innate immune recognition of bacterial
ligands by NAIPs determines inflammasome specificity. Nature
2011;477:592–595.
29. Nour AM, Yeung YG, Santambrogio L, et al. Anthrax lethal toxin tri-
ggers the formation of a membrane-associated inflammasome com-
plex in murine macrophages. Infect Immun 2009;77:1262–1271.
30. Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage sus-
ceptibility to anthrax lethal toxin. Nat Genet 2006;38:240–244.
31. Kang TJ, Basu S, Zhang L, et al. Bacillus anthracis spores and
lethal toxin induce IL-1beta via functionally distinct signaling
pathways. Eur J Immunol 2008;38:1574–1584.
32. Terra JK, Cote CK, France B, et al. Cutting edge: resistance to
Bacillus anthracis infection mediated by a lethal toxin sensitive
allele of Nalp1b/Nlrp1b. J Immunol 2010;184:17–20.
33. Moayeri M, Crown D, Newman ZL, et al. Inflammasome sensor
Nlrp1b-dependent resistance to anthrax is mediated by caspa-
se-1, IL-1 signaling and neutrophil recruitment. PLoS Pathog
2010;6:e1001222.
34. Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates
the inflammasome in response to toxins and ATP. Nature 2006;
440:228–232.
35. Sutterwala FS, Ogura Y, Szczepanik M, et al. Critical role for
NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through
its regulation of caspase-1. Immunity 2006;24:317–327.
36. Franchi L, Kanneganti TD, Dubyak GR, et al. Differential requi-
rement of P2X7 receptor and intracellular K for caspase-1 acti-
vation induced by intracellular and extracellular bacteria. J Biol
Chem 2007;282:18810–18818.
01Inflamasomas 22 3/14/12 11:28 AM

INFLAMASOMAS EN LA INFLAMACIÓN Y EL CÁNCER INTESTINAL 23GASTROENTEROLOGY • Volumen 1 • Núm. 2
37. Martinon F, Petrilli V, Mayor A, et al. Gout-associated uric acid crys-
tals activate the NALP3 inflammasome. Nature 2006;440:237–241.
38. Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and alu-
minum salts activate the NALP3 inflammasome through phago-
somal destabilization. Nat Immunol 2008;9:847–856.
39. Dostert C, Petrilli V, Van Bruggen R, et al. Innate immune acti-
vation through Nalp3 inflammasome sensing of asbestos and
silica. Science 2008;320:674–677.
40. Franchi L, Eigenbrod T, Munoz-Planillo R, et al. The inflammasome:
a caspase-1-activation platform that regulates immune responses
and disease pathogenesis. Nat Immunol 2009;10:241–247.
41. Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NFka-
ppaB activating pattern recognition and cytokine receptors li-
cense NLRP3 inflammasome activation by regulating NLRP3
expression. J Immunol 2009;183:787–791.
42. Franchi L, Eigenbrod T, Nunez G. Cutting edge: TNF-alpha mediates
sensitization to ATP and silica via the NLRP3 inflammasome in the
absence of microbial stimulation. J Immunol 2009;183: 792–796.
43. Kahlenberg JM, Dubyak GR. Mechanisms of caspase-1 activa-
tion by P2X7 receptor-mediated K release. Am J Physiol Cell Phy-
siol 2004;286:C1100–C1108.
44. Pelegrin P, Surprenant A. Pannexin-1 mediates large pore forma-
tion and interleukin-1beta release by the ATP-gated P2X7 recep-
tor. EMBO J 2006;25:5071–5082.
45. Kanneganti TD,Lamkanfi M, Kim YG, et al. Pannexin-1-mediated
recognition of bacterial molecules activates the cryopyrin in-
flammasome independent of Toll-like receptor signaling. Immu-
nity 2007;26:433–443.
46. Qu Y, Misaghi S, Newton K, et al. Pannexin-1 is required for ATP
release during apoptosis but not for inflammasome activation. J
Immunol 2011;186:6553–6561.
47. Zhou R, Tardivel A, Thorens B, et al. Thioredoxin-interacting pro-
tein links oxidative stress to inflammasome activation. Nat Im-
munol 2010;11:136–140.
48. Zhou R, Yazdi AS, Menu P, et al. A role for mitochondria in NLRP3
inflammasome activation. Nature 2011;469:221–225.
49. Bauernfeind F, Bartok E, Rieger A, et al. Cutting edge: reactive
oxygen species inhibitors block priming, but not activation, of
the NLRP3 inflammasome. J Immunol 2011;187:613–617.
50. Masters SL, Dunne A, Subramanian SL, et al. Activation of the
NLRP3 inflammasome by islet amyloid polypeptide provides a
mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immu-
nol 2010;11:897–904.
51. Grenier JM, Wang L, Manji GA, et al. Functional screening of five
PYPAF family members identifies PYPAF5 as a novel regulator of
NF-kappaB and caspase-1. FEBS Lett 2002;530:73–78.
52. Elinav E, Strowig T, Kau AL, et al. NLRP6 inflammasome re-
gulates colonic microbial ecology and risk for colitis. Cell
2011;145:745–757.
53. Chen GY, Liu M, Wang F, et al. A functional role for nlrp6 in intestinal
inflammation and tumorigenesis. J Immunol 2011;186:7187–7194.
54. Lech M, Avila-Ferrufino A, Skuginna V, et al. Quantitative expres-
sion of RIG-like helicase, NOD-like receptor and inflammasomere-
lated mRNAs in humans and mice. Int Immunol 2010;22:717–728.
55. Normand S, Delanoye-Crespin A, Bressenot A, et al. Nod-like
receptor pyrin domain-containing protein 6 (NLRP6) controls
epithelial self-renewal and colorectal carcinogenesis upon in-
jury. Proc Natl Acad Sci U S A 2011;108:9601–9606.
56. Andoh A, Bamba S, Brittan M, et al. Role of intestinal subepithe-
lial myofibroblasts in inflammation and regenerative response in
the gut. Pharmacol Ther 2007;114:94–106.
57. Miao EA, Alpuche-Aranda CM, Dors M, et al. Cytoplasmic flage-
llin activates caspase-1 and secretion of interleukin 1beta via
Ipaf. Nat Immunol 2006;7:569–575.
58. Lara-Tejero M, Sutterwala FS, Ogura Y, et al. Role of the caspa-
se-1 inflammasome in Salmonella typhimurium pathogenesis. J
Exp Med 2006;203:1407–1412.
59. Raupach B, Peuschel SK, Monack DM, et al. Caspase-1-mediated
activation of interleukin-1beta (IL-1beta) and IL-18 contributes to
innate immune defenses against Salmonella enterica serovar
Typhimurium infection. Infect Immun 2006;74:4922–4926.
60. Alaniz RC, Cummings LA, Bergman MA, et al. Salmonella typhi-
murium coordinately regulates FliC location and reduces den-
dritic cell activation and antigen presentation to CD4 T cells. J
Immunol 2006;177:3983–3993.
61. Miao EA, Leaf IA, Treuting PM, et al. Caspase-1-induced pyropto-
sis is an innate immune effector mechanism against intracellu-
lar bacteria. Nat Immunol 2010;11:1136–1142.
62. Hueffer K, Galan JE. Salmonella-induced macrophage death:
multiple mechanisms, different outcomes. Cell Microbiol 2004;
6:1019–1025.
63. Broz P, Newton K, Lamkanfi M, et al. Redundant roles for inflam-
masome receptors NLRP3 and NLRC4 in host defense against
Salmonella. J Exp Med 2010;207:1745–1755.
64. Lamkanfi M, Dixit VM. Manipulation of host cell death pathways
during microbial infections. Cell Host Microbe 2010;8:44–54.
65. Broz P, von Moltke J, Jones JW, et al. Differential requirement for
Caspase-1 autoproteolysis in pathogen-induced cell death and
cytokine processing. Cell Host Microbe 2010;8:471–483.
66. Munoz-Planillo R, Franchi L, Miller LS, et al. A critical role for he-
molysins and bacterial lipoproteins in Staphylococcus aureusin-
duced activation of the Nlrp3 inflammasome. J Immunol 2009;
183:3942–3948.
67. Harder J, Franchi L, Munoz-Planillo R, et al. Activation of the Nlrp3
inflammasome by Streptococcus pyogenes requires streptolysin
O and NF-kappa B activation but proceeds independently of TLR
signaling and P2X7 receptor. J Immunol 2009;183:5823–5829.
68. Toma C, Higa N, Koizumi Y, et al. Pathogenic Vibrio activate
NLRP3 inflammasome via cytotoxins and TLR/nucleotide-bin-
ding oligomerization domain-mediated NF-kappa B signaling. J
Immunol 2010;184:5287–5297.
69. Gross O, Poeck H, Bscheider M, et al. Syk kinase signalling cou-
ples to the Nlrp3 inflammasome for anti-fungal host defence.
Nature 2009;459:433–436.
70. Reimer T, Shaw MH, Franchi L, et al. Experimental cerebral ma-
laria progresses independently of the Nlrp3 inflammasome. Eur
J Immunol 2010;40:764–769.
71. Loftus EV Jr. Clinical epidemiology of inflammatory bowel disea-
se: Incidence, prevalence, and environmental influences. Gas-
troenterology 2004;126:1504–1517.
72. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of in-
flammatory bowel disease. Nature 2011;474:307–317.
73. Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal
models of inflammation. Annu Rev Immunol 2002;20:495–549.
74. Strober W, Fuss I, Mannon P. The fundamental basis of inflam-
matory bowel disease. J Clin Invest 2007;117:514–521.
75. Tamura K, Fukuda Y, Sashio H, et al. IL18 polymorphism is asso-
ciated with an increased risk of Crohn’s disease. J Gastroenterol
2002;37(Suppl 14):111–116.
76. Zhernakova A, Festen EM, Franke L, et al. Genetic analysis of
innate immunity in Crohn’s disease and ulcerative colitis identi-
fies two susceptibility loci harboring CARD9 and IL18RAP. Am J
Hum Genet 2008;82:1202–1210.
77. Villani AC, Lemire M, Fortin G, et al. Common variants in the
NLRP3 region contribute to Crohn’s disease susceptibility. Nat
Genet 2009;41:71–76.
78. Lewis GJ, Massey DC, Zhang H, et al. Genetic association bet-
ween NLRP3 variants and Crohn’s disease does not replicate in
a large UK panel. Inflamm Bowel Dis 2011;17:1387–1391.
79. Cooper HS, Murthy SN, Shah RS, et al. Clinicopathologic study
of dextran sulfate sodium experimental murine colitis. Lab In-
vest 1993;69:238–249.
80. Okayasu I, Hatakeyama S, Yamada M, et al. A novel method in
the induction of reliable experimental acute and chronic ulcera-
tive colitis in mice. Gastroenterology 1990;98:694–702.
81. Ni J, Chen SF, Hollander D. Effects of dextran sulphate sodium
on intestinal epithelial cells and intestinal lymphocytes. Gut
1996;39:234–241.
82. Axelsson LG, Landstrom E, Goldschmidt TJ, et al. Dextran sul-
fate sodium (DSS) induced experimental colitis in immunodefi-
cient mice: effects in CD4() -cell depleted, athymic and NK-cell
depleted SCID mice. Inflamm Res 1996;45:181–191.
01Inflamasomas 23 3/14/12 11:28 AM

24 INFLAMASOMAS EN LA INFLAMACIÓN Y EL CÁNCER INTESTINAL GASTROENTEROLOGY • Volumen 1 • Núm. 2
83. Shintani N, Nakajima T, Okamoto T, et al. Involvement of CD4 T
cells in the development of dextran sulfate sodium-induced ex-
perimental colitis and suppressive effect of IgG on their action.
Gen Pharmacol 1998;31:477–481.
84. Chen Y, Chou K, Fuchs E, et al. Protection of the intestinal muco-
sa by intraepithelial gamma delta T cells. Proc Natl Acad Sci U S
A 2002;99:14338–14343.
85. Kim TW, Seo JN, Suh YH, et al. Involvement of lymphocytes in
dextran sulfate sodium-induced experimental colitis. World J
Gastroenterol 2006;12:302–305.
86. Street ME, de’Angelis G, Camacho-Hubner C, et al. Relationships
between serum IGF-1, IGFBP-2, interleukin-1beta and interleukin-6
in inflammatory bowel disease. Horm Res 2004;61:159–164.
87. Li J, Moran T, Swanson E, Julian C, et al. Regulation of IL-8
and IL-1beta expression in Crohn’s disease associated NOD2/
CARD15 mutations. Hum Mol Genet 2004;13:1715–1725.
88. Siegmund B, Lehr HA, Fantuzzi G, et al. IL-1 beta -converting
enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad
Sci U S A 2001;98:13249–13254.
89. Ishikura T, Kanai T, Uraushihara K, et al. Interleukin-18 over-
production exacerbates the development of colitis with marke-
dly infiltrated macrophages in interleukin-18 transgenic mice. J
Gastroenterol Hepatol 2003;18:960–969.
90. Bauer C, Loher F, Dauer M, et al. The ICE inhibitor pralnacasan
prevents DSS-induced colitis in C57BL/6 mice and suppresses
IP-10 mRNA but not TNF-alpha mRNA expression. Dig Dis Sci
2007;52:1642–1652.
91. Siegmund B, Fantuzzi G, Rieder F, et al. Neutralization of inter-
leukin-18 reduces severity in murine colitis and intestinal IFN-
gamma and TNF-alpha production. Am J Physiol Regul Integr
Comp Physiol 2001;281:R1264–R1273.
92. Dupaul-Chicoine J, Yeretssian G, Doiron K, et al. Control of intes-
tinal homeostasis, colitis, and colitis-associated colorectal can-
cer by the inflammatory caspases. Immunity 2010;32:367–378.
93. Zaki MH, Boyd KL, Vogel P, et al. The NLRP3 inflammasome
protects against loss of epithelial integrity and mortality during
experimental colitis. Immunity 2010;32:379–391.
94. Pizarro TT, Michie MH, Bentz M, et al. IL-18, a novel immuno-
regulatory cytokine, is up-regulated in Crohn’s disease: expres
sion and localization in intestinal mucosal cells. J Immunol
1999;162:6829–6835.
95. Nakanishi K, Yoshimoto T, Tsutsui H, et al. Interleukin-18 regulates
both Th1 and Th2 responses. Annu Rev Immunol 2001;19:423–474.
96. Fukata M, Chen A, Klepper A, et al. Cox-2 is regulated by Toll-like
receptor-4 (TLR4) signaling: Role in proliferation and apoptosis
in the intestine. Gastroenterology 2006;131:862–877.
97. Brown SL, Riehl TE, Walker MR, et al. Myd88-dependent positio-
ning of Ptgs2-expressing stromal cells maintains colonic epithe-
lial proliferation during injury. J Clin Invest 2007;117:258–269.
98. Kampfer H, Paulukat J, Muhl H, et al. Lack of interferon-gamma
production despite the presence of interleukin-18 during cuta-
neous wound healing. Mol Med 2000;6:1016–1027.
99. Hu B, Elinav E, Huber S, et al. Inflammation-induced tumorigene-
sis in the colon is regulated by caspase-1 and NLRC4. Proc Natl
Acad Sci U S A 2010;107:21635–21640.
100. Allen IC, TeKippe EM, Woodford RM, et al. The NLRP3 inflam-
masome functions as a negative regulator of tumorigenesis du-
ring colitis-associated cancer. J Exp Med 2010;207:1045–1056.
101. Hirota SA, Ng J, Lueng A, et al. NLRP3 inflammasome plays a key
role in the regulation of intestinal homeostasis. Inflamm Bowel
Dis 2011;17:1359–1372.
102. Bauer C, Duewell P, Mayer C, et al. Colitis induced in mice with
dextran sulfate sodium (DSS) is mediated by the NLRP3 inflam-
masome. Gut 2010;59:1192–1199.
103. Rosenberg DW, Giardina C, Tanaka T. Mouse models for the stu-
dy of colon carcinogenesis. Carcinogenesis 2009;30:183–196.
104. Dove WF, Clipson L, Gould KA, et al. Intestinal neoplasia in the
ApcMin mouse: independence from the microbial and natural
killer (beige locus) status. Cancer Res 1997;57:812–814.
105. Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous
intestinal tumorigenesis through the adaptor protein MyD88.
Science 2007;317:124–127.
106. Lee SH, Hu LL, Gonzalez-Navajas J, et al. ERK activation dri-
ves intestinal tumorigenesis in Apc(min/) mice. Nat Med
2010;16:665–670.
107. Coussens LM, Werb Z. Inflammation and cancer. Nature
2002;420:860–867.
108. Tanaka T, Kohno H, Suzuki R, et al. A novel inflammation-related
mouse colon carcinogenesis model induced by azoxymethane
and dextran sodium sulfate. Cancer Sci 2003;94:965–973.
109. Suzuki R, Kohno H, Sugie S, et al. Sequential observations on
the occurrence of preneoplastic and neoplastic lesions in mou-
se colon treated with azoxymethane and dextran sodium sulfa-
te. Cancer Sci 2004;95:721–727.
110. Tarmin L, Yin J, Harpaz N, et al. Adenomatous polyposis coli
gene mutations in ulcerative colitis-associated dysplasias and
cancers versus sporadic colon neoplasms. Cancer Res 1995;
55:2035–2038.
111. Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal can-
cer in inflammatory bowel disease: the role of inflammation. Am
J Physiol Gastrointest Liver Physiol 2004;287:G7–G17.
112. Salcedo R, Worschech A, Cardone M, et al. MyD88-mediated
signaling prevents development of adenocarcinomas of the co-
lon: role of interleukin 18. J Exp Med 2010;207:1625–1636.
113. Zaki MH, Vogel P, Body-Malapel M, et al. IL-18 production downs-
tream of the Nlrp3 inflammasome confers protection against
colorectal tumor formation. J Immunol 2010;185:4912–4920.
114. Hu B, Elinav E, Flavell RA. Inflammasome-mediated suppression
of inflammation-induced colorectal cancer progression is media-
ted by direct regulation of epithelial cell proliferation. Cell Cycle
2011;10:1936–1939.
115. Abdelaziz DH, Gavrilin MA, Akhter A, et al. Asc-dependent and
independent mechanisms contribute to restriction of legionella
pneumophila infection in murine macrophages. Front Microbiol
2011;2:18.
Recibido el 13 de julio de 2011. Aceptado el 8 de septiembre de 2011.
Solicitud de reimpresosDirigir las solicitudes de reimpresos a: Grace Y. Chen, MD, PhD, Divi-
sion of Hematology and Oncology, Department of Internal Medicine,
and Comprehensive Cancer Center, University of Michigan, Ann Arbor,
Michigan 48109; correo electrónico: [email protected]; or Gabriel
Núñez, MD, Department of Pathology and Comprehensive Cancer Cen-
ter, University of Michigan, Ann Arbor, Michigan 48109; correo electró-
nico: [email protected].
AgradecimientosDedicado a la memoria de Jürg Tschopp, quien falleció repentina-
mente el 22 de marzo de 2011. Los autores ofrecen disculpas a los
autores y colaboradores cuyo trabajo no fue citado o se citó a través de
artículos de revisión de lotross, debido al espacio limitado, y gracias a
Luigi Franchi por sus críticas al manuscrito.
Conflictos de interésLos autores no declararon conflictos de intereses.
RecursosEl trabajo de los autores fue apoyado por los National Institutes of Health sub-
venciones CA133185 y P30 CA46592-22S3 y el American Cancer Society Research Scholar subvención (a G.Y.C.) y National Institutes of Health sub-
vención DK61707, AR051790, AI06331, AR059688, y DK091191 (a G.N.).
01Inflamasomas 24 3/14/12 11:28 AM

GASTROENTEROLOGY 2011;141:2000–2008
25
CLÍNICA — TRACTO ALIMENTARIO
ANTECEDENTES Y OBJETIVOS: la incidencia de esófago de Barrett y adenocarcinoma esofágico ha aumentado, a pesar de la vigilancia de los pacientes con esófago de Barrett. Existen pocos datos que demuestran que el uso de fármacos antiinfl amatorios no esteroideos (AINE) y estatinas disminuyen el riesgo de adenocarcinoma esofágico. Nosotros investigamos si el uso de AINE y/o estatinas disminuye el riesgo de progresión neoplásica del esófago de Barrett. MÉTODOS: llevamos a cabo un estudio prospectivo de 570 pacientes con esófago de Barrett en tres hospitales académicos y 12 hospitales regionales holandeses. La información del uso de medicamentos se documentó de encuestas realizadas a los pacientes durante las citas de seguimiento y se verifi có con los registros de la farmacia. Los individuos completaron un cuestionario sobre el uso de medicamentos que se obtienen sin receta. Los casos incidentes de displasia de alto grado y de adenocarcinoma se identifi caron durante el periodo de seguimiento. RESULTADOS: durante una media de seguimiento de 4.5 años, 38 pacientes (7%) desarrollaron displasia de alto grado o adenocarcinoma. Después que se diagnosticó esófago de Barrett, 318 pacientes (56%) utilizaron AINE con una mediana de duración de dos meses, 161 (28%) utilizaron ácido acetilsalicílico una mediana de duración de cinco años, 209 (37%) utilizaron estatinas una mediana deduración de cinco años, y 107 (19%) utilizaron AINE y estatinas. El uso de AINE y estatinas se asoció con un riesgo menor de progresión neoplásica (índice de riesgo [IR]. 0.47; P = .030 e IR, 0.46; P = .048, respectivamente). El uso de la combinación de AINE y estatinas aumentó el efecto protector (IR, 0.22; P = .028). CONCLUSIONES: el uso de AINE y estatinas disminuye el riesgo de progresión neoplásica en pacientes con esófago de Barrett. El uso de la combinación de AINE y estatinas parece tener un efecto protector aditivo.
Palabras clave: Enfermedad por reflujo gastroesofágico; Quimioprevención; Riesgo de cáncer; Tumor.
El esófago de Barrett (EB) es un trastorno premaligno en el cual el epitelio escamoso normal del esófago dis-
tal es reemplazado por epitelio columnar metaplásico que contiene células en anillo de sello.1 Es un trastorno relati-vamente común con una prevalencia estimada de 1 a 2% en los países occidentales.2-4 La enfermedad crónica por reflu-jo gastroesofágico desempeña un papel central en el desa-rrollo del epitelio de Barrett, y cerca de 10% de los pacien-tes con enfermedad por reflujo gastroesofágico de forma eventual desarrollará EB.5 Los pacientes con Barrett tienen un riesgo aumentado 30 a 125 veces de desarrollar ade-nocarcinoma esofágico (ACE), con una incidencia anual aproximada de 0.5%.6,7 Desafortunadamente, todavía no es posible predecir qué pacientes tienen un mayor riesgo de desarrollar ACE. Como resultado se recomienda el segui-miento endoscópico en todos los pacientes con EB.8,9
Las estrategias para prevenir el desarrollo de adenocar-cinoma en el EB se han enfocado sobre todo en la reversión del epitelio de Barrett y la detección temprana del adeno-carcinoma durante la vigilancia. Sin embargo, a pesar de la vigilancia de los pacientes con Barrett, la incidencia de ACE ha aumentando rápidamente.10,11 Por lo tanto, se requieren nuevas estrategias para prevenir el desarrollo de adenocar-cinoma. Múltiples estudios han apoyado el uso de la qui-mioprevención en el tratamiento de varios cánceres, inclui-do el cáncer esofágico.12 Estudios observacionales sugieren que el uso de antiinflamatorios no esteroides (AINE) y estatinas disminuiyen el riesgo de progresión neoplásica en los pacientes con EB.13-15 La quimioprevención con una combinación de AINE y estatinas podría proporcionar una disminución todavía mayor del riesgo.16,17 Sin embargo sólo estudios limitados han investigado el efecto del uso de AINE y estatinas sobre el desarrollo de displasia de alto grado (DAG) y ACE en el EB. La mayoría de los estudios sólo incluyeron números pequeños de pacientes y carecen de información clínica. Hasta donde sabemos no se ha pu-
*Departamento de Gastroenterología y Hepatología, ‡Departamento de Patología, §Departamento de Salud Pública, y IIDepartamento de Medicina Interna, Centro Médico
Universeitario Erasmus, Rotterdam, Países Bajos
FLORINE KASTELEIN,* MANON C. W. SPAANDER,* KATHARINA BIERMANN,‡ EWOUT W. STEYERBERG,§ ERNST J. KUIPERS,II
y MARCO J. BRUNO* en nombre del Grupo de Estudio Probar
Los fármacos antiinflamatorios no esteroides y las estatinas tienen efectos quimiopreventivos en pacientes con esófago de Barrett
Abreviaturas en este artículo: EB, esófago de Barrett; IC, intervalo de confianza; COX, ciclooxigenasa; ACE, adenocarcinoma esofágico; DAG, displasia de alto grado; DBG, displasia de bajo grado; AINE, antiinfla-matorios no esteroidos; AINEns, antiinflamatorios no esteroideos no
selectivos; IBP, inhibidores de la bomba de protones.© 2011 by the AGA Institute
doi:10.1053/j.gastro.2011.08.036
02Nosteroidal 25 3/14/12 11:30 AM

26 LOS FÁRMACOS ANTIINFLAMATORIOS NO ESTEROIDEOS Y LAS ESTATINAS GASTROENTEROLOGY Vol. 1 No. 2
blicado ningún estudio prospectivo de cohorte grande que investigue el uso de la combinación de AINE y estatinas. El objetivo de este estudio fue, por lo tanto, investigar si el uso de AINE y estatinas disminuye el riesgo de progresión neoplásica en pacientes con EB.
Pacientes y métodos
Diseño del estudioLlevamos a cabo un estudio prospectivo multicéntrico de cohorte en tres centros médicos universitarios y 12 hospi-tales regionales en Holanda (Apéndice 1). Entre noviembre de 2003 y diciembre de 2004 se incluyeron 786 pacientes que acudieron a la unidad endoscópica con EB conocido o diagnosticado. Excluimos individuos con EB menor de 2 cm, a menores de 18 años y a aquellos con DAG o ACE previo o en la endoscopia índice. No hubo restricciones res-pecto al uso de medicamentos. El diagnóstico endoscópico de EB se confirmó en todos los pacientes por la presencia demetaplasia intestinal. Los casos incidentales de DAG o ACE se identificaron durante el seguimiento. Doscientos dieciseis sujetos se retiraron del estudio debido a comorbi-lidad grave (n = 30), muerte por causas no relacionadas con el EB (n = 18), migración (n = 10), negativa para participar (n = 155) o progresión neoplásica en los nueve meses siguien-tes a la inclusión (n = 3). Los individuos que se retiraron fueron mayores que los que participaron en la vigilancia (mediana, 66.4 vs. 60.4 años, respectivamente). No obstan-te no hubo diferencias en el género, la duración del EB, la histología basal y el uso de medicamentos.
Vigilancia endoscópicaUn coordinador central del estudio controló el seguimiento de todos los pacientes con EB que participaron en este estu-dio. La vigilancia se llevó a cabo según las guías del American College of Gastroenterology: en los pacientes sin displasia se practicó gastroscopia de seguimiento cada tres años, y en los sujetos con displasia de bajo grado (DBG) cada año.18 Se consideró que los pacientes que desarrollaron DAG o ACE durante el seguimiento habían alcanzado un objetivo final y recibieron tratamiento endoscópico o quirúrgico apropia-do. Las endoscopias se practicaron por gastroenterólogos con experiencia en los 15 hospitales participantes, de acuer-do con un protocolo estandarizado. Durante la gastrosco-pia se determinaron límites endoscópicos como diafragma, unión gastroesofágica y unión escamocolumnar. También documentamos la presencia y el grado de esofagitis con-forme a la Clasificación de Los Ángeles y reportamos las anormalidades de la mucosa, incluidos nódulos, úlceras y erosiones.19 Se tomaron biopsias de las anormalidades de la mucosa, y además se tomaron biopsias de cuatro cuadran-tes cada 2 cm de la parte más distal a la parte más proximal del epitelio de Barrett.
En cada visita de vigilancia los pacientes llenaron un cuestionario sobre factores demográficos; talla; peso; taba-quismo previo y actual; uso de alcohol previo y actual; fecha en que se estableció el diagnóstico de EB; parientes con EB o ACE; síntomas como reflujo, regurgitación y disfagia; y
el uso de medicamentos. Los hallazgos endoscópicos y los datos clínicos se registraron de manera prospectiva en for-mas de registro individuales y se procesaron en una base de datos central por el coordinador del estudio.
Examen histológicoLas biopsias se fijaron en formaldehído al 10% y se coloca-ron en parafina. Se hicieron cortes seriados de 4 µm con H&E. Las laminillas histológicas fueron examinadas prime-ro por un patólogo local en el hospital participante. La dis-plasia se clasificó según los criterios del consenso de 1988, con los ajustes que se propusieron en 2001.20,21 Estudios previos muestran una alta variabilidad interobservador en la interpretación de la displasia en el EB.22 Todas las biop-sias fueron, por lo tanto, revisadas por uno o dos patólogos gastrointestinales expertos. Estos patólogos expertos no co-nocían el diagnóstico del patólogo local. Se estableció un diagnóstico final sólo si al lo menos dos patólogos estaban de acuerdo con el grado de displasia. Si había desacuerdo entre los patólogos, un panel de patólogos expertos tam-bién revisó las laminillas, y el diagnóstico final se estableció en base al consenso.
Uso de medicamentosLa información sobre el uso de medicamentos después que se había diagnosticado el EB se recogió durante el interroga-torio a los pacientes en cada una de las visitas de vigilancia. Además, los pacientes llenaron un cuestionario sobre el uso de medicamentos que se obtienen sin receta. Este cuestiona-rio incluyó datos sobre el uso de los AINE que se obtienen sin receta, como el nombre, la dosis y la frecuencia de uso de los medicamentos. La información recogida en las entre-vistas con los pacientes y en los cuestionarios se verificó con los registros de la farmacia. Todos los pacientes proporciona-ron consentimiento escrito para solicitar los registros com-pletos de la farmacia. Los registros de la farmacia contienen información de todos los medicamentos proporcionados incluyendo dosis, fecha de prescripción y medicamentos que se obtienen sin receta. Se exige legalmente que las farmacias conserven estos registros por al menos 15 años. Con los re-gistros de la farmacia inspeccionamos las prescripciones surtidas de inhibidores de la bomba de protones (IBP) (ome-prazol, lansoprazol, rabeprazol, pantoprazol, esomeprazol), AINE no selectivos (AINEns) (ácido acetilsalicílico > 325 mg al día, carbasalato de calcio > 325 mg al día, diclofenaco, flur-biprofeno, ibuprofeno, indometacina, ketoprofeno, nabume-tona, naproxeno, aceclofenaco, dexibufeno, dexketoprofeno, fenilbutazona, piroxicam, sulindac, tolmetin), inhibidores de la ciclooxigenasa (COX)-2 (celecoxib, rofecoxib, etoricoxib, meloxicam), ácido acetilsalicílico en dosis bajas (ácido ace-tilsalicílico ≤ 100 mg al día, carbasalato de calcio ≤ 100 mg al día), y estatinas (simvastatina, lovastatina, atorvastatina, fluvastatina, pravastatina) desde el diagnóstico de EB hasta la endoscopia más reciente o cuando la endoscopia reveló DAG o ACE. En cada paciente se calculó la duración total de las prescripciones surtidas de IBP, AINE, ácido acetilsali-cílico y estatinas al sumar la duración de las prescripciones individuales y sustraer la sobreposición de fechas. Los suje-
02Nosteroidal 26 3/14/12 11:30 AM
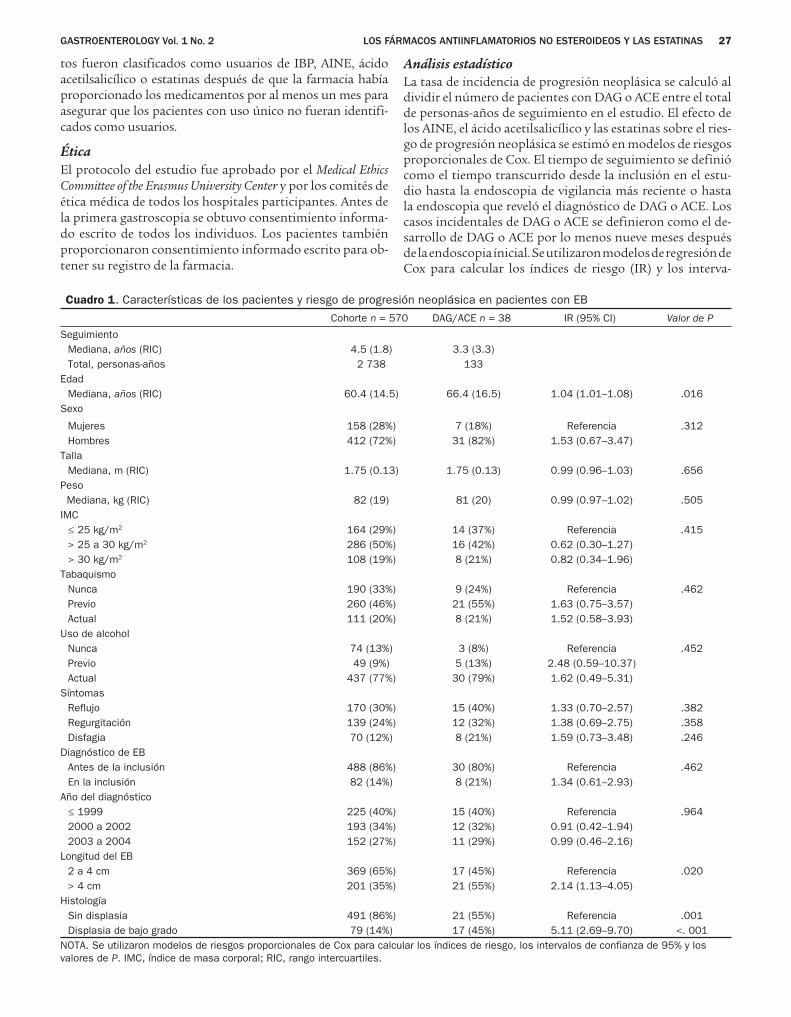
LOS FÁRMACOS ANTIINFLAMATORIOS NO ESTEROIDEOS Y LAS ESTATINAS 27GASTROENTEROLOGY Vol. 1 No. 2
tos fueron clasificados como usuarios de IBP, AINE, ácido acetilsalicílico o estatinas después de que la farmacia había proporcionado los medicamentos por al menos un mes para asegurar que los pacientes con uso único no fueran identifi-cados como usuarios.
ÉticaEl protocolo del estudio fue aprobado por el Medical Ethics Committee of the Erasmus University Center y por los comités de ética médica de todos los hospitales participantes. Antes de la primera gastroscopia se obtuvo consentimiento informa-do escrito de todos los individuos. Los pacientes también proporcionaron consentimiento informado escrito para ob-tener su registro de la farmacia.
Análisis estadísticoLa tasa de incidencia de progresión neoplásica se calculó al dividir el número de pacientes con DAG o ACE entre el total de personas-años de seguimiento en el estudio. El efecto de los AINE, el ácido acetilsalicílico y las estatinas sobre el ries-go de progresión neoplásica se estimó en modelos de riesgos proporcionales de Cox. El tiempo de seguimiento se definió como el tiempo transcurrido desde la inclusión en el estu-dio hasta la endoscopia de vigilancia más reciente o hasta la endoscopia que reveló el diagnóstico de DAG o ACE. Los casos incidentales de DAG o ACE se definieron como el de-sarrollo de DAG o ACE por lo menos nueve meses después de la endoscopia ínicial. Se utilizaron modelos de regresión deCox para calcular los índices de riesgo (IR) y los interva-
Cuadro 1. Características de los pacientes y riesgo de progresión neoplásica en pacientes con EB
Cohorte n = 570 DAG/ACE n = 38 IR (95% CI) Valor de P
Seguimiento
Mediana, años (RIC) 4.5 (1.8) 3.3 (3.3)
Total, personas-años 2 738 133
Edad
Mediana, años (RIC) 60.4 (14.5) 66.4 (16.5) 1.04 (1.01–1.08) .016
Sexo
Mujeres 158 (28%) 7 (18%) Referencia .312
Hombres 412 (72%) 31 (82%) 1.53 (0.67–3.47)
Talla
Mediana, m (RIC) 1.75 (0.13) 1.75 (0.13) 0.99 (0.96–1.03) .656
Peso
Mediana, kg (RIC) 82 (19) 81 (20) 0.99 (0.97–1.02) .505
IMC
≤ 25 kg/m2 164 (29%) 14 (37%) Referencia .415
> 25 a 30 kg/m2 286 (50%) 16 (42%) 0.62 (0.30–1.27)
> 30 kg/m2 108 (19%) 8 (21%) 0.82 (0.34–1.96)
Tabaquismo
Nunca 190 (33%) 9 (24%) Referencia .462
Previo 260 (46%) 21 (55%) 1.63 (0.75–3.57)
Actual 111 (20%) 8 (21%) 1.52 (0.58–3.93)
Uso de alcohol
Nunca 74 (13%) 3 (8%) Referencia .452
Previo 49 (9%) 5 (13%) 2.48 (0.59–10.37)
Actual 437 (77%) 30 (79%) 1.62 (0.49–5.31)
Síntomas
Reflujo 170 (30%) 15 (40%) 1.33 (0.70–2.57) .382
Regurgitación 139 (24%) 12 (32%) 1.38 (0.69–2.75) .358
Disfagia 70 (12%) 8 (21%) 1.59 (0.73–3.48) .246
Diagnóstico de EB
Antes de la inclusión 488 (86%) 30 (80%) Referencia .462
En la inclusión 82 (14%) 8 (21%) 1.34 (0.61–2.93)
Año del diagnóstico
≤ 1999 225 (40%) 15 (40%) Referencia .964
2000 a 2002 193 (34%) 12 (32%) 0.91 (0.42–1.94)
2003 a 2004 152 (27%) 11 (29%) 0.99 (0.46–2.16)
Longitud del EB
2 a 4 cm 369 (65%) 17 (45%) Referencia .020
> 4 cm 201 (35%) 21 (55%) 2.14 (1.13–4.05)
Histología
Sin displasia 491 (86%) 21 (55%) Referencia .001
Displasia de bajo grado 79 (14%) 17 (45%) 5.11 (2.69–9.70) <. 001
NOTA. Se utilizaron modelos de riesgos proporcionales de Cox para calcular los índices de riesgo, los intervalos de confianza de 95% y los
valores de P. IMC, índice de masa corporal; RIC, rango intercuartiles.
02Nosteroidal 27 3/14/12 11:30 AM
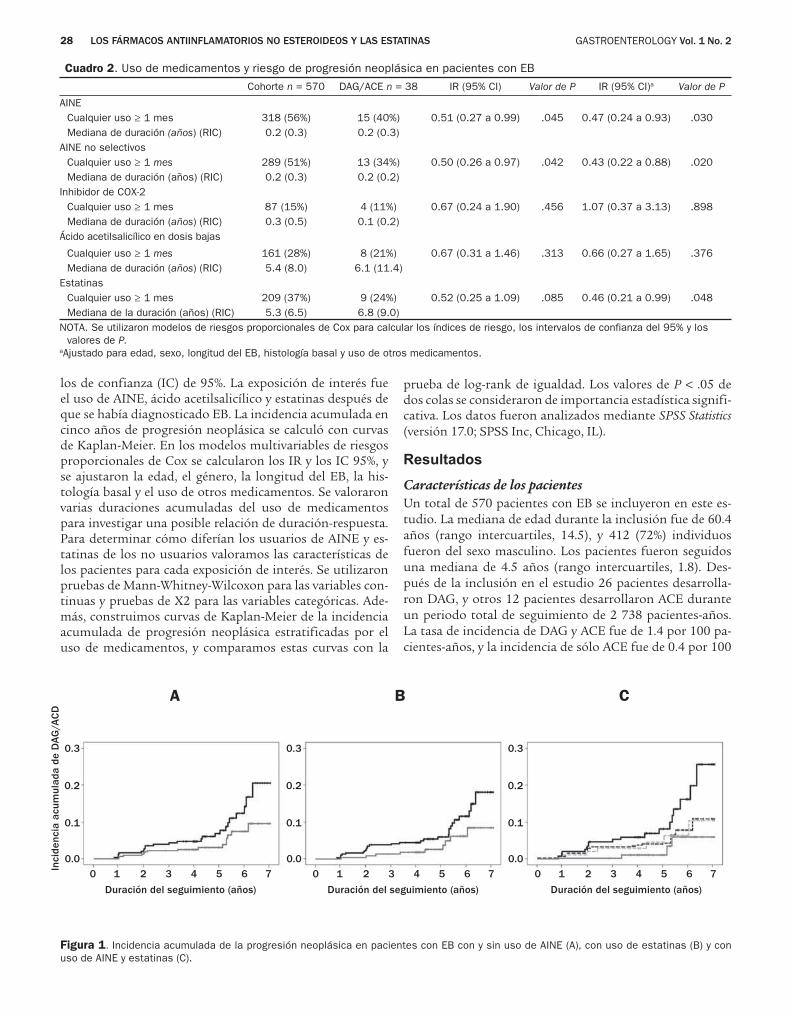
28 LOS FÁRMACOS ANTIINFLAMATORIOS NO ESTEROIDEOS Y LAS ESTATINAS GASTROENTEROLOGY Vol. 1 No. 2
los de confianza (IC) de 95%. La exposición de interés fue el uso de AINE, ácido acetilsalicílico y estatinas después de que se había diagnosticado EB. La incidencia acumulada en cinco años de progresión neoplásica se calculó con curvas de Kaplan-Meier. En los modelos multivariables de riesgos proporcionales de Cox se calcularon los IR y los IC 95%, y se ajustaron la edad, el género, la longitud del EB, la his-tología basal y el uso de otros medicamentos. Se valoraron varias duraciones acumuladas del uso de medicamentos para investigar una posible relación de duración-respuesta. Para determinar cómo diferían los usuarios de AINE y es-tatinas de los no usuarios valoramos las características de los pacientes para cada exposición de interés. Se utilizaron pruebas de Mann-Whitney-Wilcoxon para las variables con-tinuas y pruebas de X2 para las variables categóricas. Ade-más, construimos curvas de Kaplan-Meier de la incidencia acumulada de progresión neoplásica estratificadas por el uso de medicamentos, y comparamos estas curvas con la
prueba de log-rank de igualdad. Los valores de P < .05 de dos colas se consideraron de importancia estadística signifi-cativa. Los datos fueron analizados mediante SPSS Statistics (versión 17.0; SPSS Inc, Chicago, IL).
Resultados
Características de los pacientesUn total de 570 pacientes con EB se incluyeron en este es-tudio. La mediana de edad durante la inclusión fue de 60.4 años (rango intercuartiles, 14.5), y 412 (72%) individuos fueron del sexo masculino. Los pacientes fueron seguidos una mediana de 4.5 años (rango intercuartiles, 1.8). Des-pués de la inclusión en el estudio 26 pacientes desarrolla-ron DAG, y otros 12 pacientes desarrollaron ACE durante un periodo total de seguimiento de 2 738 pacientes-años. La tasa de incidencia de DAG y ACE fue de 1.4 por 100 pa-cientes-años, y la incidencia de sólo ACE fue de 0.4 por 100
Figura 1. Incidencia acumulada de la progresión neoplásica en pacientes con EB con y sin uso de AINE (A), con uso de estatinas (B) y con
uso de AINE y estatinas (C).
Cuadro 2. Uso de medicamentos y riesgo de progresión neoplásica en pacientes con EB
Cohorte n = 570 DAG/ACE n = 38 IR (95% CI) Valor de P IR (95% CI)a Valor de P
AINE
Cualquier uso ≥ 1 mes 318 (56%) 15 (40%) 0.51 (0.27 a 0.99) .045 0.47 (0.24 a 0.93) .030
Mediana de duración (años) (RIC) 0.2 (0.3) 0.2 (0.3)
AINE no selectivos
Cualquier uso ≥ 1 mes 289 (51%) 13 (34%) 0.50 (0.26 a 0.97) .042 0.43 (0.22 a 0.88) .020
Mediana de duración (años) (RIC) 0.2 (0.3) 0.2 (0.2)
Inhibidor de COX-2
Cualquier uso ≥ 1 mes 87 (15%) 4 (11%) 0.67 (0.24 a 1.90) .456 1.07 (0.37 a 3.13) .898
Mediana de duración (años) (RIC) 0.3 (0.5) 0.1 (0.2)
Ácido acetilsalicílico en dosis bajas
Cualquier uso ≥ 1 mes 161 (28%) 8 (21%) 0.67 (0.31 a 1.46) .313 0.66 (0.27 a 1.65) .376
Mediana de duración (años) (RIC) 5.4 (8.0) 6.1 (11.4)
Estatinas
Cualquier uso ≥ 1 mes 209 (37%) 9 (24%) 0.52 (0.25 a 1.09) .085 0.46 (0.21 a 0.99) .048
Mediana de la duración (años) (RIC) 5.3 (6.5) 6.8 (9.0)
NOTA. Se utilizaron modelos de riesgos proporcionales de Cox para calcular los índices de riesgo, los intervalos de confianza del 95% y los
valores de P.aAjustado para edad, sexo, longitud del EB, histología basal y uso de otros medicamentos.
0.3
0.2
0.1
0.0
0.3
0.2
0.1
0.0
0.3
0.2
0.1
0.0
Incid
encia
acum
ula
da d
e D
AG
/AC
D
Duración del seguimiento (años)
A B C
Duración del seguimiento (años) Duración del seguimiento (años)
0 1 2 3 4 5 6 7 0 1 2 3 4 5 6 7 0 1 2 3 4 5 6 7
02Nosteroidal 28 3/14/12 11:30 AM
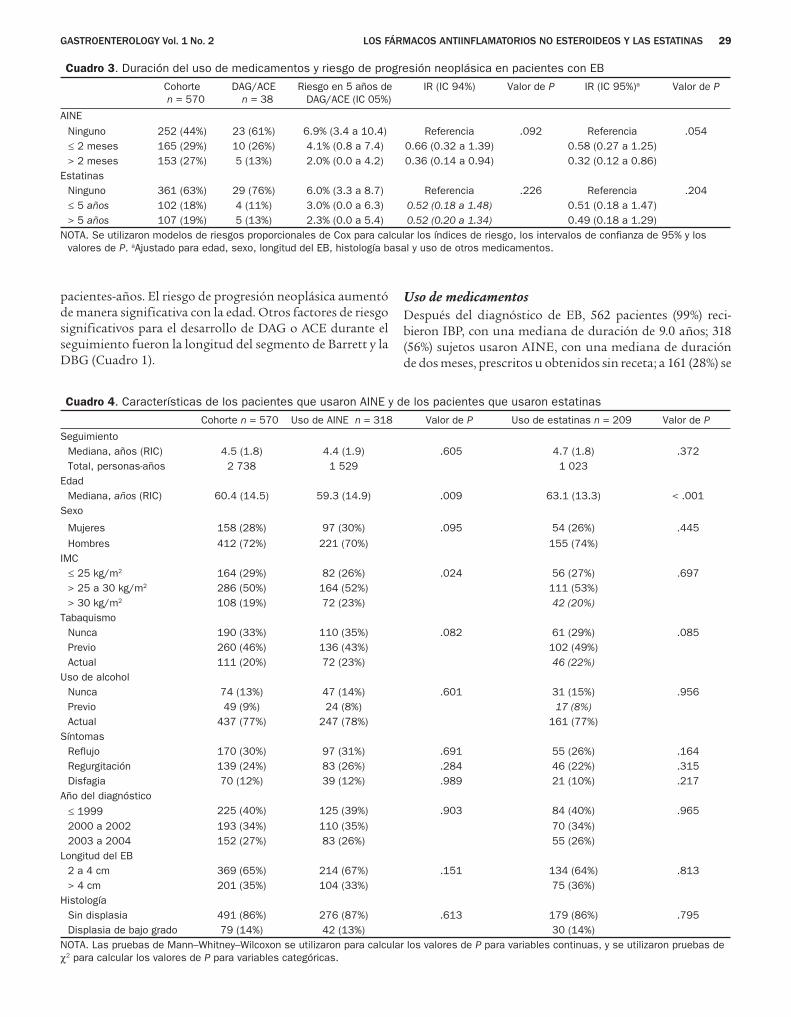
LOS FÁRMACOS ANTIINFLAMATORIOS NO ESTEROIDEOS Y LAS ESTATINAS 29GASTROENTEROLOGY Vol. 1 No. 2
pacientes-años. El riesgo de progresión neoplásica aumentó de manera significativa con la edad. Otros factores de riesgo significativos para el desarrollo de DAG o ACE durante el seguimiento fueron la longitud del segmento de Barrett y la DBG (Cuadro 1).
Uso de medicamentosDespués del diagnóstico de EB, 562 pacientes (99%) reci-bieron IBP, con una mediana de duración de 9.0 años; 318 (56%) sujetos usaron AINE, con una mediana de duración de dos meses, prescritos u obtenidos sin receta; a 161 (28%) se
Cuadro 3. Duración del uso de medicamentos y riesgo de progresión neoplásica en pacientes con EB
Cohorte
n = 570
DAG/ACE
n = 38
Riesgo en 5 años de
DAG/ACE (IC 05%)
IR (IC 94%) Valor de P IR (IC 95%)a Valor de P
AINE
Ninguno 252 (44%) 23 (61%) 6.9% (3.4 a 10.4) Referencia .092 Referencia .054
≤ 2 meses 165 (29%) 10 (26%) 4.1% (0.8 a 7.4) 0.66 (0.32 a 1.39) 0.58 (0.27 a 1.25)
> 2 meses 153 (27%) 5 (13%) 2.0% (0.0 a 4.2) 0.36 (0.14 a 0.94) 0.32 (0.12 a 0.86)
Estatinas
Ninguno 361 (63%) 29 (76%) 6.0% (3.3 a 8.7) Referencia .226 Referencia .204
≤ 5 años 102 (18%) 4 (11%) 3.0% (0.0 a 6.3) 0.52 (0.18 a 1.48) 0.51 (0.18 a 1.47)
> 5 años 107 (19%) 5 (13%) 2.3% (0.0 a 5.4) 0.52 (0.20 a 1.34) 0.49 (0.18 a 1.29)
NOTA. Se utilizaron modelos de riesgos proporcionales de Cox para calcular los índices de riesgo, los intervalos de confianza de 95% y los
valores de P. aAjustado para edad, sexo, longitud del EB, histología basal y uso de otros medicamentos.
Cuadro 4. Características de los pacientes que usaron AINE y de los pacientes que usaron estatinas
Cohorte n = 570 Uso de AINE n = 318 Valor de P Uso de estatinas n = 209 Valor de P
Seguimiento
Mediana, años (RIC) 4.5 (1.8) 4.4 (1.9) .605 4.7 (1.8) .372
Total, personas-años 2 738 1 529 1 023
Edad
Mediana, años (RIC) 60.4 (14.5) 59.3 (14.9) .009 63.1 (13.3) < .001
Sexo
Mujeres 158 (28%) 97 (30%) .095 54 (26%) .445
Hombres 412 (72%) 221 (70%) 155 (74%)
IMC
≤ 25 kg/m2 164 (29%) 82 (26%) .024 56 (27%) .697
> 25 a 30 kg/m2 286 (50%) 164 (52%) 111 (53%)
> 30 kg/m2 108 (19%) 72 (23%) 42 (20%)
Tabaquismo
Nunca 190 (33%) 110 (35%) .082 61 (29%) .085
Previo 260 (46%) 136 (43%) 102 (49%)
Actual 111 (20%) 72 (23%) 46 (22%)
Uso de alcohol
Nunca 74 (13%) 47 (14%) .601 31 (15%) .956
Previo 49 (9%) 24 (8%) 17 (8%)
Actual 437 (77%) 247 (78%) 161 (77%)
Síntomas
Reflujo 170 (30%) 97 (31%) .691 55 (26%) .164
Regurgitación 139 (24%) 83 (26%) .284 46 (22%) .315
Disfagia 70 (12%) 39 (12%) .989 21 (10%) .217
Año del diagnóstico
≤ 1999 225 (40%) 125 (39%) .903 84 (40%) .965
2000 a 2002 193 (34%) 110 (35%) 70 (34%)
2003 a 2004 152 (27%) 83 (26%) 55 (26%)
Longitud del EB
2 a 4 cm 369 (65%) 214 (67%) .151 134 (64%) .813
> 4 cm 201 (35%) 104 (33%) 75 (36%)
Histología
Sin displasia 491 (86%) 276 (87%) .613 179 (86%) .795
Displasia de bajo grado 79 (14%) 42 (13%) 30 (14%)
NOTA. Las pruebas de Mann–Whitney–Wilcoxon se utilizaron para calcular los valores de P para variables continuas, y se utilizaron pruebas de
χ2 para calcular los valores de P para variables categóricas.
02Nosteroidal 29 3/14/12 11:30 AM

30 LOS FÁRMACOS ANTIINFLAMATORIOS NO ESTEROIDEOS Y LAS ESTATINAS GASTROENTEROLOGY Vol. 1 No. 2
les prescribió ácido acetilsalicílico, con una mediana de 5.4 años de duración; y a 209 (37%) les prescribieron estatinas, con una mediana de duración de 5.3 años. De los pacientes que usaron AINE, 87 (15%) recibieron inhibidores de COX-2, y 289 (51%) usaron AINEns. De los 532 pacientes sin progre-sión neoplásica, 303 (57%) usaron AINE, 153 (29%) utiliza-ron ácido acetilsalicílico y 200 (38%) usaron estatinas. De los 38 pacientes que desarrollaron DAG o ACE durante el seguimiento, 15 (40%) pacientes usaron AINE, 8 (21%) ácido acetilsalicílico y nueve (24%) estatinas (Cuadro 2).
Efectos del uso de AINEEn un modelo de riesgos proporcionales de Cox no ajusta-do, el uso de AINE se asoció con un riesgo menor de progre-sión neoplásica en pacientes con EB (IR, 0.51; IC 95%: 0.27 a 0.99). El uso continuo de AINE se asoció con disminución del riesgo de desarrollar DAG o ACE (IR, 0.47; IC 95%: 0.24 a 0.93) después de ajustar la edad, el género, la longitud del EB, la histología basal y el uso de otros medicamentos. Este efecto del uso de AINE fue el mismo en los hombres que en las mujeres, y en los pacientes más jóvenes o mayores de 60 años de edad. Cuando se consideró el uso de inhibidores de COX-2 y AINEns por separado, sólo el uso de AINEns se relacionó con disminución significativa del riesgo de pro-gresión neoplásica en pacientes con EB (IR, 0.43; IC 95%: 0.22 a 0.88). La Figura 1A muestra la incidencia acumulada de progresión neoplásica durante el seguimiento, estratifi-cada según el uso de AINE. Los individuos que no usaron AINE tuvieron un riesgo mayor de desarrollar DAG o ACE que aquellos que los usaron (P = .041).
Para investigar una posible relación de duración–respues-ta, valoramos diferentes duraciones acumuladas del uso de AINE. El uso de AINE mayor a dos meses se asoció con una tendencia menor de progresión neoplásica que el uso de AINE de dos meses o menos (Cuadro 3). Para determinar si los usuarios de AINE difirieron de los no usuarios valo-ramos las características de los pacientes de ambos grupos. Los usuarios de AINE fueron ligeramente más jóvenes y tu-vieron un índice de masa corporal mayor que los pacientes que no usaron AINE. No hubo diferencias en el género, laedad al establecer el diagnóstico de EB, la longitud del EB, la histología basal y la duración del seguimiento (Figura 4).
Efecto del uso de ácido acetilsalicílico en dosis bajasEl uso de ácido acetilsalicílico en dosis bajas no cambió el riesgo de progresión neoplásica en un modelo no ajustado
(IR, 0.67; IC 95%: 0.31 a 1.46) y en un modelo ajustado para edad, género, longitud del BE, histología basal y uso de otros medicamentos (IR, 0.66: IC 95%: 0.27 a 1.65). Aunque elefecto del uso de ácido acetilsalicílico no fue significativo, el IR apuntó en la dirección de un efecto protector.
Efecto del uso de estatinasEn un modelo no ajustado el uso de estatinas se asoció con una tendencia hacia un riesgo disminuido de progresión neoplásica en los pacientes con EB (IR, 0.52; IC 95%: 0.25 a 1.09). La Figura 1B muestra la incidencia acumulada de DAG y ACE en pacientes con EB durante el seguimiento, estratificados por el uso de estatinas. Se observó una ten-dencia hacia un mayor riesgo de progresión neoplásica en los sujetos que no recibieron estatinas que en aquellos que usaron estatinas (P = .079). En un modelo miultivariable el uso de estatinas se vinculó con un riesgo mucho menor de progresión neoplásica (IR, 0.46; IC 95%: 0.21 a 0.99) después de ajustar la edad, el género, la longitud del EB, la histología basal y el uso de otros medicamentos. Este efecto del uso de estatinas se observó sólo en los hombres y en pacientes mayores de 60 años de edad.
Para investigar una posible relación de la duración-res-puesta valoramos diferentes duraciones acumuladas del uso de estatinas. No hubo diferencia en el efecto del uso de estatinas durante más de cinco años y el uso de estatinas de cinco años o menos (Cuadro 3). No obstante, la mayoría depacientes usaron estatinas varios años. Para determinar si los usuarios de estatinas fueron diferentes de los no usua-rios valoramos las características de los pacientes de ambos grupos. Los usuarios de estatinas tuvieron mayor edad que los pacientes que no usaron estatinas. Sin embargo no hubo diferencias en el género, el índice de masa corporal, el año del diagnóstico de EB, la longitud del EB, la histología basal y la duración del seguimiento (Cuadro 4).
Efecto del uso de AINE y estatinas Después de haberse diagnosticado EB, 150 pacientes (26%) no usaron AINE ni estatinas, 211 (37%) sólo utilizaron AINE, 102 (18%) usaron sólo estatinas, y 107 (19%) utilizaron tanto AINE como estatinas (Cuadro 5). En un modelo de regresión de Cox no ajustado el uso de AINE o estatinas se asoció con una tendencia hacia un riesgo menor de progresión neoplá-sica que no usar AINE ni estatinas (IR, 0.48; IC 95%: 0.23 a 1.01 para los AINE solos e IR, 0.48; IC 95%: 0.19 a 1.21 para las estatinas solas). El uso combinado de AINE y estatinas se
Cuadro 5. Uso de AINE y estatinas, y riesgo de progresión neoplásica en pacientes con EB
Cohorte n = 570 DAG/ACE n
= 38
Riesgo en 5 años de
DAG/ACE (IC 95%)
IR (IC 95%) Valor de P IR (IC 95%)a Valor de P
AINE y estatinas
Ninguno 150 (26%) 17 (45%) 8.5% (3.4 a 13.6) Referencia .050 Referencia .065
Sólo AINE 211 (37%) 12 (31%) 4.1% (1.2 a 7.0) 0.48 (0.23 a 1.01) 0.46 (0.22 a 0.99)
Sólo estatinas 102 (18%) 6 (16%) 4.5% (0.2 a 8.8) 0.48 (0.19 a 1.21) 0.51 (0.18 a 1.42)
AINE y estatinas 107 (19%) 3 (8%) 0.9% (0.0 a 2.7) 0.24 (0.07 a 0.82) 0.22 (0.06 a 0.85)
NOTA. Se utilizaron modelos de riesgos proporcionales de Cox para calcular los índices de riesgo, los intervalos de confianza de 95% y los valores de P.aAjustado para edad, sexo, longitud del EB, histología basal y uso de IBP.
02Nosteroidal 30 3/14/12 11:30 AM

LOS FÁRMACOS ANTIINFLAMATORIOS NO ESTEROIDEOS Y LAS ESTATINAS 31GASTROENTEROLOGY Vol. 1 No. 2
asoció con un riesgo incluso más bajo de desarrollar DAG o ACE durante el seguimiento (IR, 0.24; IC 95%: 0.07 a 0.82). En un modelo multivariable el uso de ambos, AINE y estatinas, también se asoció con un efecto protector aditivo (IR, 0.22; IC 95%: 0.06 a 0.85) después de ajustar la edad, el género, la lon-gitud del EB, la histología basal y el uso de IBP. La Figura 1C muestra la incidencia acumulada de progresión neoplásica en pacientes con EB durante el seguimiento, estratificados por el uso de ambos, AINE y estatinas. La incidencia acumulada de DAG o ACE fue menor en pacientes que usaron un AINE o estatina que en aquellos que no usaron ninguno (log rank P = .048 para los AINE solos y log rank P = .113 para las estatinas solas). En los pacientes que usaron ambos, AINE y estatinas, la incidencia acumulada de progresión neoplásica fue todavía más baja (long rank, P = .014).
DiscusiónEn este estudio prospectivo, de cohorte grande, el uso de AINE y estatinas se vinculó con 50% de disminución del riesgo de progresión neoplásica en pacientes con EB. El uso de ambos, AINE y estatinas, tuvo un efecto protector aditi-vo y se asoció con una disminución aproximada de 75% del riesgo de desarrollar DAG o ACE. Estas asociaciones fueron independientes de la edad, el género, la longitud del EB, la histología basal y el uso de otros medicamentos.
En nuestro estudio la disminución del riesgo de progre-sión neoplásica con el uso de AINE se relacionó con la dura-ción del uso del medicamento. Sin embargo, que este efecto ocurría con el uso de AINE con una mediana de duración re-lativamente corta de dos meses, lo que sugiere la posibilidad de factores de confusión no controlados. Además, el uso de ácido acetilsalicílico en dosis bajas se relacionó con una dis-minución menor no significativa del riesgo de progresión neoplásica que el uso de AINE, indicando una relación de dosis-respuesta. La presencia de la relación duración-res-puesta y dosis-respuesta apoya una asociación causal entre el uso de AINE y el riesgo de neoplásia en el EB. Aunque la quimioprevención con AINE parece ser más efectiva que la quimioprevención con dosis bajas de ácido acetilsalicílico, el uso de AINE también tiene efectos adversos serios.
Los resultados de nuestro estudio son compatibles con otros previamente publicados que investigaron el efecto del uso de AINE sobre el riesgo del desarrollo de ACE en pacien-tes con EB. Un estudio de casos y controles con 114 sujetos con ACE y 382 individuos con EB encontró que el uso de AINE fue mucho más prevalente en pacientes con EB (38%) que en aquellos con ACE (26%) (P = .02).23 Además, en un estudio prospectivo de cohorte de 350 pacientes con EB, los usuarios actuales de AINE tuvieron menor riesgo de desarro-llar ACE que los que nunca habían utilizado AINE (IR, 0.20; IC 95%: 0.10 a 0.41).15 Ninguno de estos estudios investigó el uso concomitante de IBP o estatinas. En un estudio obser-vacional reciente de 344 pacientes con EB, el uso de AINE se asoció con una tendencia no significativa hacia una menor incidencia de DAG o ACE (IR, 0.56; IC 95%: 0.27 a 1.18).24 El mismo grupo de estudio llevó a cabo un estudio de casos y controles de 116 pacientes con ACE y 696 pacientes pareados
con EB, que confirmó que el uso de AINE se relacionó de ma-nera significativa con un riesgo disminuido de ACE (índice de densidad de incidencia, 0.64; IC 95%: 0.42 a 0.97).13
Estudios previos también han investigado el mecanismo de los AINE en la quimioprevención. Los AINE inhiben las enzimas COX, COX-1 y COX-2. La COX-1 se expresa de for-ma constitutiva en el tejido humano, mientras que la COX-2es inducida en respuesta a citocinas, factores de crecimien-to y mitógenos. La inhibición de COX-2 produce apoptosis, inhibe el crecimiento celular, disminuye la proliferación ce-lular, e inhibe la angiogénesis en tejidos humanos.25 En los pacientes con Barrett la inhibición de las enzimas COX por los AINE pueden, por lo tanto, llevar a disminución del ries-go de desarrollar DAG o adenocarcinoma.
El uso de estatinas también se asoció con disminución del riesgo de progresión neoplásica en nuestro estudio. Sólo dos estudios previos han investigado el efecto del uso de es-tatinas en pacientes con EB. En un primer estudio obser-vacional, el uso de estatinas no tuvo efectó en el riesgo de desarrollar displasia o ACE (IR, 0.73; IC 95%: 0.30 a 1.78), pero este estudio no tuvo poder suficiente, ya que sólo 87 individuos recibieron estatinas.24 Los resultados de nuestro estudio fueron compatibles con los de otro estudio de casos y controles en el cual el uso de estatinas disminuyó el riesgo de ACE (índice de densidad de incidencia, 0.55; IC 95%: 0.36 a 0.86).13 Los sujetos incluidos en este estudio fueron vete-ranos que recibieron cuidados en el sistema de atención de la salud de Asuntos de Veteranos. Estos pacientes tuvieron, por lo tanto, mayor probabilidad de ser hombres de edad relativamente mayor que el total de la población de EB. Esto puede limitar la generalización de estos resultados.
El mecanismo de las estatinas en la quimioprevención se investigó en estudios previos. Las estatinas inhiben com-petitivamente la 3-hidroxi-3-metilglutaril coenzima A re-ductasa, la enzima limitante de la biosíntesis del colesterol. Aunque esta es la acción biológica más apreciada, las esta-tinas desempeñan otros papeles. En la vía de la biosíntesis del colesterol se forma farnesil y geranilgeranil, esenciales para la activación de las proteínas intracelulares a través de prenilación. Varias proteínas importantes involucradas en la señal intracelular como Ras, Rho y Rac son dependien-tes de la prenilación. La proteína Ras es el activador de la cinasa regulada por señales extracelulares, la vía activada por mitógenos y posiblemente activador también de la vía Akt. Ambas cascadas proporcionan proliferación celular y señales de supervivencia celular en las células humanas. Como resultado, las estatinas inhiben la proliferación e in-ducir apoptosis en el epitelio de Barrett, lo que lleva a dis-minución del riesgo de progresión neoplásica.14,26,27
En el estudio actual el uso de AINE y estatinas tuvo una disminución del riesgo más fuerte que el uso de AINE o es-tatinas solos. Este es el primer estudio que investiga el efecto de ambos sobre el riesgo de progresión neoplásica del EB. Un estudio previo en pacientes con cáncer colorrectal también mostró que la quimioprevención con la combinación de áci-do acetilsalicílico en dosis bajas y estatinas disminuyó el ries-go que el uso de cualquiera de los fármacos solos.16
02Nosteroidal 31 3/14/12 11:30 AM

32 LOS FÁRMACOS ANTIINFLAMATORIOS NO ESTEROIDEOS Y LAS ESTATINAS GASTROENTEROLOGY Vol. 1 No. 2
Varios estudios in vitro han investigado el efecto de la quimioprevención con la combinación de AINE y estatinas. Estos estudios demuestran efectos sinérgicos de los AINE y las estatinas en la inhibición del crecimiento celular e in-ducción de apoptosis en las células cancerosas. No se ha re-suelto en gran parte la forma en que los AINE y las estatinas funcionan de forma sinérgica.16,17
Este estudio tiene varias fortalezas, incluyendo el gran tamaño de la muestra de pacientes con EB y el seguimiento prolongado. Todos los sujetos que se presentaron con EB en la unidad endoscópica de tres hospitales académicos y 12 hospitales regionales fueron incluidos en el estudio. Como resultado, nuestra cohorte es representativa del total de la población de Barrett en los Países Bajos. La incidencia de ACE durante el seguimiento fue de 0.4 por 100 pacientes-años, lo que es igual a la incidencia reportada en la mayoría de estudios y que apoya que nuestra población es represen-tativa de la población holandesa con EB.
Tuvimos criterios estrictos para el diagnóstico de EB y para la inclusión en el estudio. Además, hubo un esquema de seguimiento estricto, un protocolo de endoscopia estan-darizado y un protocolo de biopsia estandarizado. Todas las biopsias fueron revisadas por al menos dos patólogos para establecer un diagnóstico con base en el consenso. Durante el seguimiento la información clínica se obtuvo de forma prospectiva y el coordinador del estudio la registró en una base de datos central computarizada.
La información sobre el uso de medicamentos se obtuvo de manera prospectiva, y los pacientes también llenaron un cuestionario sobre el uso de medicamentos que se obtienen sin receta. Toda la información del uso de medicamentos se verificó con los registros de la farmacia. Debido a que el uso de IBP es uno de los pilares del tratamiento de los pacientes con EB, actúa como un posible factor de confusión. Por lo tanto, también obtuvimos información detallada sobre el uso de IBP.
Nuestro estudio también tiene algunas limitaciones. De-bido a que es un estudio observacional no podemos excluir factores de confusión no controlados. No obstante hemos recogido mucha información sobre los posibles factores de confusión durante el seguimiento, y corregimos estos fac-tores en el análisis final. Por desgracia no tuvimos informa-ción sobre la indicación del uso de AINE y estatinas. Como resultado no pudimos ajustar este posible confusor. Los usuarios de AINE y estatinas pueden haber diferido en otras formas de otros pacientes con EB que en el uso de estos fár-macos. Para determinar si los usuarios de AINE y estatinas fueron diferentes de los no usuarios, valoramos las carac-terísticas de los pacientes para cada exposición de interés. Los usuarios de AINE fueron ligeramente más jóvenes y los usuarios de estatinas fueron ligermente mayores que otros pacientes con EB, pero por lo demás no hubo diferencias mayores. Debido a que ajustamos la edad en nuestros mo-delos, las diferencias entre los usuarios y los no usuarios no pueden explicar por completo el efecto de los AINE y de las estatinas observado en este estudio.
A pesar del tamaño grande de la muestra de esta cohorte, el número de eventos fue pequeño en algunos estratos de
exposición, lo que limitó la interpretación. Este fue espe-cialmente el caso cuando se examinó el efecto de la dura-ción del tratamiento y la interacción entre el uso de AINE y estatinas. Además, se informó a los pacientes de cualquier cambio observado durante la endoscopia o la valoración histológica. En teoría esto pudo haber influido en su cum-plimiento o su estilo de vida.
En conclusión, este estudio prospectivo grande de cohor-te muestra que el uso de AINE y estatinas se asocia con una disminución significativa del riesgo de progresión neoplási-ca en pacientes con EB. El uso de ambos, AINE y estatinas, parece tener un efecto aditivo de protección. Como resulta-do, la quimioprevención con AINE y estatinas tiene un pa-pel en el tratamiento de los pacientes con EB.
APÉNDICE 1. Progresión del esófago de Barret (proBar) en el grupode estudioCentros, departamentos e investigadores. Erasmus Universi-ty Medical Center, Rotterdam: Departamento de Gastroen-terología y Hepatología: F. Kastelein, M.C.W. Spaander, E.J. Kuipers, M.J. Bruno; Departamento de Patología: K. Bier-mann; Departamento de Salud Pública: E.W. Steyerberg. IJsselland Hospital, Capelle aan den Ijssel: Departamento de Gastroenterología y Hepatología: H. Geldof. Ikazia Hos-pital, Rotterdam: Departamento de Gastroenterología y Hepatología: P.C.J. ter Borg. VU University Medical Center, Amsterdam: Departamento de Gastroenterología y Hepa-tología: E.C. Klinkenberg. Albert Schweitzer Hospital, Dor-drecht: Departamento de Gastroenterología y Hepatología: W. Lesterhuis. Deventer Hospital, Deventer: Departamento de Gastroenterología y Hepatología: F. ter Borg. Medical Spectrum Twente, Enschede: Departamento de Gastroente-rología y Hepatología: J.J. Kolkman. ZGT Hospital, Hengelo: Departamento de Gastroenterología y Hepatología: G. Tan; Rijnstate Hospital, Arnhem: Departamento de Gastroentero-logía y Hepatología: B. den Hartog. Sint Franciscus Gasthuis, Rotterdam: Departamento de Gastroenterología y Hepatolo-gía: A.J.P. van Tilburg. Orbis Medical Center, Sittard: Depar-tamento de Gastroenterología y Hepatología: L.G.J.B Engels; University Medical Center, Groningen: Departamento de Gastroenterología y Hepatología: F.T.M Peters. Isala Clinics, Zwolle: Departamento de Gastroenterología y Hepatología: B.E. Schenk. Zaans Medical Center, Zaandam: Departamen-to de Gastroenterología y Hepatología: R. Loffeld. Franciscus Hospital, Roosendaal: Departamento de Gastroenterología y Hepatología: H. van Roermund.
Referencias1. Haggitt RC. Barrett’s esophagus, dysplasia, and adenocarcinoma.
Hum Pathol 1994;25:982–993.
2. Fan X, Snyder N. Prevalence of Barrett’s esophagus in patients
with or without GERD symptoms: role of race, age, and gender.
Dig Dis Sci 2009;54:572–577.
3. van Blankenstein M, Looman CW, Johnston BJ, et al. Age and
sex distribution of the prevalence of Barrett’s esophagus found
in a primary referral endoscopy center. Am J Gastroenterol
2005;100:568–576.
4. Ronkainen J, Aro P, Storskrubb T, et al. Prevalence of Barrett’s
esophagus in the general population: an endoscopic study. Gas-
troenterology 2005;129:1825–1831.
02Nosteroidal 32 3/14/12 11:30 AM

LOS FÁRMACOS ANTIINFLAMATORIOS NO ESTEROIDEOS Y LAS ESTATINAS 33GASTROENTEROLOGY Vol. 1 No. 2
5. Avidan B, Sonnenberg A, Schnell TG, et al. Hiatal hernia size,
Barrett’s length, and severity of acid reflux are all risk factors for
esophageal adenocarcinoma. Am J Gastroenterol 2002;97:1930–
1936.
6. Sikkema M, de Jonge PJ, Steyerberg EW, et al. Risk of esophageal
adenocarcinoma and mortality in patients with Barrett’s esopha-
gus: a systematic review and meta-analysis. Clin Gastroenterol
Hepatol 2010;8:235–244, quiz e32.
7. Hage M, Siersema PD, van Dekken H, et al. Oesophageal cancer
incidence and mortality in patients with long-segment Barrett’s
oesophagus after a mean follow-up of 12.7 years. Scand J Gas-
troenterol 2004;39:1175–1179.
8. Fitzgerald RC, Triadafilopoulos G. Recent developments in the
molecular characterization of Barrett’s esophagus. Dig Dis 1998;
16:63–80.
9. American Gastroenterological Association. American Gastroentero-
logical Association Medical position statement on the management
of barrett’s esophagus. Gastroenterology 2011;140:1084–1091.
10. van Soest EM, Dieleman JP, Siersema PD, et al. Increasing in-
cidence of Barrett’s oesophagus in the general population. Gut
2005;54:1062–1066.
11. Devesa SS, Blot WJ, Fraumeni JF Jr. Changing patterns in the inci-
dence of esophageal and gastric carcinoma in the United States.
Cancer 1998;83:2049–2053.
12. Mehta S, Johnson IT, Rhodes M. Systematic review: the chemo-
prevention of oesophageal adenocarcinoma. Aliment Pharmacol
Ther 2005;22:759–768.
13. Nguyen DM, Richardson P, El-Serag HB. Medications (NSAIDs, sta-
tins, proton pump inhibitors) and the risk of esophageal adeno-
carcinoma in patients with Barrett’s esophagus. Gastroenterology
2010;138:2260–2266.
14. Ogunwobi OO, Beales IL. Statins inhibit proliferation and induce
apoptosis in Barrett’s esophageal adenocarcinoma cells. Am J
Gastroenterol 2008;103:825–837.
15. Vaughan TL, Dong LM, Blount PL, et al. Non-steroidal antiinflam-
matory drugs and risk of neoplastic progression in Barrett’s oe-
sophagus: a prospective study. Lancet Oncol 2005;6:945–952.
16. Hoffmeister M, Chang-Claude J, Brenner H. Individual and joint use
of statins and low-dose aspirin and risk of colorectal cancer: a po-
pulation-based case-control study. Int J Cancer 2007;121:1325–
1330.
17. Xiao H, Yang CS. Combination regimen with statins and NSAIDs:
a promising strategy for cancer chemoprevention. Int J Cancer
2008;123:983–990. 18. Sampliner RE. Practice Parameters
Committee of the American College of Gastroenterology Updated
guidelines for the diagnosis, Surveillance, and therapy of Barrett’s
esophagus. Am J Gastroenterol 2002;97:1888–1895.
19. Armstrong D, Bennett JR, Blum AL, et al. The endoscopic as-
sessment of esophagitis: a progress report on observer agree-
ment. Gastroenterology 1996;111:85–92.
20. Montgomery E, Bronner MP, Goldblum JR, et al. Reproducibility of
the diagnosis of dysplasia in Barrett esophagus: a reaffirmation.
Hum Pathol 2001;32:368–378.
21. Reid BJ, Haggitt RC, Rubin CE, et al. Observer variation in the
diagnosis of dysplasia in Barrett’s esophagus. Hum Pathol 1988;
19:166–178.
22. Kerkhof M, van Dekken H, Steyerberg EW, et al. Grading of dyspla-
sia in Barrett’s oesophagus: substantial interobserver variation
between general and gastrointestinal pathologists. Histopatholo-
gy 2007;50:920–927.
23. Tsibouris P, Hendrickse MT, Isaacs PE. Daily use of non-steroidal
anti-inflammatory drugs is less frequent in patients with Barrett’s
oesophagus who develop an oesophageal adenocarcinoma. Ali-
ment Pharmacol Ther 2004;20:645–655.
24. Nguyen DM, El-Serag HB, Henderson L, et al. Medication usage
and the risk of neoplasia in patients with Barrett’s esophagus.
Clin Gastroenterol Hepatol 2009;7:1299–1304.
25. Anderson LA, Johnston BT, Watson RG, et al. Nonsteroidal anti-
inflammatory drugs and the esophageal inflammationmetaplasia-
adenocarcinoma sequence. Cancer Res 2006;66:4975–4982.
26. Hawk ET, Viner JL. Statins in esophageal cancer cell lines: promi-
sing lead? Am J Gastroenterol 2008;103:838–841.
27. Konturek PC, Burnat G, Hahn EG. Inhibition of Barret’s adeno-
carcinoma cell growth by simvastatin: involvement of COX-2 and
apoptosis-related proteins. J Physiol Pharmacol 2007;58(Suppl
3):141–148.
Recibido el 14 marzo de 2011. Aceptado 15 de agosto de 2011
Solicitud de reimpresosDirigir la solicitud de reimpresos a: Florine Kastelein, MD, De-
partment of Gastroenterology and Hepatology, Erasmus Medical Cen-
ter, P.O. Box 2040, 3000 CA Rotterdam, The Netherlands.correo elec-
trónico: [email protected]; fax: (31) 10-7034 682.
Conflictos de interésLos autores no declararon conflictos.
02Nosteroidal 33 3/14/12 11:30 AM

GASTROENTEROLOGY 2011;141:2047-2055
34
CLÍNICA — HÍGADOEficacia del inhibidor de proteasa BI 201335, el inhibidor de polimerasa BI 207127 y la ribavirina en pacientes con infección crónica por VHC
STEFAN ZEUZEM,* TARIK ASSELAH,‡ PETER ANGUS,§ JEAN–PIERRE ZARSKI,II DOMINIQUE LARREY,¶ BEAT MÜLLHAUPT,#
ED GANE,** MARCUS SCHUCHMANN,‡‡ ANSGAR LOHSE,§§ STANISLAS POL,II II JEAN–PIERRE BRONOWICKI,¶¶ STUART
ROBERTS,## KEIKAWUS ARASTEH,*** FABIEN ZOULIM,‡‡‡ MARKUS HEIM,§§§ JERRY O. STERN,II II II GEORGE KUKOLJ,¶¶¶
GERHARD NEHMIZ,### CARLA HAEFNER,### y WULF OTTO BOECHER###
*J. W. Goethe University Hospital, Frankfurt, Germany; ‡Service d’Hépatologie, Hôpital Beaujon, Université Diderot-Paris 7, e INSERM U773, CRB3, Clichy, France; §Austin Hospital, Heidelberg, Victoria, Australia; IIHôpital Albert Michallon, Grenoble, France; ¶Hôpital Saint-Eloi, Montpellier, France; *University Hospital of Zürich, Zürich,
Switzerland; **Auckland City Hospital, Auckland, New Zealand; ‡‡University Medical Center Mainz, Mainz, Germany; §§University Hospital Hamburg- Eppendorf, Hamburg,
Germany; II IIHôpital Cochin, Paris, France; ¶¶University Hospital of Nancy, Vandoeuvre-lès-Nancy, France; ##Alfred Hospital, Melbourne, Victoria, Australia; ***Epimed
GmbH, Berlin, Germany; ‡‡‡Hepatology Department, Hospices Civils de Lyon, Lyon University, Lyon, France; §§§University Hospital Basel, Basel, Switzerland; II II IIBoehringer
Ingelheim, Ridgefield, Connecticut; ¶¶¶Boehringer Ingelheim (Canada) Ltd, Laval, Quebec, Canada; y ###Boehringer-Ingelheim Pharma GmbH & Co KG, Biberach, Germany
ANTECEDENTES Y OBJETIVOS: se estudian esquemas terapéuticos para los pacientes con infección por el virus de la hepatitis C (VHC) que no incluyan la combinación de peginterferón alfa y ribavirina. Investigamos el efecto antiviral y la seguridad del BI 201335 (un inhibidor de la proteasa NS3/4A) y del BI 207127 (inhibidor no nucleósido de la polimerasa NS5B) con ribavirina. MÉTODOS: 32 pacientes con infección crónica por el genotipo 1 del VHC vírgenes al tratamiento fueron asignados de forma aleatoria a grupos que recibieron 400 o 600 mg de BI 207127 tres veces al día más 120 mg de BI 201335 una vez al día y 1 000 a 1 200 mg/día de ribavirina durante cuatro semanas. El objetivo de la efi cacia primaria fue la respuesta virológica (nivel del RNA del VHC < 25 UI/mL en la semana 4). Treinta y dos pacientes recibieron tratamiento; 31 completaron las cuatro semanas con el tratamiento de combinación asignado. RESULTADOS: la tasa de respuesta virológica en el grupo que recibió BI 207127 400 mg tres veces al día fueron de 47, 67 y 73% en los días 15, 22 y 29; se observó una tasa de respuesta más alta en los pacientes con el genotipo-1b en comparación con la infección por el genotipo-1a. En el grupo que recibió BI 207127 600 mg tres veces al día, las tasas de respuesta virológica fueron de 82, 100 y 100%, respectivamente, y no difi rieron entre los genotipos. Un paciente del grupo que recibió 400 mg tres veces al día tuvo resurgimiento virológico (≥ 1 log10 de rebote del RNA del VHC) el día 22. Los eventos adversos más frecuentes fueron trastornos gastrointestinales leves, exantema y fotosensibilidad. No se reportaron eventos adversos graves; ningún paciente interrumpió el tratamiento en forma prematura. CONCLUSIONES: la combinación del inhibidor de proteasa BI 201335, el inhibidor de polimerasa BI 207127 y la ribavirina tuvo una actividad rápida y potente contra VHC de genotipo-1 y no causó eventos adversos serios o graves.
Palabras clave: fármacos; estudio clínico; antivirales de acción directa; libres de peginterferón.
La infección crónica por el virus de la hepatitis C (VHC) representa un problema importante de salud pública a
nivel mundial. Con el tratamiento estándar actual para los pacientes infectados con el genotipo (GT)-1 del VHC con-sistente en peginterferón alfa (PegIFN) y ribavirina (RBV) durante 48 semanas, ~ 60% de los pacientes no alcanza una respuesta virólógica sostenida (RVS), definida como un RNA del VHC indetectable en el plasma 24 semanas después de completar el tratamiento. Incluso se reportan tasas más bajas de RVS en diferentes poblaciones como los pacientes coinfectados con VHC/virus de la inmunodeficiencia hu-mana o aquellos con cirrosis hepática descompensada.
Debido al perfil de efectos secundarios de ambos fárma-cos, muchos pacientes no son elegibles para el tratamiento con interferón. Además, de los individuos que empiezan el tratamiento con PegIFN y RBV, una proporción sustancial no toleran estos efectos secundarios e interrumpe el trata-miento de forma prematura.
Con el desarrollo de los inhibidores de la serina protea-sa NS3/4A del VHC, las tasas de RVS en los pacientes in-fectados con el GT-1 del VHC han aumentado de manera sustancial. No obstante, se observa una rápida selección de variantes asociadas con resistencia cuando los inhibidores de proteasa del VHC se administran en monoterapia. Por lo tanto, los inhibidores de proteasa deben combinarse con PegIFN y RBV para reducir las fallas del tratamiento debi-das a la selección de variantes virales resistentes. Con esta triple combinación las tasas de RVS en los pacientes con GT-1 vírgenes al tratamiento han aumentado a 65 a 84%.1-4 Sin embargo las limitaciones asociadas con el PegIFN y la RBV, incluidas toxicidad en múltiples órganos, intolerabi-lidad, conveniencia y contraindicaciones, seguirán hasta que se disponga de tratamientos libres de PegIFN/RBV.
Los estudios in vitro e in vivo sugieren que las combina-ciones de agentes antivirales de acción directa (AAD) con
Abreviaturas utilizadas en este artículo: EA, evento adverso; AAD, agen-te antiviral directo; GT, genotipo; LIDD, límite inferior de detección; LIDC, límite inferior de cuantificación; IN, inhibidor nucleósido de polimerasa; INN, inhibidor no nucleósido; PegIFN, peginterferón alfa; RBV, ribavirina;
RVS, respuesta virológica sostenida. © 2011 by the AGA Institute
doi:10.1053/j.gastro.2011.08.051

EFICACIA DEL INHIBIDOR DE PROTEASA BI 201335 35GASTROENTEROLOGY • Volumen 1 • Núm. 2
diferentes modos de acción y con perfiles de resistencia que no se sobreponen disminuyen la selección de varian-tes resistentes, y por lo tanto mejoran la respuesta al trata-miento antiviral.5,6 El primer estudio de fase clínica 1b que investigó un tratamiento de combinación libre de PegIFN/RBV, que comprendió el inhibidor de proteasa NS3/4A danoprevir (RG7227) y el inhibidor nucleósido (IN) de la polimerasa NS5B mericitabina (RG7128) en pacientes in-fectados con el VHC GT-1, mostró una potencia antiviral significativa y suprimió el rebote viral hasta 14 días.7
El BI 201335 es un inhibidor de la proteasa NS3/4A del VHC de segunda generación con actividad in vitro alta-mente potente contra los subtipos GT-1a/1b (mediana de los valores de concentración efectiva de 6.5 y 3.1 mol/L) y una farmacocinética mejorada para dosificación una vez al día. En la actualidad el BI 201335 está en la fase 3 de desarrollo.4,8,9 El BI 207127 es un inhibidor no nucleósido (INN) biodisponible por vía oral, reversible, del dominio 1 “thumb” de la polimerasa NS5B del VHC con actividad antiviral potente y específica in vitro (valores de la mediana de la concentración efectiva de la replicación del VHC de 23 y 11 nmol/L contra el GT-1a y el GT-1b, respectivamente). Hoy en día el BI 207127 está en la fase 2 de su desarrollo.10
Los estudios de resistencia a fármacos en cultivos celulares muestran que el BI 201335 y el BI 207127 tienen diferentes perfiles de resistencia, y observaciones previas con inhi-bidores de la proteasa NS3/4A y compuestos INN del do-minio 1 “thumb” NS5B han mostrado que las combinacio-nes de dos fármacos disminuyen en gran medida la selec-ción devariantes de resistencia a los fármacos.11
Aquí reportamos los resultados de un estudio clínico de fase 1b (SOUND-C1: Safety and antiviral effect of Oral com-binations withoUt iNterferon in patients Diagnosed with chronic hepatitis C, seguridad y efecto antiviral de la combinación oral libre de interferón en pacientes diagnosticados con hepatitis C crónica) que investigó la seguridad, el efecto antiviral y la farmacocinética del BI 207127 en combina-ción con el BI 201335 y RBV durante cuatro semanas en pacientes con infección crónica por el VHC GT-1 vírgenes al tratamiento.
Pacientes y métodos
Pacientes Los pacientes fueron reclutados en mayo de 2010 en Aus-tralia, Francia, Alemania, Nueva Zelanda y Suiza. Éstos fueron elegibles si tenían 18 a 75 años de edad, con infec-ción crónica por VHC GT-1 y vírgenes al tratamiento con interferón, PegIFN, RBV o cualquier AAD para la infección crónica por hepatitis C. Los pacientes tenían niveles plas-máticos de RNA de VHC × 100 000 UI/mL en el escrutinio y una biopsia hepática dentro de dos años, o un FibroScan (ECHOSENS, París, Francia) dentro de seis meses antes del escrutinio que excluía cirrosis. Se excluyeron sujetos con VHC de GT mixto, virus de la hepatitis B, virus de la inmu-nodeficiencia humana, antecedente de exantema previo oactual o fotosensibilidad, hepatopatía descompensada o hiperbilirrubinemia (> 1.5 × el límite superior normal) (se aceptaron pacientes con polimorfismo de Gilbert). Todos los individuos proporcionaron consentimiento informado escrito antes de su participación en el estudio.
Diseño del estudioEste fue un estudio de fase Ib, multicéntrico, abierto, alea-torizado. Treinta y cuatro pacientes vírgenes al tratamiento con infección crónica por VHC GT-1 fueron distribuidos de forma aleatoria 1:1 para recibir 400 o 600 mg de BI 207127 tres veces al día más 120 mg de BI 201335 una vez al día y 1 000 mg (peso < 75 kg) o 1 200 mg (peso ≥ 75 kg) de RBV al día durante cuatro semanas (Figura 1). BI 201335, BI 207127 y RBV se administraron con alimentos. Se permitió el uso de eritropoyetina y disminuciones de la dosis de RBV a dis-creción del investigador. Treinta y dos pacientes empezaron el tratamiento. La distribución aleatoria se estratificó según el nivel basal del RNA del VHC plasmático (< 800 000 y ≥ 800 000 UI/mL) y el subtipo del VHC (1a y 1b). Se admi-nistró una dosis de impregnación de 240 mg de BI 201335 elprimer día, seguida de 120 mg una vez al día empezando el día 2. Se administró una dosis de inducción de 1 200 mg de BI 207127 el primer día con el objetivo de alcanzar una disminución temprana y profunda de la carga viral,10 segui-
Figura 1. Esquema del estu-
dio. eRVR, respuesta virológi-
ca rápida extendida (nivel de
RNA del VHC ≤ 25 UI/mL en
la semana 4, y RNA del VHC
indetectable de la semana 5 a
la semana 18); QD, una vez al
día; TID, tres veces al día.
1
2
120 mg QD BI 2011335/400 mg TID BI 201127/RBV
120 mg QD BI 201335/ PegIFN/RBV
PegIFN/RBV
Seguimiento
Seguimiento
PegIFN/RBV
eRVR–
eRVR+
eRVR–
eRVR+
120 mg QD BI 2011335/ 600 mg TID BI 201127/RBV
Día 1 Día 29 Semana 24 Semana 48
120 mg QD BI 201335/PegIFN/RBV

36 EFICACIA DEL INHIBIDOR DE PROTEASA BI 201335 GASTROENTEROLOGY • Volumen 1 • Núm. 2
da de la dosis asignada. El tratamiento concomitante con los siguientes sustratos de P-gp, UGT1A1, CYP3A4 o 2C9, con un rango terapéutico estrecho, no se permitió: alfenta-nilo, astemizol, cisaprida, mosaprida, di/ergotamina, ciclos-porina, everolimus, sirolimus, tacrolimus, fentanilo, irino-tecán, pimozida, quinidina y terfenadina.
El día 29 todos los pacientes se les cambió su tratamiento y se les asignó a 120 mg de BI 201335 una vez al día, PgIFN alfa-2a y RBV hasta la semana 24 o 48, según la respuesta virológica rápida extendida (nivel de RNA del VHC ≤ 25 UI/mL en la se-mana 4 y RNA del VHC indetectable de la semana 5 a 18). Los sujetos con resurgimiento virológico antes del día 29, definido como rebote del RNA del VHC ≥ 1 log10 de un nadir cuantifi-cable del tratamiento con BI 207127/BI 201335/RBV, y confir-mado por una segunda determinación plasmática consecutiva del RNA del VHC, se les cambió de inmediato a un esquema de tratamiento con PegIFN alfa-2a y RBV durante 48 semanas.
Nuestro estudio se realizó de conformidad con los prin-cipios de la Buena Práctica Clínica y fue aprobado por los comités de revisión institucional apropiados y las agencias reguladoras (NCT01132313).
Valoración de la eficaciaSe obtuvieron muestras de plasma para determinación del RNA del VHC al tiempo del escrutinio y los días 1, 2, 4, 8, 10, 15, 22 y 29. Las muestras fueron procesadas en forma central en un laboratorio con el ensayo de Roche COBAS TaqMan HCV/HPS (versión 2.0; Roche, Basilea, Suiza), con un lími-te inferior de cuantificación (LIDC) de 25 UI/mL y un límite inferior de detección (LIDD) de 17 UI/mL.
El GT del VHC en el escrutinio y en la distribución alea-toria se determinó mediante el ensayo Trugene HCV (Bayer, Leverkusen, Alemania); debido a limitaciones técnicas de este
ensayo de genotipificación, el GT del VHC definitivo utiliza-do para todos los análisis se basó en la secuenciación comple-ta del NS5B y los análisis filogenéticos de todos los pacientes aleatorizados. Se obtuvieron muestras para secuenciación de la proteasa NS3/4A del VHC y de la polimerasa NS5B en el escrutinio y en todas las visitas de los pacientes. La genotipifi-cación viral se practicó en todos los pacientes de forma basal y en aquellos en que el nivel plasmático de RNA del VHC se estancó por arriba de ~ 1 000 UI/mL, o valores plasmáticos del RNA del VHC que indicaban resurgimiento viral, durante el periodo de estudio.
El RNA viral del plasma se aisló con el Estuche QiaAmp Viral RNA Extraction (Qiagen, Hilden, Alemania). El DNA complementario se sintetizó con el Sistema SuperScript III One-Step RT-PCR y Platinum Taq DNA Polymerase median-te cebadores específicos del GT (Qiagen). El LIDD del método de amplificación de la reacción en cadena de la transcripción inversa de la polimerasa restringió el análisis a las muestras de los pacientes con niveles de RNA del VHC ≥ 1 000 UI/mL. Las secuencias de la proteasa NS3/4A y el nucleósido de po-limerasa NSSB se obtuvieron por secuenciación directa del DNA del producto amplificado con el Sistema ABI 3730 Gene-tic Analyzer Detection (Applied Biosystems, San Francisco, CA). La base con software ABI permitió la detección de variantes presentes en ≥ 30% de la población total.
Valoraciones de la seguridadTodos los eventos adversos (EA) que ocurrieron durante el curso del estudio se documentaron y reportaron por el in-vestigador al patrocinador. Como se definió en un plan de manejo del exantema, la intensidad del exantema se graduó como leve (localizado), moderada (difuso, 30 a < 70% de la superficie corporal), o severa (difuso generalizado, afección
Figura 2. Distribución de los pacientes.
Reclutados(N = 56)
Aleatorizados(n = 34)
No entraron (n = 22)
No tratados (n = 2)
Completaron el tratamiento
(n = 14)
Completaron el tratamiento
(n = 17)
BI 207127 600 mg/BI 201335 120 mg/
RBV(n = 17)
BI 207127 400 mg/BI 201335 120 mg/
RBV(n = 15)
Interrumpieron (n = 1); falta de eficacia

EFICACIA DEL INHIBIDOR DE PROTEASA BI 201335 37GASTROENTEROLOGY • Volumen 1 • Núm. 2
de membranas mucosas, signos de anafilaxia o peligro de muerte). La intensidad de todos los demás EA se juzgó con base en la tolerabilidad del paciente del evento como leve (fácil de tolerar), moderada (interferencia con las activida-des habituales) o grave (incapacitante o causando incapaci-dad para trabajar o llevar a cabo las actividades habituales). También se valoraron electrocardiogramas, signos vitales y parámetros de rutina del laboratorio. El estudio incluyó revisiones periódicas de los datos por un comité de monito-rización de datos externo al patrocinador.
Análisis estadísticosEl objetivo primario de eficacia de este estudio fue la res-puesta serológica rápida, definida como un nivel de RNA del VHC por debajo del LIDC (< 25 UI/mL) en la semana 4. Los objetivos de la seguridad incluyeron signos vitales, ex-ploración física, interrupción debida a EA y anormalidades del laboratorio. Se calcularon las estadísticas descriptivas de la eficacia, seguridad y farmacocinética. Se planeó un análisis preliminar después que todos los pacientes hubie-ran terminado el día 29.
Resultados
Distribución y características basales de los pacientesCincuenta y seis individuos fueron reclutados para el estu-dio, de los cuales 34 fueron distribuidos de forma aleatoria y 24 no empezaron el tratamiento debido a que retiraron el consentimiento (n = 3), no cumplieron los criterios de inclusión/exclusión (n = 9), o faltaron al corte predefinido de aleatorización debido a la inclusión rápida (n = 12) (Figura 2). A estos últimos 12 pacientes se les ofreció la distribu-ción aleatoria en la segunda parte del estudio, que se está llevando a cabo. Por lo tanto, 32 pacientes recibieron por lo menos una dosis del tratamiento asignado (Figura 2). De estos 32 sujetos, 31 completaron las cuatro semanas de tratamiento asignado; un paciente con resurgimiento vi-rológico confirmado se cambió a PegIFN y RBV el día 29 como estaba definido en el protocolo.
Los pacientes fueron distribuidos de manera uniforme en ambos grupos de dosis con respecto al nivel basal del RNA del VHC, etnia, edad, sexo, índice de masa corporal y sub-tipo (Cuadro 1). La media de la edad fue de 51 ± 11 años, la media del índice de masa corporal fue de 23.8 ± 3.5 kg/m2 y la media del nivel de RNA del VHC fue de 6.48 log10 UI/mL. Un número ligeramente mayor de pacientes tuvo infección del GT-1a, y todos excepto un paciente fueron caucásicos.
EficaciaDurante los dos primeros días de tratamiento todos los pa-cientes tuvieron una disminución rápida y abrupta de los niveles de RNA del VHC. Esta disminución fue seguida de una segunda fase de declinación más lenta hasta el día 29 en todos los individuos que recibieron BI 207127 600 mg tres veces al día y en todos excepto dos pacientes que recibieron 400 mg tres veces al día (Figura 3). En el grupo que recibió 600 mg tres veces al día, las declinaciones de los niveles del RNA del VHC fueron un poco más abruptas, las curvas
del RNA del VHC se agruparon más estrechamente, y una mayor proporción de pacientes alcanzó un RNA del VHC por debajo del LIDC o del LIDD hacia el final del tratamiento en comparación con el grupo que recibió 400 mg tres veces al día.
Las tasas de respuesta virológica (nivel del RNA del VHC < 25 UI/mL) en el grupo que recibió BI 207127 400 mg tres veces al día fueron de 27, 47, 67 y 73% en los días 8, 15, 22, y 29, respectivamente; se observaron tasas más altas de res-puesta en los sujetos infectados con el GT-1b en compara-ción con los pacientes infectados con el GT-1a (Cuadro 2). En contraste, las tasas de respuesta correspondientes en el grupo que recibió BI 207127 600 mg tres veces al día fueron de 18, 82, 100 y 100%, sin diferencias de subtipos (Cuadro 2). La mayoría de los pacientes del grupo que recibió 600 mg tres veces al día tuvo incluso niveles indetectables de RNA del VHC los días 22 y 29 (53 y 71%, respectivamente), mientras que estas tasas no excedieron de 20% en el grupo que recibió 400 mg tres veces al día.
Se observó un resurgimiento virológico durante el tra-tamiento el día 22, que se confirmó el día 29, y el paciente se cambió a PegIFN más RBV según el protocolo; el suje-to mostró una buena respuesta virológica cuatro semanas más tarde (disminución del RNA del VHC de 2 log10). Otro paciente tuvo un aumento del nivel del RNA del VHC de un nadir de 0.7 log10 UI/mL. El RNA del VHC se quedó en este nivel hasta que el sujeto se cambió a terapia triple con BI 201335/PegIFN/RBV el día 29; después de 10 días de
Cuadro 1. Resumen de las características basales
BI 207127 400 mg
3 veces al día +
BI201335 120 mg
una vez al día + RBV
(n = 15)
BI 207127 600 mg
3 veces al día +
BI201335 120 mg
una vez al día + RBV
(n = 17)
Edad (a)
Media 51 51
DE 10.0 11.5
Índice de masa
corporal (kg/m2)
Media 23 24
DE 2.9 3.8
Sexo, n (%)
Hombres 8 (53.3) 10 (58.8)
Mujeres 7 (46.7) 7 (41.2)
Etnia, n (%)
Asiáticos 0 (0.0) 1 (5.9)
Caucásicos 15 (100.0) 16 (94.1)
RNA del VHC
(log10
UI/mL)
Media 6.45 6.51
DE 0.51 0.66
Genotipo, n (%)
1a 10 (66.7) 8 (47.1)
1b 5 (33.3) 8 (47.1)
Otro 0 1a (5.9)
aEl análisis retrospectivo del GT basado en la secuencia de NS5B
identificó una muestra del GT-1 como GT-6e.
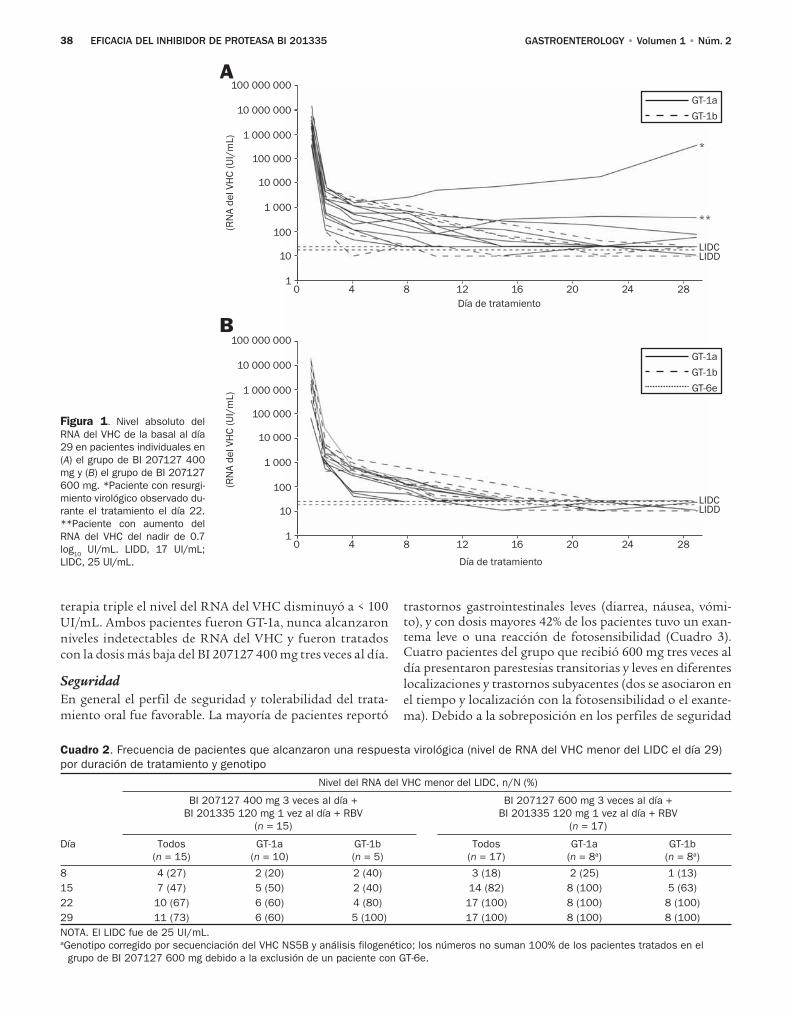
38 EFICACIA DEL INHIBIDOR DE PROTEASA BI 201335 GASTROENTEROLOGY • Volumen 1 • Núm. 2
terapia triple el nivel del RNA del VHC disminuyó a < 100 UI/mL. Ambos pacientes fueron GT-1a, nunca alcanzaron niveles indetectables de RNA del VHC y fueron tratados con la dosis más baja del BI 207127 400 mg tres veces al día.
SeguridadEn general el perfil de seguridad y tolerabilidad del trata-miento oral fue favorable. La mayoría de pacientes reportó
trastornos gastrointestinales leves (diarrea, náusea, vómi-to), y con dosis mayores 42% de los pacientes tuvo un exan-tema leve o una reacción de fotosensibilidad (Cuadro 3). Cuatro pacientes del grupo que recibió 600 mg tres veces al día presentaron parestesias transitorias y leves en diferentes localizaciones y trastornos subyacentes (dos se asociaron en el tiempo y localización con la fotosensibilidad o el exante-ma). Debido a la sobreposición en los perfiles de seguridad
Figura 1. Nivel absoluto del
RNA del VHC de la basal al día
29 en pacientes individuales en
(A) el grupo de BI 207127 400
mg y (B) el grupo de BI 207127
600 mg. *Paciente con resurgi-
miento virológico observado du-
rante el tratamiento el día 22.
**Paciente con aumento del
RNA del VHC del nadir de 0.7
log10
UI/mL. LIDD, 17 UI/mL;
LIDC, 25 UI/mL.
100 000 000
10 000 000
1 000 000
100 000
10 000
1 000
100
10
1
A
B
(RN
A de
l VH
C (U
I/m
L)(R
NA
del V
HC
(UI/
mL)
0 4 8 12 16 20 24 28Día de tratamiento
GT-1a
GT-1b
LIDCLIDD
LIDCLIDD
**
*
Día de tratamiento
100 000 000
10 000 000
1 000 000
100 000
10 000
1 000
100
10
10 4 8 12 16 20 24 28
GT-1a
GT-1b
GT-6e
Cuadro 2. Frecuencia de pacientes que alcanzaron una respuesta virológica (nivel de RNA del VHC menor del LIDC el día 29)
por duración de tratamiento y genotipo
Nivel del RNA del VHC menor del LIDC, n/N (%)
BI 207127 400 mg 3 veces al día +
BI 201335 120 mg 1 vez al día + RBV
(n = 15)
BI 207127 600 mg 3 veces al día +
BI 201335 120 mg 1 vez al día + RBV
(n = 17)
Día Todos
(n = 15)
GT-1a
(n = 10)
GT-1b
(n = 5)
Todos
(n = 17)
GT-1a
(n = 8a)
GT-1b
(n = 8a)
8 4 (27) 2 (20) 2 (40) 3 (18) 2 (25) 1 (13)
15 7 (47) 5 (50) 2 (40) 14 (82) 8 (100) 5 (63)
22 10 (67) 6 (60) 4 (80) 17 (100) 8 (100) 8 (100)
29 11 (73) 6 (60) 5 (100) 17 (100) 8 (100) 8 (100)
NOTA. El LIDC fue de 25 UI/mL.aGenotipo corregido por secuenciación del VHC NS5B y análisis filogenético; los números no suman 100% de los pacientes tratados en el
grupo de BI 207127 600 mg debido a la exclusión de un paciente con GT-6e.
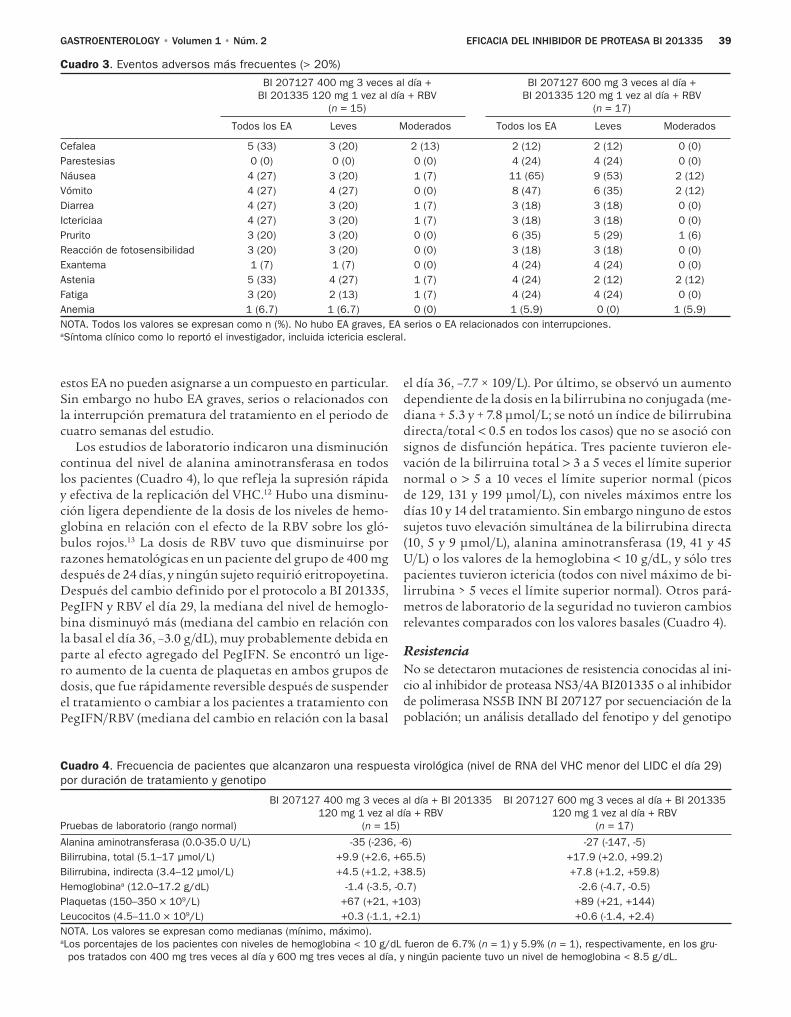
EFICACIA DEL INHIBIDOR DE PROTEASA BI 201335 39GASTROENTEROLOGY • Volumen 1 • Núm. 2
estos EA no pueden asignarse a un compuesto en particular. Sin embargo no hubo EA graves, serios o relacionados con la interrupción prematura del tratamiento en el periodo de cuatro semanas del estudio.
Los estudios de laboratorio indicaron una disminución continua del nivel de alanina aminotransferasa en todos los pacientes (Cuadro 4), lo que refleja la supresión rápida y efectiva de la replicación del VHC.12 Hubo una disminu-ción ligera dependiente de la dosis de los niveles de hemo-globina en relación con el efecto de la RBV sobre los gló-bulos rojos.13 La dosis de RBV tuvo que disminuirse por razones hematológicas en un paciente del grupo de 400 mg después de 24 días, y ningún sujeto requirió eritropoyetina. Después del cambio definido por el protocolo a BI 201335, PegIFN y RBV el día 29, la mediana del nivel de hemoglo-bina disminuyó más (mediana del cambio en relación con la basal el día 36, –3.0 g/dL), muy probablemente debida en parte al efecto agregado del PegIFN. Se encontró un lige-ro aumento de la cuenta de plaquetas en ambos grupos de dosis, que fue rápidamente reversible después de suspender el tratamiento o cambiar a los pacientes a tratamiento con PegIFN/RBV (mediana del cambio en relación con la basal
el día 36, –7.7 × 109/L). Por último, se observó un aumento dependiente de la dosis en la bilirrubina no conjugada (me-diana + 5.3 y + 7.8 µmol/L; se notó un índice de bilirrubina directa/total < 0.5 en todos los casos) que no se asoció con signos de disfunción hepática. Tres paciente tuvieron ele-vación de la bilirruina total > 3 a 5 veces el límite superior normal o > 5 a 10 veces el límite superior normal (picos de 129, 131 y 199 µmol/L), con niveles máximos entre los días 10 y 14 del tratamiento. Sin embargo ninguno de estos sujetos tuvo elevación simultánea de la bilirrubina directa (10, 5 y 9 µmol/L), alanina aminotransferasa (19, 41 y 45 U/L) o los valores de la hemoglobina < 10 g/dL, y sólo tres pacientes tuvieron ictericia (todos con nivel máximo de bi-lirrubina > 5 veces el límite superior normal). Otros pará-metros de laboratorio de la seguridad no tuvieron cambios relevantes comparados con los valores basales (Cuadro 4).
ResistenciaNo se detectaron mutaciones de resistencia conocidas al ini-cio al inhibidor de proteasa NS3/4A BI201335 o al inhibidor de polimerasa NS5B INN BI 207127 por secuenciación de la población; un análisis detallado del fenotipo y del genotipo
Cuadro 3. Eventos adversos más frecuentes (> 20%)
BI 207127 400 mg 3 veces al día +
BI 201335 120 mg 1 vez al día + RBV
(n = 15)
BI 207127 600 mg 3 veces al día +
BI 201335 120 mg 1 vez al día + RBV
(n = 17)
Todos los EA Leves Moderados Todos los EA Leves Moderados
Cefalea 5 (33) 3 (20) 2 (13) 2 (12) 2 (12) 0 (0)
Parestesias 0 (0) 0 (0) 0 (0) 4 (24) 4 (24) 0 (0)
Náusea 4 (27) 3 (20) 1 (7) 11 (65) 9 (53) 2 (12)
Vómito 4 (27) 4 (27) 0 (0) 8 (47) 6 (35) 2 (12)
Diarrea 4 (27) 3 (20) 1 (7) 3 (18) 3 (18) 0 (0)
Ictericiaa 4 (27) 3 (20) 1 (7) 3 (18) 3 (18) 0 (0)
Prurito 3 (20) 3 (20) 0 (0) 6 (35) 5 (29) 1 (6)
Reacción de fotosensibilidad 3 (20) 3 (20) 0 (0) 3 (18) 3 (18) 0 (0)
Exantema 1 (7) 1 (7) 0 (0) 4 (24) 4 (24) 0 (0)
Astenia 5 (33) 4 (27) 1 (7) 4 (24) 2 (12) 2 (12)
Fatiga 3 (20) 2 (13) 1 (7) 4 (24) 4 (24) 0 (0)
Anemia 1 (6.7) 1 (6.7) 0 (0) 1 (5.9) 0 (0) 1 (5.9)
NOTA. Todos los valores se expresan como n (%). No hubo EA graves, EA serios o EA relacionados con interrupciones.aSíntoma clínico como lo reportó el investigador, incluida ictericia escleral.
Cuadro 4. Frecuencia de pacientes que alcanzaron una respuesta virológica (nivel de RNA del VHC menor del LIDC el día 29)
por duración de tratamiento y genotipo
Pruebas de laboratorio (rango normal)
BI 207127 400 mg 3 veces al día + BI 201335
120 mg 1 vez al día + RBV
(n = 15)
BI 207127 600 mg 3 veces al día + BI 201335
120 mg 1 vez al día + RBV
(n = 17)
Alanina aminotransferasa (0.0-35.0 U/L) -35 (-236, -6) -27 (-147, -5)
Bilirrubina, total (5.1–17 μmol/L) +9.9 (+2.6, +65.5) +17.9 (+2.0, +99.2)
Bilirrubina, indirecta (3.4–12 μmol/L) +4.5 (+1.2, +38.5) +7.8 (+1.2, +59.8)
Hemoglobinaa (12.0–17.2 g/dL) -1.4 (-3.5, -0.7) -2.6 (-4.7, -0.5)
Plaquetas (150–350 × 109/L) +67 (+21, +103) +89 (+21, +144)
Leucocitos (4.5–11.0 × 109/L) +0.3 (-1.1, +2.1) +0.6 (-1.4, +2.4)
NOTA. Los valores se expresan como medianas (mínimo, máximo).aLos porcentajes de los pacientes con niveles de hemoglobina < 10 g/dL fueron de 6.7% (n = 1) y 5.9% (n = 1), respectivamente, en los gru-
pos tratados con 400 mg tres veces al día y 600 mg tres veces al día, y ningún paciente tuvo un nivel de hemoglobina < 8.5 g/dL.

40 EFICACIA DEL INHIBIDOR DE PROTEASA BI 201335 GASTROENTEROLOGY • Volumen 1 • Núm. 2
será reportado en otra publicación. La secuenciación de áci-dos nucleicos del virus aislado el día 30 (y comparado con la secuencia obtenida de forma basal) del paciente con resurgi-miento viral confirmado identificó R155K en NS3 y P495L en NS5B como cambios mayores en la secuencia de aminoá-cidos que representaron la selección del virus doble mutante confiriendo resistencia al BI 201335 y al BI 207127. La geno-tipificación integral del NS3/4A y NS5B no pudo practicarse en las muestras del paciente que aumentó 0.7 log10 UI/mL porque el nivel del RNA del VHC era menor de 1 000 UI/mL y demasiado bajo para generar ampliaciones de longitud com-pleta. No obstante obtuvimos secuencias parciales del domi-nio de la proteasa NS3 del virus aislado el día 17, y en relación con la secuencia basal identificamos un cambio de aminoáci-do R155K en el NS3; un amplicon que abarcó el extremo 3’ de la región NSSN del virus del día 17 no detectó ningún cam-bio canónico en los residuos P495 o P496 que son conocidos por conferir resistencia al dominio 1 del “thumb” del INN.
DiscusiónMuchos pacientes con hepatitis C crónica que requieren tra-tamiento antiviral no toleran el PegIFN y constituyen 13% de las suspensiones del tratamiento.14,15 Además se estima que 30 a 60% de pacientes con VHC no reciben el tratamiento debido a contraindicaciones médicas al PegIFN/RBV,16,17 lo que destaca la necesidad clínica de regímenes libres de inter-ferón. En la actualidad varias combinaciones libres de Peg-IFN de dos AAD con diferentes modos de acción, con o sin RBV, están en investigación clínica.7,18-21 Los inhibidores de la proteasa NS3/4A, los inhibidores de NS5A y los INN NS5B tienen una barrera genética más baja para la resis-tencia en comparación con los IN NS5B.22 Hasta ahora la mayoría de combinaciones dobles de AAD con una barrera baja a la resistencia han fallado para mantener la supresión de la replicación viral.18,20 Parecen preexistir variantes con resistencia a dos AAD y son seleccionadas durante el trata-miento de doble combinación de AAD.23 Las estrategias po-tenciales para superar este problema son 1) combinaciones de AAD que incluyan inhibidores de polimerasa con una barrera alta a la resistencia, 2) tratamiento triple con AAD y 3) combinaciones de dos AAD con una barrera genética baja a la resistencia más RBV.
El resurgimiento virológico con la selección de varian-tes resistentes no se ha observado todavía en los estudios clínicos de corto plazo del inhibidor de proteasa NS3/4A danoprevir más el IN mericitabina,7 lo que sugiere que la inclusión de un IN en el tratamiento de combinación con AAD es una estrategia atractiva. Sin embargo todavía se requieren datos de eficacia (RVS) y seguridad de los trata-mientos a más largo plazo. El enfoque de combinar tres AAD sin resistencia cruzada con una barrera más baja a la resistencia (es decir, un INN más un inhibidor de la pro-teasa NS3/4A y un inhibidor de NS5A) está bien apoyado por análisis matemáticos. Rong y colaboradores mostra-ron que es poco probable que existan variantes resistentes contra las tres clases de fármacos antes del inicio del tra-tamiento, y es poco probable que ocurra la emergencia du-
rante el tratamiento.24 No obstante las interacciones entre los medicamentos y los perfiles de seguridad que se sobre-ponen aún son un problema por examinar.
Una tercera estrategia atractiva es combinar dos AAD con un una barrera genética baja más RBV. Un estudio que valoró el GS-9256 más regobuvir con o sin RBV mostró el importante papel de la RBV en la disminución del RNA del VHC, y la reducción del resurgimiento viral de las com-binaciones de AAD con una barrera baja a la resistencia.18
En el presente estudio todos los pacientes recibieron el in-hibidor de proteasa NS3/4A BI 201335 y el INN NS5B BI 207127 en combinación con RBV. Esta combinación libre de PegIFN mostró una actividad antiviral rápida y fuerte en sujetos crónicamente infectados con el VHC GT-1. Sólo un paciente, tratado con la dosis más baja de BI 207127, presentó resurgimiento virológico y se seleccionó para vi-rus doblemente resistentes. Este fue un individuo con el GT-1a en el grupo de dosis más baja, lo que refleja activi-dad antiviral ligeramente disminuida del BI 207127 contra este subtipo, que se observó también en monoterapia.10 En contraste, no ocurrió resurgimiento virológico en el grupo que recibió BI 207127 600 mg tres veces al día, lo que de-muestra que la selección rápida y uniforme de mutaciones de resistencia asociados con la monoterapia con el inhibi-dor de proteasa NS3/4A disminuye o se retrasa en este régi-men libre de PegIFN. La razón de la alta actividad antivi-ral relacionada con una tasa baja de resurgimiento de esta combinación en comparación con otras combinaciones de inhibidores de proteasas/INN con sólo dos de 32 pacien-tes desarrollando una mutante única o doble al BI 207127 y/o al BI 20133518,19 puede deberse al efecto de la RBV, que es tal vez causado por una actividad antiviral directa débil así como por la inducción de genes sensibles al interferón.12
Es importante notar que el BI 207127 pertenece a la cla-se 1 del dominio “thumb” del INN que desplaza al asa λ1 “finger” del dominio “thumb” superior e interfiere con el cambio conformacional requerido para iniciar la síntesis de RNA. En contraste con los otros sitios que están expues-tos en la superficie de la polimerasa NS5B, la unión al sitio del “thumb” 1 comprende una interfase entre los dominios funcionales que restringen la secuencia del polimorfismo y pueden proporcionar una barrera de resistencia más alta en relación con otros INN.11 Los resultados del estudio defase 2b que se está realizando que compara regímenes de dosis del BI 201335 y BI 207127 con o sin RBV propor-cionará mayor información.
Los tratamientos con BI 201335 y BI 207127 más RBV mostraron un perfil global en seguridad y tolerabilidad. Como se esperaba para los resultados de ambos compuestos en los estudios de fase 1 y 2,4,9,10 una proporción sustancial de pacientes presentó un exantema leve a moderado, foto-sensibilidad, o síntomas gastrointestinales, que no impacta-ron en la continuación del tratamiento. La ictericia fue más común pero por lo regular leve y siempre debida a hiperbi-lirrubinemia predominantemente no conjugada, sin signos de toxicidad hepática. Los mecanismos involucrados, como se ha propuesto en estudios extensos in vitro, incluyen la in-

EFICACIA DEL INHIBIDOR DE PROTEASA BI 201335 41GASTROENTEROLOGY • Volumen 1 • Núm. 2
hibición mediada por el BI 201335 de los transportadores de bilirrubina (p. ej, OATP1B1, MRP-2) y las enzimas conjugan-tes (p. ej., UGT1A1)25 en presencia de hemólisis inducida por RBV. La ligera elevación simultánea del nivel de bilirrubina conjugada encontrada en algunos pacientes puede explicar-se por la sobreestimación relacionada con el ensayo diazo de la bilirrubina directa26 y el efecto inhibidor del BI 201335 sobre el transportador de bilirrubina MRP-2.25 Se han des-crito mecanismos similares de interacción con el metabolis-mo de la bilirrubina para otros inhibidores de proteasa del VHC27 y del virus de la inmunodeficiencia humana.28,29 En confirmación de estos reportes, la ictericia asociada con el BI 201335 no se relacionó con otros síntomas en los estudios de fase 2 del BI 201335 en combinación con PegIFN/RBV.25 La ligera elevación de plaquetas observada estuvo en un ran-go similar al encontrado con la monoterapia con RBV.30 Sin embargo no hubo eventos trombóticos reportados en nues-tro estudio y el efecto fue rápidamente reversible con una disminución incremental de la cuenta de plaquetas después de cambiar a los pacientes a PegIFN/RBV. De forma similar, es probable que la disminución de los niveles de hemoglo-bina y las tasas de anemia encontradas después de cuatro semanas de tratamiento de VHC con BI 201335, BI 207127 y RBV sean causadas por la hemólisis inducida por RBV. El estudio más grande de monoterapia con RBV describió una media de disminución del nivel de hemoglobina después de cuatro semanas de RBV de > 2 g/dL, donde 20% de los sujetos presentó una disminución ≥ 4 g/dL.13 Sin embargo el estudio de fase 2b que se está realizando está valorando varios regímenes de dosificación a más largo plazo de esta combinación oral, incluido un régimen sin RBV, y por lo tanto mostrará si la combinación de AAD tiene un efecto adicional sobre la hemoglobina o la cuenta de plaquetas. En resumen, el perfil general de seguridad de esta combinación de AAD es comparable con otros compuestos de ambas cla-ses de fármacos. La ausencia de EA graves e interrupciones del régimen muestran que el tratamiento por lo general es seguro y los efectos secundarios son manejables.
En fecha reciente se reportó la prueba de concepto para alcanzar RVS con un régimen de tratamiento sin PegIFN en pacientes con VHC. La combinación de un inhibidor de NS5A y un inhibidor de la proteasa NS3/4A en pacientes con GT-1 y respuesta nula a tratamientos previos con PegIFN/RBV alcanzaron una RVS en 4 de 11 pacientes que fueron tratados durante 24 semanas, con sólo una recaída en esta cohorte.20 Esto indica que el VHC es erradicable en pacientes infectados de forma crónica con un régimen de combina-ción de AAD libre de PegIFN y apoya las investigaciones con varias combinaciones de AAD que pueden ser capaces de mejorar las tasas de RVS sin la necesidad de PegIFN.
El régimen oral con BI 201335 y BI 207127 más RBV mos-tró actividad antiviral potente en pacientes con VHC GT-1 que fue generalmente seguro y tolerable sin la selección de resistencia en el grupo de dosis más altas. El paso crucial si-guiente en el desarrollo clínico es la evaluación de la seguri-dad y la RVS con el tratamiento a más largo plazo con esta combinación sin PegIFN. Además, al eliminar no sólo el Peg-
IFN sino también la RBV del tratamiento futuro del VHC sin duda mejoraría la tolerabilidad y potencialmente permitiría el tratamiento de pacientes con contraindicaciones para la RBV. En la actualidad se lleva a cabo un estudio prospectivo de fase 2b que valora la sustentabilidad de las respuestas viro-lógicas a diferentes tratamientos de más largo plazo.
Referencias1. Poordad F, McCone J Jr, Bacon BR, et al. Boceprevir for un-
treated chronic HCV genotype 1 infection. N Engl J Med 2011;364:1195–1206.
2. Jacobson IM, McHutchison JG, Dusheiko GM, et al. Telaprevir in combination with peginterferon and ribavirin in genotype 1 HCV treatment-naïve patients: final results of phase 3 ADVANCE study (abstr 211). Hepatology 2010;52(Suppl 1):427A.
3. Sherman KE, Flamm SL, Afdhal NH, et al. Telaprevir in combina-tion with peginterferon alfa2a and ribavirin for 24 or 48 weeks in treatment-naïve genotype 1 HCV patients who achieved an ex-tended rapid viral response: final results of phase 3 ILLUMINATE study (abstr LB-2). Hepatology 2010;52(Suppl 1):401A–402A.
4. Sulkowski MS, Ceasu E, Asselah T, et al. SILEN-C1: sustained virologic response (SVR) and safety of BI201335 combined with peginterferon alfa-2a and ribavirin (P/R) in treatment-naïve pa-tients with chronic genotype 1 HCV infection (abstr 60). J Hepa-tol 2011;54(Suppl 1):S27.
5. Tan H, Rajyaguru S, Wu T, et al. Combination of the NS3/4A protease inhibitor ITMN-191 (R7227) with the active moiety of the NS5B inhibitors R1626 or R7128 enhances replicon clearan-ce and reduces the emergence of drug resistant variants (abstr 1885). Hepatology 2008;48(Suppl 1):1153A.
6. Lacolla M, Lallos LB, Serra I, et al. A triple combination of direc-tacting antiviral agents demonstrates robust anti-HCV activity in vitro (abstr 769). J Hepatol 2010;52(Suppl 1):S299.
7. Gane EJ, Roberts SK, Stedman CA, et al. Oral combination the-rapy with a nucleoside polymerase inhibitor (RG7128) and dano-previr for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 2010;376:1467–1475.
8. Manns MP, Bourlière M, Benhamou Y, et al. Potency, safety and pharmacokinetics of the NS3/4A protease inhibitor BI201335 in patients with chronic HCV genotype-1 infection. J Hepatol 2011; 54:1114–1122.
9. Sulkowski MS, Bourliere M, Bronowicki J-P, et al. SILEN-C2: sus-tained virologic response (SVR) and safety of BI201335 com-bined with peginterferon alfa-2a and ribavirin (P/R) in chronic HCV genotype-1 patients with non-response to P/R (abstr 66). J Hepatol 2011;54(Suppl 1):S30.
10. Larrey D, Lohse A, de Ledinghen V, et al. 4 week therapy with the non-nucleoside polymerase inhibitor BI 207127 in combination with peginterferon-alfa2a and ribavirin in treatment naïve and treatment experienced chronic HCV GT1 patients (abstr 2007). J Hepatol 2010;52(Suppl 1):S466.
11. Kukolj G, McGibbon GA, McKercher G, et al. Binding site cha-racterization and resistance to a class of non-nucleoside inhi-bitors of the hepatitis C virus NS5B polymerase. J Biol Chem 2005;280:39260–39267.
12. Noureddin M, Rotman Y, Feld J, et al. Effects of ribavirin mo-notherapy on ALT, HCV viral levels, serum cytokines, and hepa-tic interferon-stimulated gene expression in chronic hepatitis C (abstr 908). Hepatology 2010;52(Suppl 1):756A.
13. Bodenheimer HC Jr, Lindsay KL, Davis GL, et al. Tolerance and efficacy of oral ribavirin treatment of chronic hepatitis C: a mul-ticenter trial. Hepatology 1997;26:473–477.
14. Fried MW. Side effects of therapy of hepatitis C and their mana-gement. Hepatology 2002;36(Suppl 1):S237–S244.
15. McHutchison JG, Lawitz EJ, Shiffman ML, et al. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infec-tion. N Engl J Med 2009;361:580–593.
16. Falck-Ytter Y, Kale H, Mullen KD, et al. Surprisingly small effect of antiviral treatment in patients with hepatitis C. Ann Intern Med 2002;136:288–292.
17. Kanwal F, Hoang T, Spiegel BM, et al. Predictors of treatment in patients with chronic hepatitis C infection—role of patient versus nonpatient factors. Hepatology 2007;46:1741–1749.

42 EFICACIA DEL INHIBIDOR DE PROTEASA BI 201335 GASTROENTEROLOGY • Volumen 1 • Núm. 2
18. Zeuzem S, Buggisch P, Agarwal K, et al. Dual, triple, and quadru-ple combination treatment with a protease inhibitor (GS-9256) and a polymerase inhibitor (GS-9190) alone and in combination with ribavirin (RBV) or PegIFN/RBV for up to 28 days in treatment naïve, genotype 1 HCV subjects (abstr LB-1). Hepatology 2010; 52(Suppl 1):400A–401A.
19. Di Bisceglie AM, Nelson DR, Gane E, et al. VX-222 with TVR alo-ne or in combination with peginterferon alfa-2a and ribavirin in treatment-naive patients with chronic hepatitis C: ZENITH study interim results (abstr 1363). J Hepatol 2011;54(Suppl 1):S540.
20. Lok A, Gardiner D, Lawitz E, et al. Quadruple therapy with BMS- 790052, BMS-650032 and Peg-IFN/RBV for 24 weeks results in 100% SVR12 in HCV genotype 1 null responders (abstr 1356). J Hepatol 2011;54(Suppl 1):S536.
21. Lawitz E, Rodriguez-Torres M, Denning J, et al. Once daily dual-nucleotide combination of PSI-938 and PSI-7977 provides 94% HCV RNA LOD at Day 14: first purine/pyrimidine clinical combi-nation data (the NUCLEAR study) (abstr 1370). J Hepatol 2011; 54(Suppl 1):S543.
22. McCown MF, Rajyaguru S, Le Pogam S, et al. The hepatitis C virus replicon presents a higher barrier to resistance to nucleosi-de analogs than to nonnucleoside polymerase or protease inhi-bitors. Antimicrob Agents Chemother 2008;52:1604–1612.
23. Sarrazin C, Zeuzem S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 2010; 138:447–462.
24. Rong L, Dahari H, Ribeiro RM, et al. Rapid emergence of pro-tease inhibitor resistance in hepatitis C virus. Sci Transl Med 2010;2:30–32.
25. Sane R, Podila L, Mathur A, et al. Mechanisms of isolated un-conjugated hyperbilirubinemia induced by the HCV NS3/4A pro-tease inhibitorBI201335 (abstr 1236). J Hepatol 2011;54(Suppl 1):S488.
26. Chowdhury JR, Chowdhury NR, Jansen PLM. Bilirubin metabo-lism and its disorders. In: Zakim D, Boyer TD, eds. Hepatology: a textbook of liver disease. 4th ed. Philadelphia, PA: WB Saun-ders, 2003:233–270.
27. Huisman MT, Snoeys J, Monbaliu J, et al. In vitro studies investi-gating the mechanism of interaction between TMC435 and hepa-tic transporters (abstr 278). Hepatology 2010;52(Suppl 1):461A.
28. Lankisch TO, Moebius U, Wehmeier M, et al. Gilbert’s disease
and atazanavir: from phenotype to UDP-glucuronosyltransferase
haplotype. Hepatology 2006;44:1324–1332.
29. Sulkowski, MS. Drug-induced liver injury associated with anti-
retroviral therapy that includes HIV-1 protease inhibitors. Clin
Infect Dis 2004;38(Suppl 2):S90–S97.
30. Fueloep B, Rohde P, Buggisch P, et al. The antiviral efficacy of
ribavirin priming. A follow up of 93 patients treated with ribavirin
monotherapy followed by standard combination treatment (abstr
980). Hepatology 2010;52(Suppl 1):793A.
Recibido el 3 de junio de 2011. Aceptado el 30 de agosto de 2011.
Solicitud de reimpresosDirigir la solicitud de reimpresos a: Stefan Zeuzem, MD, Johann Wol-
fgang Goethe University Hospital, Frankfurt, Germany. correo electróni-
co: [email protected]; fax: (49) 696 301 6448.
AgradecimientosLos autores agradecen a Yves Benhamou su contribución al diseño del es-
tudio y al reclutamiento de pacientes.
La asistencia editorial fue proporcionada por StemScientific, con recur-
sos de Boehringer Ingelheim.
Conflictos de interésLos autores declaran lo siguiente: S.Z. es consultor de Abbott Laboratories,
Achillion Pharmaceuticals, Anadys Pharmaceuticals, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, iTherX, Janssen/Tibotec, Merck, Novartis, Pfizer, Pharmasset, Roche/Genentech, Santaris Pharma, y Ver-
tex Pharmaceuticals. T.A. es consultor de Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck, Novartis, y Roche. J.-P.Z. es
cconsultor de Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences,
Roche, Janssen, y Merck/Schering-Plough. D.L. es consultor de Boerhinger Ingelheim, Bristol-Myers Squibb, Roche, Janssen, Merck, y Cytheris. B.M. es
consultor de MSD, Roche, Janssen, y Gilead Sciences. M.S. es consultor de Bristol-Myers Squibb y Roche y es conferencista de Bristol-Myers Squibb, Falk, Gilead Sciences, Merck, y Roche. A.L. ha participado en estudios de Boehringer Ingelheim, Roche, MSD, Janssen, y Falk y ha recibido honorarios
por conferencias de MSD, Roche, y Falk. S.P. es consultor de Bristol-Myers Squibb, Boehringer Ingelheim, Tibotec/Janssen-Cilag, Gilead Sciences, Ro-che, Merck/Schering-Plough, y Abbott Laboratories; es conferencista de GlaxoSmithKline, Bristol-Myers Squibb, Boehringer Ingelheim, Tibotec/Jans-sen-Cilag, Gilead Sciences, Roche, y Schering-Plough; y ha recibido subven-ciones de Bristol-Myers Squibb, Gilead Sciences, Roche, y Merck/Schering-
Plough. J.-P.B. es consultor de Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Janssen, Merck, Novartis, y Roche. F.Z.
es consultor de Janssen, Schering-Plough, y Vertex Pharmaceuticals. M.H.
es consultor de Novartis, MSD, Roche, Jansen, y Gilead Sciences. J.O.S., G.K., G.N., C.H., y W.O.B. son empleados de Boehringer Ingelheim. El resto de los autores no declararon conflictos.

GASTROENTEROLOGY 2012;142:152–164
43
ANTECEDENTES Y OBJETIVOS: algunos estudios indican que el colesterol de la dieta es importante en la progresión de la fi brosis hepática. Nosotros investigamos los mecanismos por los cuales el colesterol de la dieta podría contribuir a la fi brogénesis hepática. MÉTODOS: ratones C57BL/6 fueron alimentados con una dieta rica en colesterol o una dieta control durante cuatro semanas; enseguida se indujo fi brosis hepática mediante ligadura del conducto biliar o la administración de tetracloruro de carbono. Se aislaron las células estelares hepáticas (CEH) de los ratones alimentados con dietas en colesterol o de ratones tipo Niemann Pick con defi ciencia C1, que acumulan colesterol libre intracelular. RESULTADOS: después de la ligadura del conducto biliar o la administración de tetracloruro de carbono, los ratones alimentados con dietas ricas en colesterol tuvieron aumento signifi cativo de fi brosis hepática y activación de las CEH en comparación con los ratones con la dieta control. No hubo diferencia signifi cativa en el grado de daño hepatocelular o en la infl amación hepática, incluido la apoptosis de los hepatocitos o activación de las células de Kupffer, entre los ratones con dietas ricas en colesterol o con dietas control. Los niveles de colesterol libre fueron mayores en las CEH de los ratones alimentados con dietas ricas en colesterol que con la dieta control. En las CEH cultivadas, la acumulación de colesterol libre en las CEHaumentaron los niveles del receptor 4 semejante a Toll (TLR-4), lo que llevó a una regulación negativa de la proteína morfogenética ósea y el inhibidor de la activina unido a la membrana (un seudorreceptor del factor transformador del crecimiento [TGF]ß); las CEH se sensibilizaron ante la activación inducida por el TGFß. La fi brosis hepática no se agravó con la dieta rica en colesterol en los ratones C3H/HeJ, que expresan una forma mutante del TLR4; las CEH que expresan el TLR4 mutante no se activaron por la acumulación de colesterol libre. CONCLUSIONES: el colesterol de la dieta agrava la fibrosis hepática porque el colesterol libre se acumula en las CEH, lo que resulta en aumento de las señales del TLR4, con regulación negativa de la proteína morfogenética ósea y del inhibidor de la activina unida a la membrana, así como en la sensibilización de las CEH al TGFß. Esta vía podría ser el blanco de tratamientos antifibróticos.
Palabras clave: enfermedad hepática; modelo en el ratón; dislipidemia; lipopolisacáridos.
La fibrosis hepática es un trastorno que indica la progre-sión de enfermedades hepáticas, puede llevar a cirrosis
o carcinoma hepatocelular.1 Por esta razón es importante determinar los mecanismos patológicos asociados con este trastorno.
Los factores alimenticios son, en cuestión, determinantes significativas del desarrollo de la fibrosis hepática. Datos derivados de 9 221 participantes en la encuesta denomina-da National Health and Nutrition Examination Survey en Esta-dos Unidos mostraron que el mayor consumo de colesterol en la alimentación se asocia con un mayor riesgo de cirrosis o cáncer hepático tanto en análisis de variables no ajustados como ajustados.2
Varios estudios reportan que las estatinas y la ezetimi-ba (agentes hipocolesterolemiantes) mejoran la fibrosis hepática en sujetos con hipercolesterolemia.3,4 En recientes estudios de laboratorio, roedores o conejos desarrollaron fibrosis hepática después del consumo de una dieta rica en colesterol (RC) a largo plazo con contenido de ácido cólico (dieta aterogénica) o una dieta alta en grasas y RC.5,6 Sin em-bargo los experimentos que utilizaron estas dietas no son adecuados para explicar el papel exacto del colesterol en el desarrollo de la fibrosis hepática porque el ácido cólico y los ácidos grasos libres inducen genes de fibrosis hepática,7 apoptosis de los hepatocitos e inflamación hepática.8 Aun-que estos estudios son parte de una serie de evidencias que
*J. W. Goethe University Hospital, Frankfurt, Germany; ‡Service d’Hépatologie, Hôpital Beaujon, Université Diderot-Paris 7, e INSERM U773, CRB3, Clichy, France; §Austin Hospital, Heidelberg, Victoria, Australia; IIHôpital Albert Michallon, Grenoble, France; ¶Hôpital Saint-Eloi, Montpellier, France; *University Hospital of Zürich, Zürich,
Switzerland; **Auckland City Hospital, Auckland, New Zealand; ‡‡University Medical Center Mainz, Mainz, Germany; §§University Hospital Hamburg- Eppendorf, Hamburg,
Germany; II IIHôpital Cochin, Paris, France; ¶¶University Hospital of Nancy, Vandoeuvre-lès-Nancy, France; ##Alfred Hospital, Melbourne, Victoria, Australia; ***Epimed
GmbH, Berlin, Germany; ‡‡‡Hepatology Department, Hospices Civils de Lyon, Lyon University, Lyon, France; §§§University Hospital Basel, Basel, Switzerland; II II IIBoehringer
Ingelheim, Ridgefield, Connecticut; ¶¶¶Boehringer Ingelheim (Canada) Ltd, Laval, Quebec, Canada; y ###Boehringer-Ingelheim Pharma GmbH & Co KG, Biberach, Germany
TOSHIAKI TERATANI,* KENGO TOMITA,*,‡ TAKAHIRO SUZUKI,* TETSUYA OSHIKAWA,* HIROKAZU YOKOYAMA,§
KATSUYOSHI SHIMAMURA,* SUSUMU TOMINAGA, SADAYUKI HIROI, RIE IRIE,¶ YOSHIKIYO OKADA,‡
CHIE KURIHARA,‡ HIROTOSHI EBINUMA,* HIDETSUGU SAITO,# RYOTA HOKARI,‡ KAZUO SUGIYAMA,*
TAKANORI KANAI,* SOICHIRO MIURA,‡ y TOSHIFUMI HIBI*
Una dieta rica en colesterol exacerba la fibrosis hepática en ratones a través de la acumulación de colesterol libre en las células estelares hepáticas
Abreviaturas utilizadas en este artículo: AcLDL, proteínas acetil de la lipoproteína de baja densidad; ALT, alanina aminotransferasa; Bambi, BMP e inhibidor de activina unido a la membrana; LCB, ligadura del conducto biliar; CCl
4, tetracloruro de carbono: EC, éster de colesterol;
compuesto 58035, inhibidor de la aciltransferasa del colesterol acil-CoA 58035; CL, colesterol libre; RC, rica en colesterol; CEH, células estelares
hepáticas; KO, knock-out; LPS, lipopolisacáridos; mRNA, RNA mensa-jero; NPC1, Niemann-Pick tipo C1; αSMA, actina-α del músculo liso; CT, colesterol total; TG, triglicéridos; TGFß, factor β transformador del
crecimiento; TLR4, receptor 4 semejante a Toll; TNFα, factor de necrosis tumoral; TS, tipo silvestre.
© 2012 by the AGA Institutedoi:10.1053/j.gastro.2011.09.049

44 UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA GASTROENTEROLOGY • Volumen 1 • Núm. 2
se acumulan y que muestra el papel del colesterol en el de-sarrollo y progresión de la fibrosis hepática, el papel exacto del colesterol en los mecanismos de la fibrosis hepática es-tán todavía en investigación.
Para aclarar el impacto preciso del colesterol en la fisio-patología de la fibrosis hepática se utilizan modelos expe-rimentales que incluyeron la administración de dietas RC sin ácido cólico o una cantidad excesiva de ácidos grasos a ratones en los que se indujo fibrosis hepática por ligadura del conducto biliar (LCB) o intoxicación con tetracloruro de carbono (CCL4). Las células estelares hepáticas (CEH), las principales productoras de matriz extracelular en el hígado fibrótico, desempeñan un papel clave en la fibrosis hepática, aunque la fibrosis hepática se asocia con daño hepático, por mecanismos en que se incluye a la muerte de hepatocitos y activación de las células de Kupffer.1 Nuestros resultados muestran que el consumo de una dieta RC causa acumula-ción de colesterol libre en las CEH, que a su vez suprimen de manera significativa la expresión del factor transformador del crecimiento-β (TGFβ), el seudorreceptor de la proteína morfogenética ósea y el inhidor de la activina unida a la membrana (Bambi) a través del reforzamiento de las señales del receptor 4 semejante a Toll (TLR4), lo que lleva al agra-vamiento de la fibrosis hepática.
Materiales y métodosPor favor consulte la sección de Materiales Suplementarios y Métodos para una descripción más detallada.
Modelo animalRatones machos de ocho semanas de edad de tipo silvestre (TS) C57BL/6, C3H/HeN o C3H/HeJ fueron alimentados con una dieta RC (1% peso/peso) (TD 92181) o una dieta control correspondiente (Teklad no. 7001; Harlan Teklad, Madison, WI) durante cuatro semanas, y enseguida se practicó LCB du-rante tres semanas, o se les administró CCl4 en una dosis de 5 μL (CCl4 al 10% en aceite de maíz)/g de peso en inyección intraperitoneal dos veces por semana durante cuatro semanas.
Análisis estadísticoTodos los datos se expresan como medias ± errores estándar de las medias. Los análisis estadísticos se realizaron con la prueba t de Student no pareada o análisis de varianza de una cola (una P < .05 fue considerada significativa).
Resultados
La dieta RC aceleró de manera significativa la fibrosis hepática inducida por LCB o CCl4
A los ratones C57BL/6 se les dio una dieta RC o una dieta control durante cuatro semanas y enseguida se dividieron en dos grupos; en un grupo se practicó LCB durante tres semanas y el otro grupo recibió tratamiento simulado. En un experimento similar, pero separado, los ratones fueron alimentados con una dieta RC o una dieta control durante cuatro semanas, y enseguida se dividieron en dos grupos: uno para cuatro semanas de tratamiento con CCl4 y el otro para tratamiento con aceite de maíz.
La dieta RC no aumentó la media del peso corporal o del peso del hígado vs. la dieta control (Cuadro 1 Suplementa-rio). Aunque la dieta RC aumentó la concentración sérica del colesterol total (CT) de manera significativa, no se observó ningún cambio en los niveles séricos de triglicéridos (TG) ni de glucosa (Cuadro 1 Suplementario). Además, la dieta RC sola no fue suficiente para causar esteatosis o fibrosis he-pática (Figuras 1 y 2). Sin embargo, como se mostró con la tinción tricrómica de Masson, la tinción inmunohistoquí-mica de la actina-α del músculo liso (αSMA) en el tejido hepático, así como con la determinación cuantitativa de hidroxiprolina hepática, la LCB exacerbó de forma signifi-cativa la fibrosis hepática en el grupo de dieta RC en compa-ración con el control (Figuras 1A y 1B). Las expresiones del RNA mensajero (mRNA) de la colágena 1α1, colágena 1α2 y αSMA estuvieron significativamente aumentadas con el de-sarrollo de la fibrosis hepática inducida por la LCB, que fue más evidente en el grupo de dieta RC que en el grupo con-trol (Figura 1C). Los niveles del mRNA del TGFß no demos-tró diferencia significativa entre los grupos de dieta (Figura 1C). De forma similar al modelo de LCB, el modelo murino de fibrosis hepática inducida por CCl4 mostró una progre-sión significativa de la fibrosis hepática en el grupo de dieta RC vs. control (Figuras 1D y E). La expresión del mRNA de la colágena 1α1, colágena 1α2, y αSMA fue favorecida en gran medida como resultado del desarrollo de fibrosis hepática in-ducida por CCl4, y esto se observó de forma más clara en el grupo de dieta RC que en el grupo de dieta control (Figura 1F). Los niveles del mRNA del TGFß no mostraron diferen-cias significativas entre los grupos (Figura 1F).
La dieta RC no aceleró el daño hepatocelular inducido por la LCB o el CCl4
Los niveles de CT hepático aumentaron de forma signifi-cativa con el consumo de la dieta RC tanto en el grupo de LCB como en grupos simulados (Figura 2A). Sin embargo los niveles de TG no mostraron diferencia significativa en-tre el grupo de dieta RC y el control, y la dieta RC no causó esteatosis hepática (Figuras 1A y 2A).
Los niveles séricos de alanina aminotransferasa (ALT), un marcador biológico del daño hepatocelular, aumentaron de manera significativa en los ratones tratados con LCB; no obstante este aumento no dependió del cosumo de coles-terol de la dieta en los grupos simulados o de LCB (Figura 2B). Además, la dieta RC no impactó de forma significativa en los potenciales de la membrana interna mitocondrial o el número de hepatocitos medido a través del marcado del extremo libre de la desoxiuridina mediada por la desoxinu-cleotidil transferasa terminal en los hígados de ratones con LCB (Figura 2C).
Los niveles hepáticos de CT aumentaron de manera sig-nificativa con el consumo de la dieta RC vs. la dieta control tanto en los grupos tratados con CCl4 como con aceite de maíz (Figura 2D). Sin embargo los niveles hepáticos de TG no tuvieron diferencia significativa entre los grupos con dieta RC y control, y la dieta RC no causó esteatosis hepáti-ca (Figuras 1D y 2D).
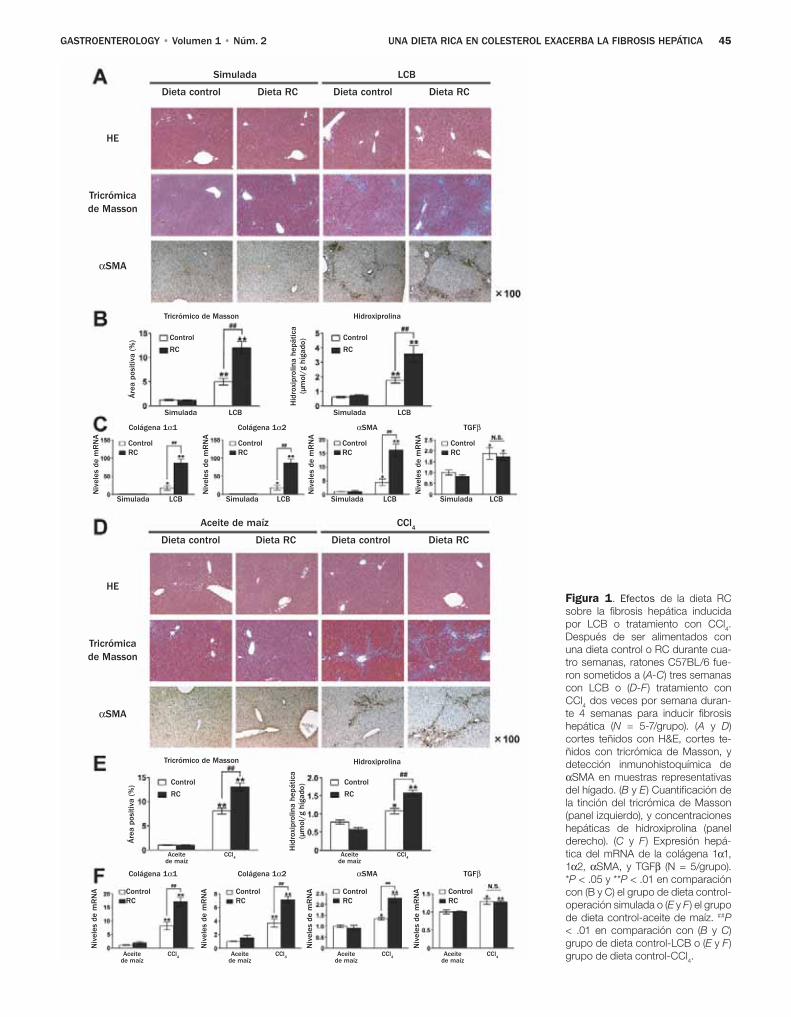
UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA 45GASTROENTEROLOGY • Volumen 1 • Núm. 2
Figura 1. Efectos de la dieta RC
sobre la fibrosis hepática inducida
por LCB o tratamiento con CCl4.
Después de ser alimentados con
una dieta control o RC durante cua-
tro semanas, ratones C57BL/6 fue-
ron sometidos a (A-C) tres semanas
con LCB o (D-F) tratamiento con
CCl4 dos veces por semana duran-
te 4 semanas para inducir fibrosis
hepática (N = 5-7/grupo). (A y D)
cortes teñidos con H&E, cortes te-
ñidos con tricrómica de Masson, y
detección inmunohistoquímica de
αSMA en muestras representativas
del hígado. (B y E) Cuantificación de
la tinción del tricrómica de Masson
(panel izquierdo), y concentraciones
hepáticas de hidroxiprolina (panel
derecho). (C y F) Expresión hepá-
tica del mRNA de la colágena 1α1,
1α2, αSMA, y TGFβ (N = 5/grupo).
*P < .05 y **P < .01 en comparación
con (B y C) el grupo de dieta control-
operación simulada o (E y F) el grupo
de dieta control-aceite de maíz. ##P
< .01 en comparación con (B y C)
grupo de dieta control-LCB o (E y F)
grupo de dieta control-CCl4.
Simulada LCB
Dieta control Dieta RC
Dieta control Dieta RC
Aceite de maíz CCl4
Dieta control Dieta RC
Dieta control Dieta RC
HE
Tricrómica
de Masson
αSMA
HE
Tricrómica
de Masson
αSMA
Simulada LCB Simulada LCB
Simulada LCB Simulada LCB Simulada LCB Simulada LCB
ControlRC
ControlRC
Control
RC
Control
RC
Control
RC
Control
RC
ControlRC
ControlRC
ControlRC
ControlRC
ControlRC
ControlRC
Niv
ele
s de m
RN
AN
ivele
s de m
RN
A
Hidroxiprolina
Hidroxiprolina
Tricrómico de Masson
Tricrómico de Masson
Colágena 1α1 Colágena 1α2 αSMA TGFβ
Colágena 1α1 Colágena 1α2 αSMA TGFβ
Áre
a p
osi
tiva
(%
)Á
rea p
osi
tiva
(%
)
Hid
roxi
pro
lina h
epática
(μ
mol/
g h
ígado)
Hid
roxi
pro
lina h
epática
(μ
mol/
g h
ígado)
Niv
ele
s de m
RN
AN
ivele
s de m
RN
A
Niv
ele
s de m
RN
AN
ivele
s de m
RN
A
Niv
ele
s de m
RN
AN
ivele
s de m
RN
A
Aceite CCl4
Aceite CCl4
Aceite CCl4
Aceite CCl4
de maíz de maíz de maíz de maíz
Aceite CCl4
Aceite CCl4
de maíz de maíz
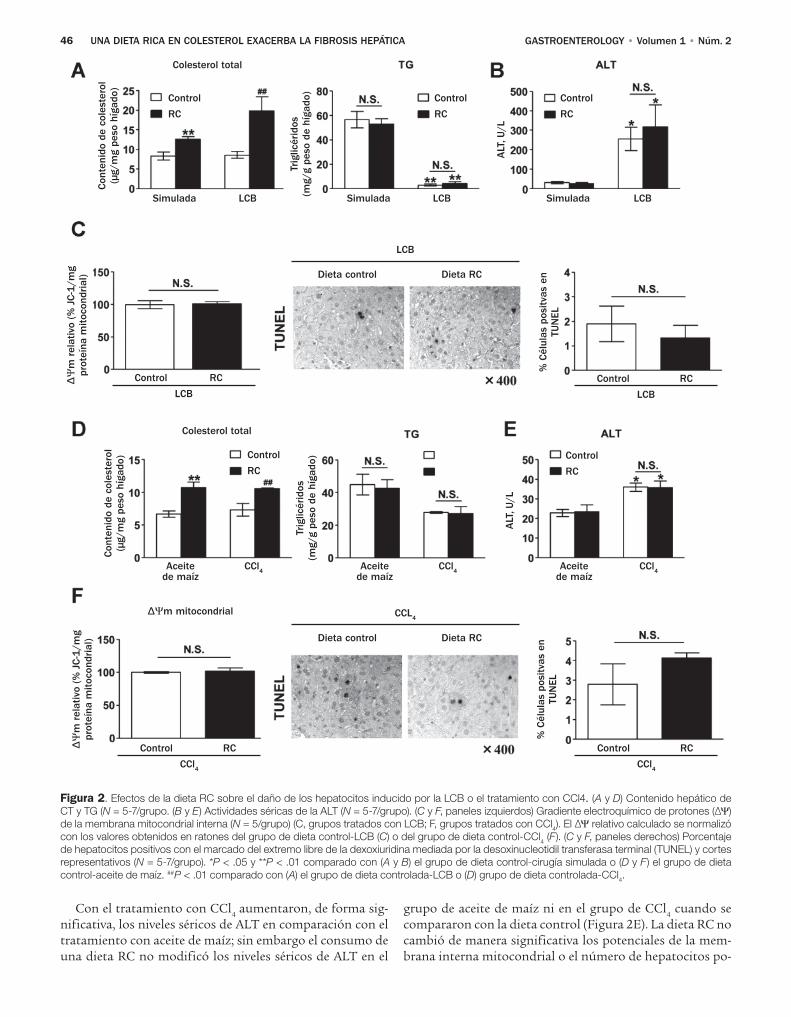
46 UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA GASTROENTEROLOGY • Volumen 1 • Núm. 2
Con el tratamiento con CCl4 aumentaron, de forma sig-nificativa, los niveles séricos de ALT en comparación con el tratamiento con aceite de maíz; sin embargo el consumo de una dieta RC no modificó los niveles séricos de ALT en el
grupo de aceite de maíz ni en el grupo de CCl4 cuando se compararon con la dieta control (Figura 2E). La dieta RC no cambió de manera significativa los potenciales de la mem-brana interna mitocondrial o el número de hepatocitos po-
Figura 2. Efectos de la dieta RC sobre el daño de los hepatocitos inducido por la LCB o el tratamiento con CCl4. (A y D) Contenido hepático de
CT y TG (N = 5-7/grupo. (B y E) Actividades séricas de la ALT (N = 5-7/grupo). (C y F, paneles izquierdos) Gradiente electroquímico de protones (ΔΨ)
de la membrana mitocondrial interna (N = 5/grupo) (C, grupos tratados con LCB; F, grupos tratados con CCl4). El ΔΨ relativo calculado se normalizó
con los valores obtenidos en ratones del grupo de dieta control-LCB (C) o del grupo de dieta control-CCl4 (F). (C y F, paneles derechos) Porcentaje
de hepatocitos positivos con el marcado del extremo libre de la dexoxiuridina mediada por la desoxinucleotidil transferasa terminal (TUNEL) y cortes
representativos (N = 5-7/grupo). *P < .05 y **P < .01 comparado con (A y B) el grupo de dieta control-cirugía simulada o (D y F) el grupo de dieta
control-aceite de maíz. ##P < .01 comparado con (A) el grupo de dieta controlada-LCB o (D) grupo de dieta controlada-CCl4.
Aceite CCl4
Aceite CCl4
Aceite CCl4
de maíz de maíz de maíz
Simulada LCB Simulada LCB Simulada LCB
Control RC
CCl4
Control RC
LCB
Control RC
LCB
Control RC
CCl4
Control
RC
Control
RC
Control
RC
Control
RC
Control
RC
% C
élu
las
posi
tvas
en
TU
NEL
% C
élu
las
posi
tvas
en
TU
NEL
ALT
, U
/LA
LT, U
/L
ΔΨm mitocondrial
Colesterol total
Colesterol totalΔ
Ψm
rela
tivo
(%
JC-1
/mg
pro
teín
a m
itoco
ndrial)
ΔΨ
m r
ela
tivo
(%
JC-1
/mg
pro
teín
a m
itoco
ndrial)
Conte
nid
o d
e c
ole
stero
l (μ
g/m
g p
eso
híg
ado)
Conte
nid
o d
e c
ole
stero
l (μ
g/m
g p
eso
híg
ado)
Trig
licé
ridos
(mg/g
peso
de h
ígado)
Trig
licé
ridos
(mg/g
peso
de h
ígado)
CCL4
Dieta control Dieta RC
LCB
Dieta control Dieta RC
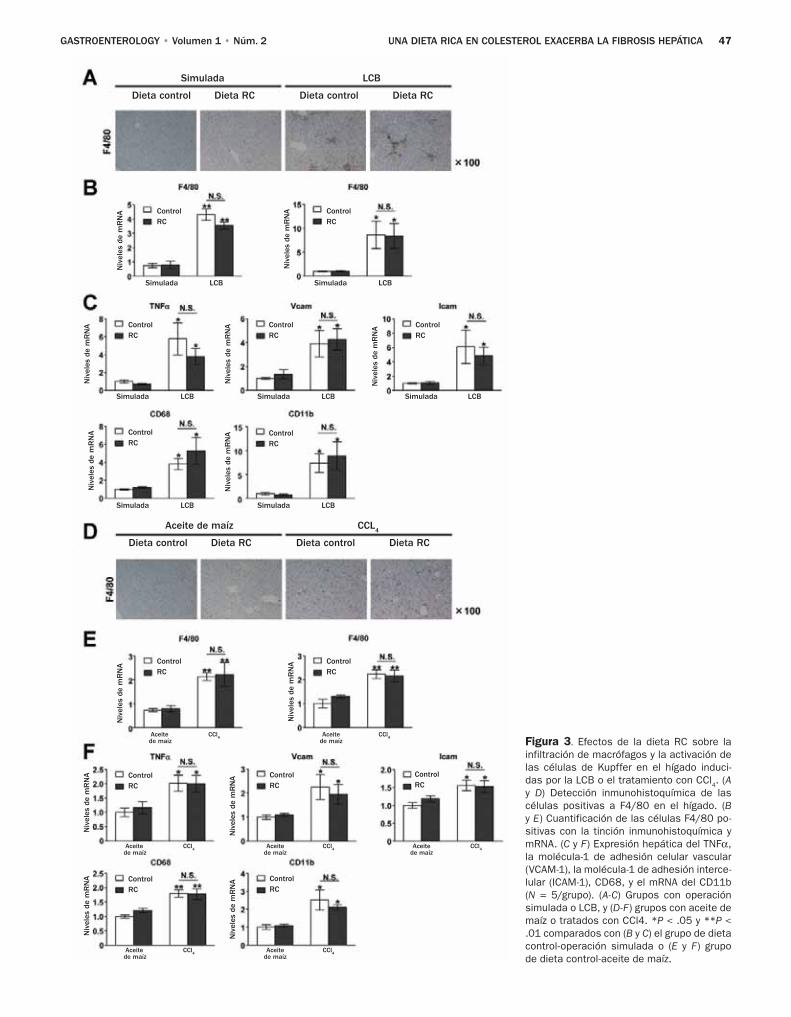
UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA 47GASTROENTEROLOGY • Volumen 1 • Núm. 2
Figura 3. Efectos de la dieta RC sobre la
infiltración de macrófagos y la activación de
las células de Kupffer en el hígado induci-
das por la LCB o el tratamiento con CCl4. (A
y D) Detección inmunohistoquímica de las
células positivas a F4/80 en el hígado. (B
y E) Cuantificación de las células F4/80 po-
sitivas con la tinción inmunohistoquímica y
mRNA. (C y F) Expresión hepática del TNFα,
la molécula-1 de adhesión celular vascular
(VCAM-1), la molécula-1 de adhesión interce-
lular (ICAM-1), CD68, y el mRNA del CD11b
(N = 5/grupo). (A-C) Grupos con operación
simulada o LCB, y (D-F) grupos con aceite de
maíz o tratados con CCl4. *P < .05 y **P <
.01 comparados con (B y C) el grupo de dieta
control-operación simulada o (E y F) grupo
de dieta control-aceite de maíz. Aceite CCl
4 Aceite CCl
4
de maíz de maíz
Aceite CCl4
Aceite CCl4
de maíz de maíz
Simulada LCB Simulada LCB
Simulada LCB Simulada LCB
Simulada LCB Simulada LCB Simulada LCB
Aceite CCl4
Aceite CCl4
Aceite CCl4
de maíz de maíz de maíz
Control
RC
Control
RC
Control
RC
Control
RC
Control
RC
Control
RC
Control
RC
Control
RC
Control
RC
Control
RC
Control
RC
Control
RC
Control
RC
Control
RC
Niv
ele
s de m
RN
AN
ivele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
AN
ivele
s de m
RN
AN
ivele
s de m
RN
AN
ivele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
AN
ivele
s de m
RN
A
Niv
ele
s de m
RN
A
Aceite de maíz CCL4
Dieta control Dieta RC
Dieta control Dieta RC
Simulada LCB
Dieta control Dieta RC
Dieta control Dieta RC

48 UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA GASTROENTEROLOGY • Volumen 1 • Núm. 2
sitivos con el marcado del extremo libre de la desoxiuridina mediada por la desoxinucleotidil transferasa terminal en los hígados de ratones tratados con CCl4 (Figura 2F).
Además, la dieta RC no modificó el daño hepatocelular agudo inducido por la LCB o el CCl4, inclusive cuando el daño hepático estuvo en su máximo (Figuras 1A y B y 2A y B Suplementarias).
Estos resultados muestran que el aumento de los niveles he-páticos de colesterol inducidos por el consumo de una dieta RC no agravan el daño hepatocelular inducido por LCB o CCl4.
La dieta RC no impactó sobre la activación de las células de Kupffer inducida por LCB o por CCl4, ni sobre la inflamación hepáticaLa infiltración hepática de macrófagos se valoró mediante tinción inmunohistoquímica con el marcador de células de Kupffer/macrófagos con anticuerpos F4/80. Los resultados muestran que aumenta el infiltrado de macrófagos por la LCB en el hígado tanto en los ratones controles como en los alimentados con dieta RC. Sin embargo, el consumo de una dieta RC no modificó el infiltrado (Figura 3A y B). Las células de Kupffer son la mayor fuente de producción del factor-α de necrosis tumoral (TNFα) en el hígado. Aunque la LCB aumentó, de forma significativa, la expresión del mRNA del TNFα en el hígado, la dieta RC no aceleró la expresión genética del TNFα. Además, aunque la LCB au-mentó de manera significativa la expresión del mRNA de la molécula-1 de adhesión celular vascular, la molécula-1 de adhesión intercelular, CD68 y CD11b, este aumento no tuvo impacto por el consumo de la dieta RC (Figura 3C).
Los ratones tratados con CCl4 mostraron aumento del infil-trado de macrófagos en el hígado, similar a aquellos tratados con LCB. Sin embargo el consumo de la dieta RC no tuvo un impacto en el infiltrado hepática de macrófagos mediada por CCl4 (Figura 3D y E). El tratamiento con CCl4 aumentó los ni-veles hepáticos de TNFα, de la molécula-1 de adhesión celular vascular, la molécula-1 de adhesión intracelular y el mRNA de CD68 y CD11b; sin embargo estos niveles no tuvieron influen-cia alguna por el consumo de la dieta RC (Figura 3F). Además, la dieta RC no alteró la inflamación hepática aguda inducida por LCB o CCl4, incluso cuando la inflamación hepática estaba en su máximo (Figuras 1C-E y 2C-E Suplementarias).
Estos resultados muestran que la dieta RC utilizada en este estudio no cambió la activación de las células de Kup-ffer o el reclutamiento de macrófagos al hígado. Además, la dieta RC no impactó sobre la infiltración de células infla-matorias mediada por la LCB o el CCL4 como las células T y los neutrófilos en el hígado (Figura 3 Suplementaria). Con la tinción de H&E y la tinción inmunohistoquímica para el CD68 se mostró que la dieta RC no induce formación de células espumosas macrofágicas hepáticas ni causa infla-mación hepática (Figura 1A y D y Figura 4 Suplementaria).
El agravamiento de la fibrosis hepática inducido por la dieta RC fue independiente de las células de KupfferPara determinar si el avance de la fibrosis hepática resul-tante por el consumo de la dieta RC requería la presencia
de células de Kupffer, ratones depletados de células de Ku-pffer con clodronato liposomal fueron tratados con LCB o intoxicación con CCl4. En el modelo de LCB el clodronato li-posomal alcanzó una depleción casi completa de las células de Kupffer (Figura 4A), junto con la supresión de citocinas proinflamatorias como el TNFα y la interleucina-1β (Figura 5A Suplementaria). El tratamiento con clodronato liposo-mal no impactó sobre el daño hepatocelular inducido por LCB (Figura 4A, fila inferior). En los ratones tratados con clodronato liposomal el consumo de la dieta RC favoreció de forma significativa el agravamiento de la fibrosis hepática inducido por LCB (Figura 4B). Acorde con estos resultados, los ratones tratados con clodronato liposomal mostraron un aumento significativo de la expresión de colágena 1α1, colágena 1α2, y αSMA inducido por la LCB en el hígado cuando se alimentaron con una dieta RC (Figura 4C).
De forma similar, el tratamiento con clodronato liposo-mal depletó casi por completo las células de Kupffer en el modelo de CCl4 (Figura 4D), y suprimió también las cito-cinas proinflamatorias como el TNFα y la interleucina-1β (Figura 5B Suplementaria). La administración de clodrona-to liposomal no modificó el daño hepatocelular inducido por CCl4 (Figura 4D, fila inferior). En los ratones en que se infundió clodronato liposomal la dieta RC reforzó la progresión de la fibrosis hepática inducida por CCl4 (Figu-ra 4E). Acorde con estos hallazgos lo ratones tratados con clodronato liposomal mostraron un aumento significativo de la expresión del RNA de la colágena 1α1, la colágena 1α2 y la αSMa inducido por CCl4 en el hígado cuando se admi-nistró la dieta RC (Figura 4F).
Estos resultados sugirieron que la dieta RC favoreció la fibrosis hepática inducida por LCB y CCl4 en una forma in-dependiente de las células de Kupffer.
La acumulación de colesterol libre sensibilizó a las CEH para la activación inducida por TGFβPara examinar los efectos de la dieta RC sobre las CEH, se aislaron de células de ratones que recibieron la dieta control o la dieta RC. Con la LCB en el grupo de dieta control, la media (± DE) del contenido de CT fue de 28.94 ± 11.55 μg/mg de proteína celular. En las CEH del grupo de dieta RC, la media (± DE) del contenido de CT aumentó de manera significativa a 59.90 ± 22.93 μg/proteína celular. Además, se determinó los niveles de colesterol libre (CL) y de ésteres decolesterol (EC) en las CEH. En consecuencia los niveles de CL fueron mayores en las CEH del gupo de dieta RC que en las del grupo control; sin embargo no hubo diferencia en el nivel de EC entre los grupos (Figura 5A).
Segundo, para investigar los efectos de la dieta RC en la activación de las CEH, éstas se aislaron de ratones de ambos grupos y se estimularon con la citocina profibrogénica TGFβ. Muestras de CEH antes del tratamiento con TGFβ, obteni-das de los ratones, mostraron que la dieta RC no modificó los niveles de mRNA de la colágena 1α1, la colágena 1α2 o la αSMA. El tratamiento con TGFβ reforzó de forma signi-ficativa los niveles de transcriptos de mRNA de la colágena 1α1, 1α2, y αSMA en las CEH. El efecto incremental se obser-
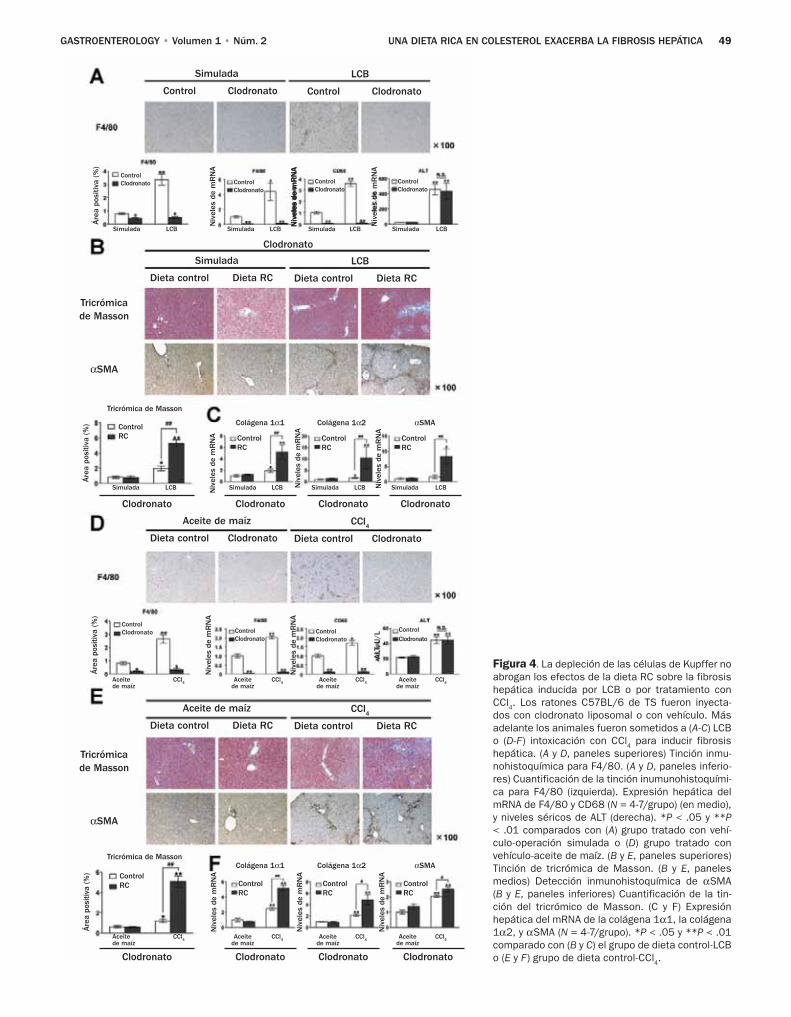
UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA 49GASTROENTEROLOGY • Volumen 1 • Núm. 2
Figura 4. La depleción de las células de Kupffer no
abrogan los efectos de la dieta RC sobre la fibrosis
hepática inducida por LCB o por tratamiento con
CCl4. Los ratones C57BL/6 de TS fueron inyecta-
dos con clodronato liposomal o con vehículo. Más
adelante los animales fueron sometidos a (A-C) LCB
o (D-F) intoxicación con CCl4 para inducir fibrosis
hepática. (A y D, paneles superiores) Tinción inmu-
nohistoquímica para F4/80. (A y D, paneles inferio-
res) Cuantificación de la tinción inumunohistoquími-
ca para F4/80 (izquierda). Expresión hepática del
mRNA de F4/80 y CD68 (N = 4-7/grupo) (en medio),
y niveles séricos de ALT (derecha). *P < .05 y **P
< .01 comparados con (A) grupo tratado con vehí-
culo-operación simulada o (D) grupo tratado con
vehículo-aceite de maíz. (B y E, paneles superiores)
Tinción de tricrómica de Masson. (B y E, paneles
medios) Detección inmunohistoquímica de αSMA
(B y E, paneles inferiores) Cuantificación de la tin-
ción del tricrómico de Masson. (C y F) Expresión
hepática del mRNA de la colágena 1α1, la colágena
1α2, y αSMA (N = 4-7/grupo). *P < .05 y **P < .01
comparado con (B y C) el grupo de dieta control-LCB
o (E y F) grupo de dieta control-CCl4.
Aceite de maíz
Dieta control Dieta RC
Aceite de maíz
Dieta control Clodronato
Simulada
Dieta control Dieta RC
Simulada
Control Clodronato
Tricrómica
de Masson
αSMA
Tricrómica
de Masson
αSMA
CCl4
Dieta control Dieta RC
CCl4
Dieta control Clodronato
LCB
Dieta control Dieta RC
LCB
Control Clodronato
Aceite CCl4
Aceite CCl4
Aceite CCl4
Aceite CCl4
de maíz de maíz de maíz de maíz
Simulada LCB
Simulada LCB
Simulada LCB
Simulada LCB
Simulada LCB
Simulada LCB
Simulada LCB
Simulada LCB
Aceite CCl4
Aceite CCl4
Aceite CCl4
Aceite CCl4
de maíz de maíz de maíz de maíz
ControlRC
ControlRC Control
RCControlRC
ControlRC
Colágena 1α2
Colágena 1α2
αSMA
αSMA
Colágena 1α1
Colágena 1α1
Tricrómica de Masson
Tricrómica de Masson
Niv
ele
s de m
RN
AN
ivele
s de m
RN
AN
ivele
s de m
RN
AN
ivele
s de m
RN
A
Niv
ele
s de m
RN
AN
ivele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
AN
ivele
s de m
RN
A
ALT
, U
/LN
ivele
s de m
RN
A
Áre
a p
osi
tiva
(%
)Á
rea p
osi
tiva
(%
)Á
rea p
osi
tiva
(%
)
Áre
a p
osi
tiva
(%
)
ControlRC
ControlRC
ControlRC
Control
ClodronatoControl
Clodronato
Control
Clodronato
Control
Clodronato Control
Clodronato
Control
Clodronato
Control
Clodronato
Control
Clodronato
Clodronato
Clodronato
Clodronato
Clodronato
Clodronato
Clodronato
Clodronato
Clodronato
Clodronato
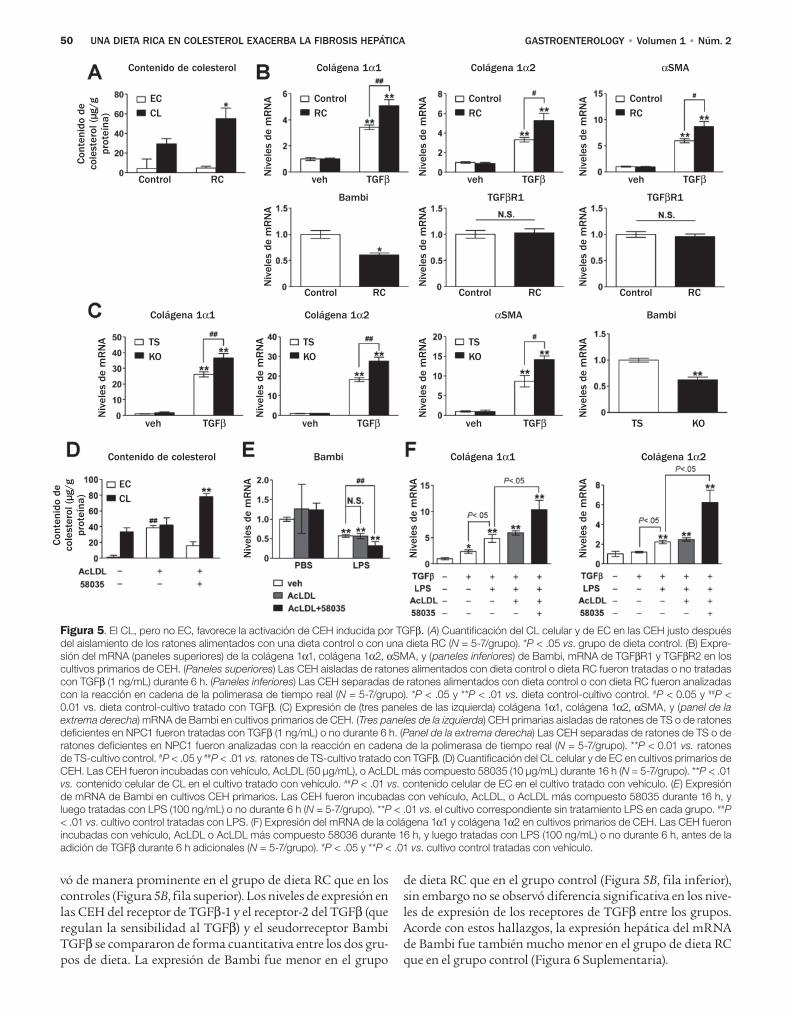
50 UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA GASTROENTEROLOGY • Volumen 1 • Núm. 2
vó de manera prominente en el grupo de dieta RC que en los controles (Figura 5B, fila superior). Los niveles de expresión en las CEH del receptor de TGFβ-1 y el receptor-2 del TGFβ (que regulan la sensibilidad al TGFβ) y el seudorreceptor Bambi TGFβ se compararon de forma cuantitativa entre los dos gru-pos de dieta. La expresión de Bambi fue menor en el grupo
de dieta RC que en el grupo control (Figura 5B, fila inferior), sin embargo no se observó diferencia significativa en los nive-les de expresión de los receptores de TGFβ entre los grupos. Acorde con estos hallazgos, la expresión hepática del mRNA de Bambi fue también mucho menor en el grupo de dieta RC que en el grupo control (Figura 6 Suplementaria).
Figura 5. El CL, pero no EC, favorece la activación de CEH inducida por TGFβ. (A) Cuantificación del CL celular y de EC en las CEH justo después
del aislamiento de los ratones alimentados con una dieta control o con una dieta RC (N = 5-7/grupo). *P < .05 vs. grupo de dieta control. (B) Expre-
sión del mRNA (paneles superiores) de la colágena 1α1, colágena 1α2, αSMA, y (paneles inferiores) de Bambi, mRNA de TGFβR1 y TGFβR2 en los
cultivos primarios de CEH. (Paneles superiores) Las CEH aisladas de ratones alimentados con dieta control o dieta RC fueron tratadas o no tratadas
con TGFβ (1 ng/mL) durante 6 h. (Paneles inferiores) Las CEH separadas de ratones alimentados con dieta control o con dieta RC fueron analizadas
con la reacción en cadena de la polimerasa de tiempo real (N = 5-7/grupo). *P < .05 y **P < .01 vs. dieta control-cultivo control. #P < 0.05 y ##P <
0.01 vs. dieta control-cultivo tratado con TGFβ. (C) Expresión de (tres paneles de las izquierda) colágena 1α1, colágena 1α2, αSMA, y (panel de la
extrema derecha) mRNA de Bambi en cultivos primarios de CEH. (Tres paneles de la izquierda) CEH primarias aisladas de ratones de TS o de ratones
deficientes en NPC1 fueron tratadas con TGFβ (1 ng/mL) o no durante 6 h. (Panel de la extrema derecha) Las CEH separadas de ratones de TS o de
ratones deficientes en NPC1 fueron analizadas con la reacción en cadena de la polimerasa de tiempo real (N = 5-7/grupo). **P < 0.01 vs. ratones
de TS-cultivo control. #P < .05 y ##P < .01 vs. ratones de TS-cultivo tratado con TGFβ. (D) Cuantificación del CL celular y de EC en cultivos primarios de
CEH. Las CEH fueron incubadas con vehículo, AcLDL (50 μg/mL), o AcLDL más compuesto 58035 (10 μg/mL) durante 16 h (N = 5-7/grupo). **P < .01
vs. contenido celular de CL en el cultivo tratado con vehículo. ##P < .01 vs. contenido celular de EC en el cultivo tratado con vehículo. (E) Expresión
de mRNA de Bambi en cultivos CEH primarios. Las CEH fueron incubadas con vehículo, AcLDL, o AcLDL más compuesto 58035 durante 16 h, y
luego tratadas con LPS (100 ng/mL) o no durante 6 h (N = 5-7/grupo). **P < .01 vs. el cultivo correspondiente sin tratamiento LPS en cada grupo. ##P
< .01 vs. cultivo control tratadas con LPS. (F) Expresión del mRNA de la colágena 1α1 y colágena 1α2 en cultivos primarios de CEH. Las CEH fueron
incubadas con vehículo, AcLDL o AcLDL más compuesto 58036 durante 16 h, y luego tratadas con LPS (100 ng/mL) o no durante 6 h, antes de la
adición de TGFβ durante 6 h adicionales (N = 5-7/grupo). *P < .05 y **P < .01 vs. cultivo control tratadas con vehículo.
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Conte
nid
o d
e
cole
stero
l (μ
g/g
pro
teín
a)
Conte
nid
o d
e
cole
stero
l (μ
g/g
pro
teín
a)
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Control
RC
Control
RC
Control
RC
EC
CL
EC
CL
TS
KO
TS
KO
TS
KO
Control RC veh TGFβ veh TGFβ veh TGFβ
veh TGFβ veh TGFβ veh TGFβ TS KO
Control RC Control RC Control RC
Contenido de colesterol Colágena 1α1 Colágena 1α2 αSMA
Contenido de colesterol Bambi Colágena 1α1 Colágena 1α2
Bambi TGFβR1 TGFβR1
Colágena 1α1 Colágena 1α2 αSMA Bambi
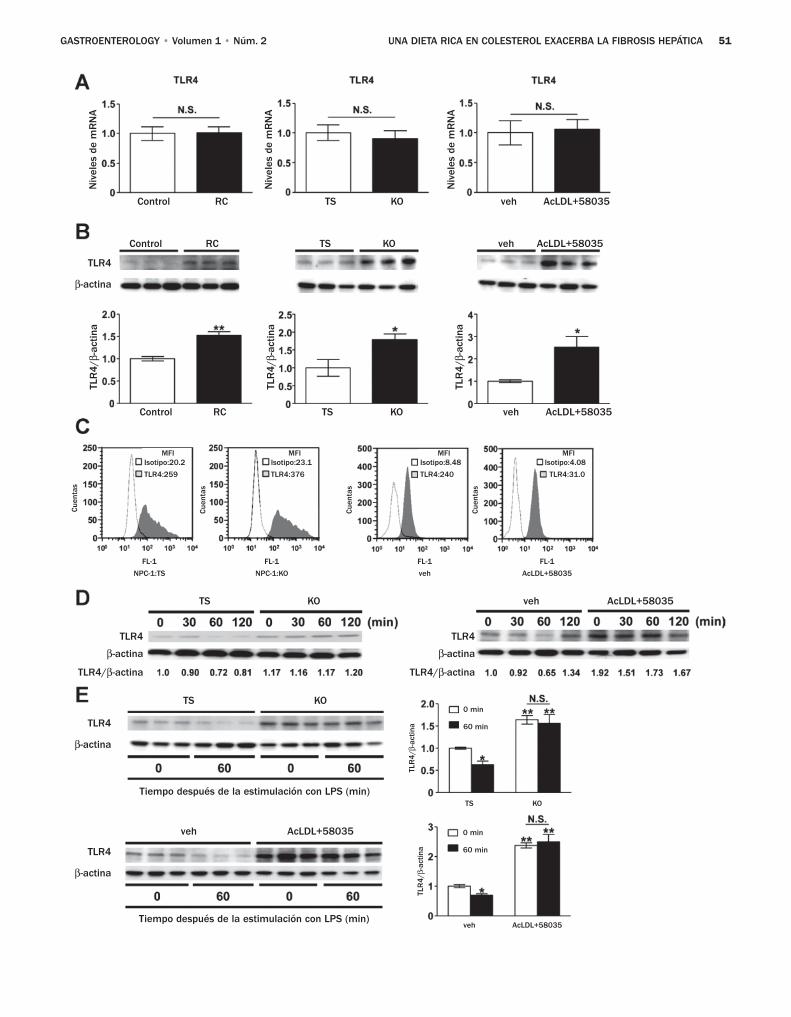
UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA 51GASTROENTEROLOGY • Volumen 1 • Núm. 2
Tiempo después de la estimulación con LPS (min)
TLR
4/β
-act
ina
TLR
4/β
-act
ina
Cuenta
s
Niv
ele
s de m
RN
ATLR
4/β
-act
ina
TLR
4/β
-act
ina
TLR
4/β
-act
ina
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Cuenta
s
Cuenta
s
Cuenta
s
0 min
60 min
0 min
60 min
MFIIsotipo:20.2
TLR4:259
FL-1 FL-1 FL-1 FL-1
NPC-1:TS NPC-1:KO veh AcLDL+58035
Control RC TS KO veh AcLDL+58035
Control RC TS KO veh AcLDL+58035
Control RC TS KO veh AcLDL+58035
MFIIsotipo:23.1
TLR4:376
MFIIsotipo:8.48
TLR4:240
MFIIsotipo:4.08
TLR4:31.0
TLR4
β-actina
TLR4
β-actina
TLR4/β-actina
TLR4
β-actina
TLR4
β-actina
TLR4/β-actina
TLR4
β-actina
veh AcLDL+58035
TS KO
Tiempo después de la estimulación con LPS (min)
veh AcLDL+58035
TS KO
TS KO veh AcLDL+58035

52 UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA GASTROENTEROLOGY • Volumen 1 • Núm. 2
Tercero, se valoró el efecto del CL sobre la sensibilidad de las CEH al TGFβ. La proteína Niemann-Pick C1 (NPC1) es una proteína endosomal tardía que regula el transporte in-tracelular de colesterol. Las células homocigotas deficientes de NPC1 muestran una acumulación de CL intracelular.9,10 Por lo tanto, las CEH aisladas de ratones noqueados (KO) de NPC1 se utilizaron para este análisis. Antes del tratamiento con TGFβ no se encontraron diferencias significativas en-tre las CEH de TS y de NPC1 KO en los niveles de expresión de la colágena 1α1, colágena 1α2 o αSMA en los transcrip-tos de RNA. El tratamiento con TGFβ aumentó los niveles de colágena 1α1, colágena 1α2 y αSMA en los transcriptos demRNA. Este efecto positivo se observó de forma marcada en las CEH de NPC1 KO que en las CEH de TS (Figura 5C, tres páneles de la izquierda). Los niveles del mRNA de Bambi fue-ron menores en las CEH de NPCI KO en comparación con las CEH de TS (Figura 5C, panel de la derecha). Se reporta que el colesterol se acumula de manera predominante en los endo-somas/lisosomas tardíos de las células en los ratones NPC1 KO.9 Nuestro estudio encontró que los niveles de CL en los endosomas/lisosomas tardíos fueron mayores en las CEH de NPC1 KO que en las CEH de TS (Figura 7A Suplementaria). De forma similar, los niveles de CL en los endosomas/lisoso-mas tardíos fueron mayores en las CEH del grupo de dieta RC que en el grupo de dieta control (Figura 7B Suplementaria).
Se encontró que el CL se acumula en las células trata-das con la combinación de acetil lipoproteínas de baja den-sidad (AcLDL) y el inhibidor de la acil-CoA:colesterol acil transferasa 58035 (compuesto 58035), mientras que el CL se acumula en las células tratadas sólo con AcLDL.11 En nuestro estudio el tratamiento de las CEH con AcLDL más el compuesto 58035 aumentó la acumulación de CL, y el tratamiento con AcLDL solas favoreció en gran medida la acumulación de CL (Figura 5D). Sin embargo los niveles de expresión de Bambi no disminuyeron cuando las CEH fueron tratadas con AcLDL solas o con la combinación de AcLDL y el compuesto 58035 (Figura 5E). Se reporta que la expresión del gen Bambi en las CEH depende de las se-ñales de TLR4 y disminuye con la adición de lipopolisacá-ridos (LPS).12 En nuestro estudio los niveles de expresión del mRNA de Bambi disminuyeron cuando las CEH fue-ron tratadas con LPS (Figura 5E). La disminución de los niveles de mRNA de Bambi se reforzó significativamente cuando las células se trataron con AcLDL más el compuesto 58035, mientras que las células tratadas con AcLDL solas no mostraron una reducción significativa en comparación con los controles (Figura 5E). La acumulación de CL en las
CEH intensificó también la traducción de la señal de TLR-4 mediada por LPS para inducir citocinas proinflamatorias como la proteína-1 de quimioatracción de monocitos y la proteína-2 inflamatoria de macrófagos (Figuras 8A y B Su-plementarias), que se sabe que son reguladas a la alta por la vía de la señal de TLR4 mediada por LPS en las CEH.12,13 Las CEH pretratadas con LPS mostraron un reforzamiento significativo de la expresión del mRNA de la colágena 1α1 y 1α2 cuando se estimularon con TGFβ (Figura 5F). Estas células mostraron otro aumento de la expresión del mRNA de estos genes cuando se cargaron con AcLDL más el com-puesto 58035, mientras que no se observó un incremento significativo cuando las células fueron incubadas sólo con AcLDL (Figura 5F). Estos resultados indican claramente que la acumulación de CL en las CEH las sensibilizó ante las señales inducidas por TGFβ mediante regulación a la baja de la expresión del gen de Bambi.
A diferencia de las CEH, no se observaron diferencias significativas en los niveles de CL en las células de Kupffer entre el grupo de dieta RC y el grupo de dieta control; no obstante los niveles de EC fueron mayores en las células de Kupffer del grupo de dieta RC que en las del grupo de die-ta control (Figura 9A Suplementaria). Una acumulación de EC no aceleró la expresión del mRNA del TNFα en las célu-las de Kupffer (Figura 9B Suplementaria), ni reforzó la ex-presión del mRNA del TNFα inducida por LPS en estas cé-lulas (Figura 9B Suplementaria). Estos resultados muestran que la dieta RC no hace que las células de Kupffer inicien la fibrosis hepática, aunque los niveles de EC aumentaron en las células de Kupffer.
La acumulación de CL en las CEH reguló a la alta la expresión de TLR4El consumo de la dieta RC no alteró los niveles de expresión del mRNA del TLR4 en las CEH. Sin embargo la dieta RC aumentó la cantidad de proteína de TLR4 de las CEH (Fi-gura 6A y 6B). Además, los mayores niveles que se expresan del gen de TLR4 en cuanto a la cantidad de proteína (pero no de mRNA) se observó también en las CEH de NPC1 KO y en las CEH estimuladas con AcLDL más el compuesto 58035 (Figura 6A y 6B). Por otra parte, las CEH NPC1 KO mostraron una expresión de proteína TLR4 de membrana mayor en comparación con las CEH del TS (Figura 6C). Se obtuvieron resultados similares con las CEH tratadas con AcLDL más el compuesto 58035 (Figura 6C).
En condiciones normales las proteínas de membrana se internan en el citoplasma por endocitosis, en donde se degra-
Figura 6. El CL aumenta la expresión de la proteína TLR4 en las CEH. TLR4 (A) mRNA y (B) expresión de la proteína (CEH aisladas de ratones alimen-
tados con dieta control o ratones alimentados con RC [izquierda], TS o CEH deficientes de NPC1 [en medio], CEH tratadas con vehículo o cargadas
con CL [derecha]). (B, paneles inferiores) Cuantificación de la expresión de la proteína TLR4. **P < .01 vs. grupo de dieta control (izquierda); *P < .05
vs. CEH TS (en medio); y *P < .05 vs. CEH tratadas con vehículo (derecha). (C) Ensayo con el separador de células activado con fluorescencia de la
expresión de TLR4 en las membranas plasmáticas de las CEH de TS o deficientes en NPC1 (izquierda) y CEH tratadas con vehículo o cargadas con
CL (derecha). La media de la intensidad de la fluorescencia (MFI) se muestra también en la esquina superior derecha de cada panel. (D) Cambios
dinámicos y (E) cuantificación de la expresión de la proteína TLR4 en las CEH de ratones TS o deficientes en NPC1 (D, paneles de la izquierda; E, pa-
neles superiores), y CEH tratadas con vehículo o cargadas con CL (D, paneles de la derecha; E, paneles inferiores) después de tratamiento con LPS
(100 ng/mL). Los niveles relativos de TLR4 con la β-actina están indicados debajo de las bandas correspondientes. *P < .05 y **P < .01 vs. CEH de
TS o CEH tratadas con vehículo antes del tratamiento con LPS. Se utilizaron CEH de ratones o ratas cultivadas 6 días después del aislamiento (C-E).
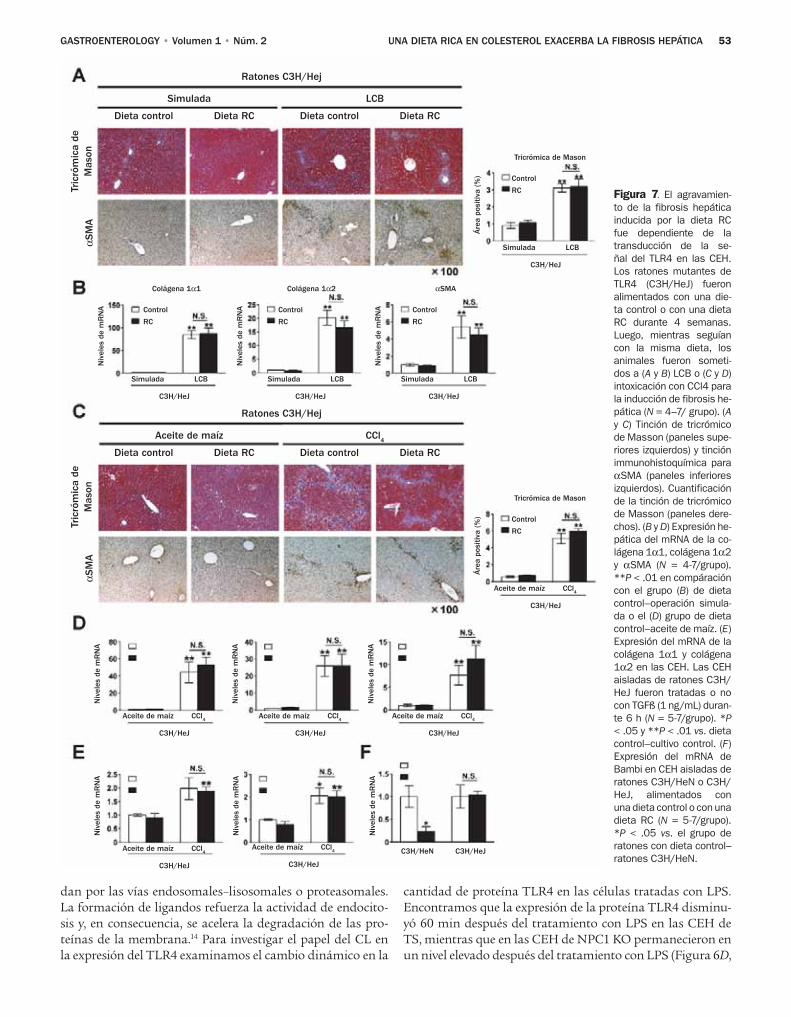
UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA 53GASTROENTEROLOGY • Volumen 1 • Núm. 2
dan por las vías endosomales–lisosomales o proteasomales. La formación de ligandos refuerza la actividad de endocito-sis y, en consecuencia, se acelera la degradación de las pro-teínas de la membrana.14 Para investigar el papel del CL en la expresión del TLR4 examinamos el cambio dinámico en la
cantidad de proteína TLR4 en las células tratadas con LPS. Encontramos que la expresión de la proteína TLR4 disminu-yó 60 min después del tratamiento con LPS en las CEH de TS, mientras que en las CEH de NPC1 KO permanecieron en un nivel elevado después del tratamiento con LPS (Figura 6D,
Figura 7. El agravamien-
to de la fibrosis hepática
inducida por la dieta RC
fue dependiente de la
transducción de la se-
ñal del TLR4 en las CEH.
Los ratones mutantes de
TLR4 (C3H/HeJ) fueron
alimentados con una die-
ta control o con una dieta
RC durante 4 semanas.
Luego, mientras seguían
con la misma dieta, los
animales fueron someti-
dos a (A y B) LCB o (C y D)
intoxicación con CCl4 para
la inducción de fibrosis he-
pática (N = 4–7/ grupo). (A
y C) Tinción de tricrómico
de Masson (paneles supe-
riores izquierdos) y tinción
immunohistoquímica para
αSMA (paneles inferiores
izquierdos). Cuantificación
de la tinción de tricrómico
de Masson (paneles dere-
chos). (B y D) Expresión he-
pática del mRNA de la co-
lágena 1α1, colágena 1α2
y αSMA (N = 4-7/grupo).
**P < .01 en compáración
con el grupo (B) de dieta
control–operación simula-
da o el (D) grupo de dieta
control–aceite de maíz. (E)
Expresión del mRNA de la
colágena 1α1 y colágena
1α2 en las CEH. Las CEH
aisladas de ratones C3H/
HeJ fueron tratadas o no
con TGFß (1 ng/mL) duran-
te 6 h (N = 5-7/grupo). *P
< .05 y **P < .01 vs. dieta
control–cultivo control. (F)
Expresión del mRNA de
Bambi en CEH aisladas de
ratones C3H/HeN o C3H/
HeJ, alimentados con
una dieta control o con una
dieta RC (N = 5-7/grupo).
*P < .05 vs. el grupo de
ratones con dieta control–
ratones C3H/HeN.
Aceite de maíz
Dieta control Dieta RC
Simulada
Dieta control Dieta RC
Ratones C3H/Hej
Ratones C3H/Hej
Simulada LCB
C3H/HeJ
Colágena 1α1
Control
RC
Niv
ele
s de m
RN
AN
ivele
s de m
RN
AN
ivele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Áre
a p
osi
tiva
(%
)Á
rea p
osi
tiva
(%
)
Colágena 1α2
Control
RC
Tricrómica de Mason
Control
RC
Tricrómica de Mason
Control
RC
Tric
róm
ica d
e
Maso
n
Tric
róm
ica d
e
Maso
nαS
MA
αSM
A
αSMA
Control
RC
Simulada LCB
C3H/HeJ
Simulada LCB
C3H/HeJ
Simulada LCB
C3H/HeJ
Aceite de maíz CCl4
C3H/HeJ
Aceite de maíz CCl4
C3H/HeJ
C3H/HeN C3H/HeJ
Aceite de maíz CCl4
C3H/HeJ
Aceite de maíz CCl4
C3H/HeJ
Aceite de maíz CCl4
C3H/HeJ
Aceite de maíz CCl4
C3H/HeJ
CCl4
Dieta control Dieta RC
LCB
Dieta control Dieta RC

54 UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA GASTROENTEROLOGY • Volumen 1 • Núm. 2
fila de la izquierda, y E, fila superior). Se obtuvieron resultados similares con las CEH tratadas con AcLDL más el compuesto 58035 (Figura 6D, fila derecha, y E, fila inferior). Estos resultados muestran claramente que la acumulación de CL en las CEH aumentó el contenido de la proteína TLR4. Conjeturamos que la acumulación de CL intercelular suprimió la degrada-ción de TLR4 mediada por ligandos.
El agravamiento de la fibrosis hepática inducido por la dieta RC fue dependiente de la transducción de la señal de TLR4 en las CEHEn la última parte del experimento utilizamos ratones C3H/HeJ que no responden a LPS (TLR4 mutantes) para valorar si el agravamiento de la fibrosis hepática inducido por la dieta RC depende de la transducción de la señal de TLR4. A diferencia delos resultados obtenidos en los ratones de TS, el consumo de una dieta RC no indujo la progresión de la fibrosis hepática inducida por la LCB en los ratones C3H/HeJ (Figura 7A y B). En forma similar, la dieta RC no aceleró la progresión de la fi-brosis hepática inducida por CCl4 (Figura 7C y D).
Enseguida examinamos si la activación de las CEH por el CL acumulado requiere la señal de TLR4 en las CEH. Se obtuvieron CEH de ratones C3H/HeJ que recibieron la die-ta control o la dieta RC durante cuatro semanas. El trata-miento con TGFβ aumentó los niveles de transcriptos del mRNA de la colágena 1α1, la colágena 1α2 y la αSMA en las CEH. Sin embargo, a diferencia de los resultados ob-tenidos con las CEH del ratón de TS, no se encontraron diferencias entre los grupos que recibieron la dieta RC o la dieta control (Figura 7E). Por otra parte, aunque el consu-mo de la dieta RC atenuó la expresión del gen de Bambi en las CEH aisladas de ratones C3H/HeN (TS control de los ratones C3H/HeJ), el nivel del mRNA de Bambi en las CEH no cambió en los ratones C3H/HeJ (Figura 7F).
Estos hallazgos muestran que el agravamiento de la fibro-sis hepática inducido por la dieta RC fue dependiente de la transducción de la señal de TLR4. Nuestro estudio sugiere que el consumo de una dieta RC atenúa la expresión de Bam-bi en las CEH a través de la señal de TLR4, lo que lleva al pro-greso de la fibrosis hepática inducida por la LCB o el CCL4.
DiscusiónNuestros resultados muestran que una dieta RC agrava la fibrosis hepática inducida por LCB o CCL4, aunque la dieta RC por si sola no es suficiente para inducir fibrosis hepática. El TGFβ, que es el factor predisponente más potente de la fibrogénesis humana, parece desempeñar un papel central en la fisiopatología de la fibrosis hepática.1 Por otra parte, estos resultados muestran que otras causas mayores de exa-cerbación de la fibrosis hepática implicaron la acumulación de colesterol en la forma de CL en las CEH, que sensibilizó a las CEH a la activación inducida por el TGFβ.
Recientes investigaciones muestran que la acumulación de CL intracelular aumenta los niveles de la proteína TLR4 en la membrana para facilitar la activación de la señal del TLR4.15 Nuestros resultados muestran que la acumulación de CL en las CEH aumenta los niveles de la proteína TLR4 unida a la cito-
membrana; no obstante, la cantidad de transcriptos de mRNA del TLR4 fue similar. En condiciones normales las moléculas de la proteína TLR4 de la citomembrana son transferidas al cito-plasma por endocitosis, y degradadas a través de la vía endoso-mal-lisosomal o proteasomal. La degradación de las proteínas TLR4 se acelera cuando la internalización de estas moléculas es favorecida por la formación de ligandos. La inhibición de las vías de la degradación intensifica la transducción de la señal del TLR4 mediada por ligandos.14 En nuestro estudio el nivel de la proteína TLR4 en las CEH disminuyó de manera signi-ficativa en las células incubadas con LPS, el ligando mayor del TLR4. Además, la disminución del nivel de la proteína TLR4 en las CEH inducida por LPS se inhibió prominentemente por el CL acumulado en las CEH. Estos resultados sugieren que el CL acumulado en las CEH inhibe la vía de degradación del TLR4, lo que aumenta los niveles de la proteína TLR4.
En el presente estudio los niveles de CL en los endosomas/lisosomas tardíos fueron mucho mayores en las CEH del gru-po de dieta RC o en los ratones NPC1 KO que en el grupo control o en los ratones de TS. Estudios recientes reportan que el CL modula la vía de endocitosis endosomal-lisosomal a través de la regulación de la motilidad del endosoma.16 Los ratones NPC1 KO, que acumulan CL intracelular predomi-nantemente en los endosomas/lisosomas en forma tardía, re-tuvieron proteínas precursoras de amiloide como resultado de la disfunción endosomal.9 Los ratones NPC1 KO tuvieron también disfunción autofágica-lisosomal en el cerebro.10 Es-tos hallazgos sugieren que la acumulación de CL en las CEH está involucrada en la disfunción endosomal-lisosomal, lo que lleva a acumulación de la proteína TLR4.
La expresión del seudorreceptor del TGFβ Bambi en las CEH fue dependiente de las señales de TLR4.12 Se reporta que la activación de las señales de TLR4 en las CEH, que regulan en forma negativa la expresión del gen de Bambi, sensibilizan a las CEH a la activación inducida por el TGFβ, lo que contribuye a la progresión de la fibrosis hepática.12 En nuestro estudio las CEH disminuyeron la expresión delgen de Bambi cuando se incubaron con el ligando LPS del TLR4. La acumulación de CL (no de EC) en las CEH fa-voreció la regulación negativa de Bambi mediada por LPS y aceleró marcadamente (mediada por LPS) el reforzamiento de la sensibilidad de las CEH contra las señales del TGFβ. Nosotros sostenemos que estos cambios activan a las CEH, favoreciendo así la fibrosis hepática.
En nuestro experimento los ratones C3H/HeJ mutantes de TLR4 que recibieron la dieta RC no mostraron mayor fibrosis hepática. La acumulación de CL en las CEH de los ratones mu-tantes de TLR4 no dio lugar a la regulación negativa de Bambi, y no hubo cambio en la sensibilidad de las CEH contra la señal del TGFβ. Basados en estos resultados concluimos que la acti-vación de la vía de la señal del TLR4 mediada por el CL que se acumula en las CEH desempeñó un papel crítico en la exacer-bación de la fibrosis hepática inducida por la dieta RC.
En los modelos murinos de fibrosis hepática reportados aquí, el consumo de una dieta RC no afectó la lesión del hepatocito ni influyó en la fisiopatología de la inflamación hepática, incluida la activación de las células de Kupffer.

UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA 55GASTROENTEROLOGY • Volumen 1 • Núm. 2
Marí y colaboradores17 encontraron que el CL acumu-lado en los hepatocitos aumentó el daño hepático agudo mediado por LPS e indujo a los hepatocitos a la apoptosis mediada por el TNFα. Sin embargo otros investigadores sostienen que la apoptosis de los hepatocitos mediada por el TNF no está relacionada en la progresión de la fibrosis hepática,18,19 y sus hallazgos parecen arrojar luz sobre la ra-zón por la que la acumulación de colesterol en los hepatoci-tos resultante del consumo de una dieta RC no aumentó el daño de los hepatocitos, como se observó en nuestro estu-dio. Wouters y colaboradores20 reportaron que la adminis-tración de una dieta RC y alta en grasas en ratones KO del receptor de lipoproteínas de baja densidad y de la apolipo-proteína E causa inflamación hepática y la transformación de las células de Kupffer en células espumosas. Sin embar-go el consumo de una dieta RC no precipitó la formación de células espumosas macrofágicas en los modelos utiliza-dos en nuestro estudio. También mostramos que una dieta RC aumenta la fibrosis hepática inducida por LCB y por CCl4 en ratones depletados de células de Kupffer con la ad-ministración de clodronato. En conjunto estos resultados sugieren que las CEH, más que los hepatocitos o las células de Kupffer, son el sitio primario de las alteraciones en la fibrosis hepática resultantes del consumo de una dieta RC.
En resumen nuestro estudio proporciona nueva infor-mación sobre los mecanismos que relacionan el consumo de una dieta RC y la fibrosis hepática. La acumulación de CL en las CEH inducida por una dieta RC favoreció la trans-ducción de la señal del TLR4, esto aumenta sus niveles de la membrana y suprime, por tanto, la expresión del gen de Bambi en las CEH. En consecuencia, aumentan las señales del TGFβ de las CEH, lo que resulta en la activación de las CEH y la progresión de la fibrosis hepática.
Este trabajo indica que en el proceso de la progresión de la fibrosis hepática, el colesterol funciona como un factor que refuerza las señales del CL, que se acumula en las CEH más que como un factor extracelular inducible por la acti-vación de las CEH. Los hallazgos en este estudio justifican más investigaciones que se enfoquen en el CL en las CEH como el blanco de nuevas estrategias terapéuticas para el tratamiento de la fibrosis hepática.
Material suplementarioNota: para acceder al material suplementario que acompa-ña a este artículo, visite la versión en línea de Gastroentero-logy en www.gastgrojournal.org, y en doi: 10.1053/j.gastro.
Referencias1. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroente-
rology 2008;134:1655–1669.2. Ioannou GN, Morrow OB, Connole ML, et al. Association bet-
ween dietary nutrient composition and the incidence of cirrho-sis or liver cancer in the United States population. Hepatology 2009;50:175–184.
3. Ekstedt M, Franzén LE, Mathiesen UL, et al. Statins in nonalco-holic fatty liver disease and chronically elevated liver enzymes: a histopathological follow-up study. J Hepatol 2007;47:135–141.
4. Yoneda M, Fujita K, Nozaki Y, et al. Efficacy of ezetimibe for the treatment of non-alcoholic steatohepatitis: an open-label, pilot study. Hepatol Res 2010;40:613–621.
5. Sumiyoshi M, Sakanaka M, Kimura Y. Chronic intake of a highcholesterol diet resulted in hepatic steatosis, focal no-dular hyperplasia and fibrosis in non-obese mice. Br J Nutr 2010;103:378–385.
6. Kainuma M, Fujimoto M, Sekiya N, et al. Cholesterol-fed rabbit as a unique model of nonalcoholic, nonobese, non-insulin-resis-tant fatty liver disease with characteristic fibrosis. J Gastroente-rol 2006;41:971–980.
7. Vergnes L, Phan J, Strauss M, et al. Cholesterol and cholate com-ponents of an atherogenic diet induce distinct stages of hepatic inflammatory gene expression. J Biol Chem 2003;278:42774–42784.
8. Malhi H, Bronk SF, Werneburg NW, et al. Free fatty acids in-duce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem 2006;281:12093–12101.
9. Jin LW, Shie FS, Maezawa I, et al. Intracellular accumulation of amyloidogenic fragments of amyloid-beta precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities. Am J Pathol 2004;164:975–985.
10. Liao G, Yao Y, Liu J, et al. Cholesterol accumulation is associa-ted with lysosomal dysfunction and autophagic stress in Npc1 –/– mouse brain. Am J Pathol 2007;171:962–975.
11. Li Y, Schwabe RF, DeVries-Seimon T, et al. Free cholestero-lloaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis. J Biol Chem 2005;280:21763–21772.
12. Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 2007;13:1324–1332.
13. Paik YH, Schwabe RF, Bataller R, et al. Toll-like receptor 4 me-diates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 2003;37:1043–1055.
14. Wang Y, Chen T, Han C, et al. Lysosome-associated small Rab GTPase Rab7b negatively regulates TLR4 signaling in ma-crophages by promoting lysosomal degradation of TLR4. Blood 2007;110:962–971.
15. Suzuki M, Sugimoto Y, Ohsaki Y, et al. Endosomal accumulation of Toll-like receptor 4 causes constitutive secretion of cytokines and activation of signal transducers and activators of transcrip-tion in Niemann-Pick disease type C (NPC) fibroblasts: a poten-tial basis for glial cell activation in the NPC brain. J Neurosci 2007;27:1879–1891.
16. Chen H, Yang J, Low PS, et al. Cholesterol level regulates endo-some motility via Rab proteins. Biophys J 2008;94:1508–1520.
17. Marí M, Caballero F, Colell A, et al. Mitochondrial free choleste-rol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab 2006;4:185–198.
18. Bohan A, Chen WS, Denson LA, et al. Tumor necrosis factor alpha-dependent up-regulation of Lrh-1 and Mrp3(Abcc3) redu-ces liver injury in obstructive cholestasis. J Biol Chem 2003;278: 36688–36698.
19. Simeonova PP, Gallucci RM, Hulderman T, et al. The role of tu-mor necrosis factor-alpha in liver toxicity, inflammation, and fi-brosis induced by carbon tetrachloride. Toxicol Appl Pharmacol 2001;177:112–120.
20. Wouters K, van Gorp PJ, Bieghs V, et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis.
Hepatology 2008;48:474–486.
Recibido el 13 de noviembre de 2010. Aceptado el 24 de septiem-
bre de 2011.
Solicitud de reimpresosDirigir la solicitud de reimpresos a: Kengo Tomita, MD, PhD, Division
of Gastroenterology and Hepatology, Department of Internal Medicine,
National Defense Medical College, 3-2 Namiki, Tokorozawa-shi, Saita-
ma 359-8513, Japan. Correo electrónico: [email protected]; fax: (81)
4-2996-5201.
AgradecimientosLos autores agradecen a Mina Kitazume y Miho Takabe (Keio University)
sus útiles consejos y asistencia técnica; y a los Dres. Shuhji Seki, Manabu
Kinoshita y Hiroyuki Nakashima (Departamento de Immunología y Micro-

56 UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA GASTROENTEROLOGY • Volumen 1 • Núm. 2
biología, National Defense Medical College) por su útil discusión y co-
mentarios críticos.
T. Teratani y K. Tomita contribuyeron por igual en este trabajo y
comparten la primera autoría.
Conflictos de interésLos autores no declararon conflictos de interés.
RecursosEste estudio fue apoyado en parte por un Grant-in-Aid para la investi-
gación científica del Ministerio de Educación, Cultura, Deporte, Ciencia
y Tecnología de Japón (a K. Tomita).
Materiales suplementarios y métodos
ReactivosLos reactivos se obtuvieron como sigue: AcLDL de Biomedical Technologies (Stoughton, MA). Compuesto 58035 y LPS de Sigma (St. Louis, MO). TGFß de R&D Systems (Minneapolis, MN). CCl4 de Wako Pure Chemical Industries (Osaka, Japón).
Modelo animal Los ratones machos WT C57BL/6, C3H/HeN y C3H/HeJ y las ratas Sprague–Dawley se compraron en Sankyo Labora-tories (Tokyo, Japón). Los ratones NPC1-/- fueron compra-dos de Jackson Laboratories (Bar Harbor, ME). Los ratones fueron criados y mantenidos en una instalación con tempe-ratura y luz controladas con acceso ilimitado al alimento y agua. Para la LCB anestesiamos a los ratones y después de laparotomía en la línea media ligamos el conducto colédoco dos veces con suturas de seda y cerramos el abdomen. Prac-ticamos la cirugía simulada en forma similar, excepto que no se ligó el conducto biliar. Los ratones fueron sacrificados tres semanas después de la LCB. Para el daño hepático agu-do los ratones fueron alimentados con una dieta control o una dieta RC durante cuatro semanas, y luego fueron sa-crificados cinco días después de la LCB o 24 horas después de una inyección de CCl4. Todos los animales recibieron cuidados humanitarios en cumplimiento con los criterios del Consejo Nacional de Investigación descritos en la “Guía para el cuidado y uso de los animales de laboratorio”, pre-parada por la National Academy of Sciences de EU y publicada por los National Institutes of Health EU (Bethesda, MD).
Análisis bioquímico e histológicoLas concentraciones séricas de ALT, TG, glucosa y colesterol se determinaron como se describió previamente. El conte-nido hepático de TG y las concentraciones hepáticas de hi-droxiprolina se determinaron como se describió antes.1 Los niveles hepáticos de colesterol o del contenido de colesterol de las CEH o de las células de Kupffer se determinaron con el estuche Cholesterol/Cholesteryl Ester Quantitation (BioVi-sion, Mountain View, CA) siguiendo las instrucciones del fabricante. Determinamos el contenido de colesterol de las CEH o de las células de Kupffer justo después de aislarlas. Los tejidos hepáticos se fijaron en paraformaldehido al 4%, se embebieron en parafina, y se tiñeron con H&E y solu-ción tricrómica de Masson. Para el análisis de proteínas o del RNA los tejidos se congelaron en nitrógeno líquido y se guardaron a -80 °C hasta que se necesitaron.
Depleción de las células de KupfferInyectamos liposomas cargados con ácido diclorometileno difosfónico (clodronato) o con salina fosfato-buffer (En-capsula NanoSciences, Nashville, TN) por vía intravenosa en los ratones (200 μL por ratón).
Aislamiento y cualtivo de las CEHLas CEH se aislaron de ratones o ratas como se describió antes.1 Cultivamos las CEH en placas de plástico para cul-tivo de tejidos en medio Eagle Dulbecco modificado con 1 o 10% de suero bovino fetal, y las utilizamos como cultivos primarios sin pasos. Para la acumulación del CL en las CEH se incubaron las CEH con AcLDL (50 μg/mL) más compues-to 58035 (10 μg/mL) durante 16 horas. Utilizamos estu-ches para ensayos inmunosorbentes ligados a enzimas para la proteína-1 de quimioatracción de monocitos del ratón (Thermo Scientific, Rodkford, IL) o para la proteína-2 infla-matoria de macrófagos (R&D Systems) para la cuantificación de la proteína-1 de quimioatracción de los monocitos y de la proteína-2 inflamatoria de macrófagos en cultivos de CEH.
Aislamiento y cultivo de las células de KupfferLas células de Kupffer se aislaron de los ratones, y se cultiva-ron como se describió previamente.2
InmunohistoquímicaLos cortes parafinizados se desparfinizaron, rehidrataron y bloquearon con suero normal de caballo y se incubaron con un anticuerpo monoclonal anti-αSMA 1A4 (Dako Japan, Kyoto, Japan), un anticuerpo monoclonal anti-F4/80 (Se-rotec Oxfoard, UK), o un anticuerpo monoclonal anti-CD3 (Abcam, Cambridge, UK) durante la noche a 4 °C. El antí-geno F4/80 del ratón es una glucoproteína de 160 kilodal-tones expresada por los macrófagos del ratón; el anticuerpo anti-ratón F4/80 se une a los monocitos/macrófagos y a las células de Kupffer del ratón. El antígeno no es expresado por los linfocitos o las células polimorfunucleares. La unión del anticuerpo se detectó mediante incubación con anticuerpos IgG biotinilados anti-ratón y se visualizó con un estuche Vec-tastatin Elite ABC (Vector Laboratories, Inc, Burlingame, CA) mediante reacción con el sustrato Vectastastin DAB (Vector).
Se hicieron cortes del hígado congelado de 6 mm de es-pesor con un criostato, se recogieron en laminillas y se se-caron de inmediato. Los cortes se fijaron con acetona. Las laminillas se incubaron durante la noche con anti-CD68 Ab (Serotec), seguido de incubación con Histofine Simple Stra-in Mouse MAX-PO (Nichirei, Tokyo, Japón) durante 1 hora.
Infiltración de neutrófilosLos neutrófilos del hígado se tiñeron mediante la técnica de AS-D cloroacetato esterasa. Los cortes de hígado embebidos en parafina se tiñeron con naftol AS-D cloroacetato (Sigma Chemical Co, St. Louis, MO) siguiendo las instrucciones del fabricante.
Detección de apoptosisSe practicó tinción con desoxinucleotidil transferasa me-diada por la del extremo libre de la desoxiuridina transfe-

UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA 57GASTROENTEROLOGY • Volumen 1 • Núm. 2
rasa terminal (Chemicon International, Temecula, CA) en los especímenes para valorar la apoptosis. La apoptosis se cuantificó al contar las células positivas teñidas en 10 cam-pos al azar a 220X de aumento. La apoptosis se determinó para cada espécimen como un porcentaje del total de célu-las por campo. La unión del anticuerpo se detectó mediante incubación con anticuerpo IgG biotinilado anti-ratón y se visualizó con Vectastain Elite ABC (Vector laboratories, Inc) mediante reacción con sustrato Vectastatin DAB (Vector).
Aislamiento y caracterización mitocondrialUna fracción mitocondrial se enriqueció con especímenes de hígado de 100 mg con el estuche Mitochondria Isolation (Sigma) con dos pasos consecutivos de centrifugación a 600 y 11 000 g. El gradiente electroquímico de protones (ΔΨ) de la membrana mitocondrial interna se probó al medir la cap-tación del colorante fluorescente carbocianina JC-1 (Sigma) en las mitocondrias, siguiendo las instrucciones del fabrican-te.1 Se calculó el ΔΨ relativo en comparación con los valores obtenidos en los ratones alimentados con una dieta control.
Análisis de la reacción en cadena de la transcripción de polimerasa cuantitativa inversa de tiempo real Se extrajo el RNA total de los homogenados de hígado o de las CEH mediante el reactivo TaKaRa RNAiso (TaKaRa Bio, Ohrsu, Japan), según las instrucciones del fabricante. La transcripción inversa se practicó como se describió antes.1
Para la cuantificación de la expresión del mRNA se practica-ron las siguientes amplificaciones de la reacción en cadena de la polimerasa de tiempo real en duplicado con el estuche SYBR Premix Ex Taq (Perfect Real Time) (TaKaRa Bio) en un sistema Thermal Cycler Dice Real Time (TaKaRa Bio).
Análisis Western blotLa preparación de los extractos de proteínas de las CEH, la elec-troforesis de los extractos de proteínas de las células (5 μg), y los blots subsecuentes se practicaron con la utilización de anti-cuerpos contra TLR4 y ß-actina, como se describió antes.1
Análisis de citometría de flujoLa expresión de TLR4 en la superficie de las CEH se detectó mediante citometría de flujo con células vivas teñidas con anticuerpos anti-TLR4 conjugados con ficoeritrina (Ab-cam) o con un control con antiinmunoglobulina G conju-gada con ficoeritrina. Se analizó un total de 10 000 células/trastorno en un FAC-Scan con el FACSCalibur (Becton Dic-kinson, Franklin Lakes, N.J.).
Aislamiento de endosomas/lisosomas tardíos de las CEHLas fracciones tardías endosomales/lisosomales se prepa-raron de CEH con el estuche de aislamiento de lisosomas (Sigma) siguiendo las instrucciones del fabricante.
Referencias1. Tomita K, Tamiya G, Ando S, et al. Tumour necrosis factor alpha
signalling through activation of Kupffer cells plays an essential
role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut
2006;55:415–424.
2. Tomita K, Azuma T, Kitamura N, et al. Leptin deficiency en-
hances sensitivity of rats to alcoholic steatohepatitis through
suppression of metallothionein. Am J Physiol Gastrointest Liver
Physiol 2004;284:G1078–G1085.
3. Paik YH, Schwabe RF, Bataller R, et al. Toll-like receptor 4 me-
diates inflammatory signaling by bacterial lipopolysaccharide in
human hepatic stellate cells. Hepatology 2003;37:1043–1055.
4. Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-
beta signaling and hepatic fibrosis. Nat Med 2007;13:1324–
1332.
Figura 1 Suplementaria. Efectos de la dieta RC sobre el daño hepático agudo inducido por la LCB. Después de ser alimentados con una dieta
control o con una dieta RC durante 4 semanas, ratones C57BL/6 fueron sometidos a daño hepático agudo inducido 5 días después de la LCB (N = 5/
grupo). (A) Actividades de la ALT sérica (N = 5/grupo). (B, paneles de la izquierda) Gradiente electroquímico de protones de la membrana mitocondrial
interna (N = 5/grupo). El ΔΨ relativo calculado se normalizó con los valores obtenidos en ratones del grupo de dieta control–LCB. (B, paneles de la
derecha) Porcentaje de hepatocitos positivos con TUNEL (N = 5/grupo) y cortes representativos.
LCB
Dieta control Dieta RC
ControlRC
ALT
, U/L
ALT
ΔΨ
rela
tiva
(%
JC-1
/mg
pro
teín
a m
itocondri
al)
% d
e c
élu
las
posi
tiva
s con T
UN
EL
Control RC
LCB
Control RC
LCB
Simulada LCB
ΔΨm mitocondrial
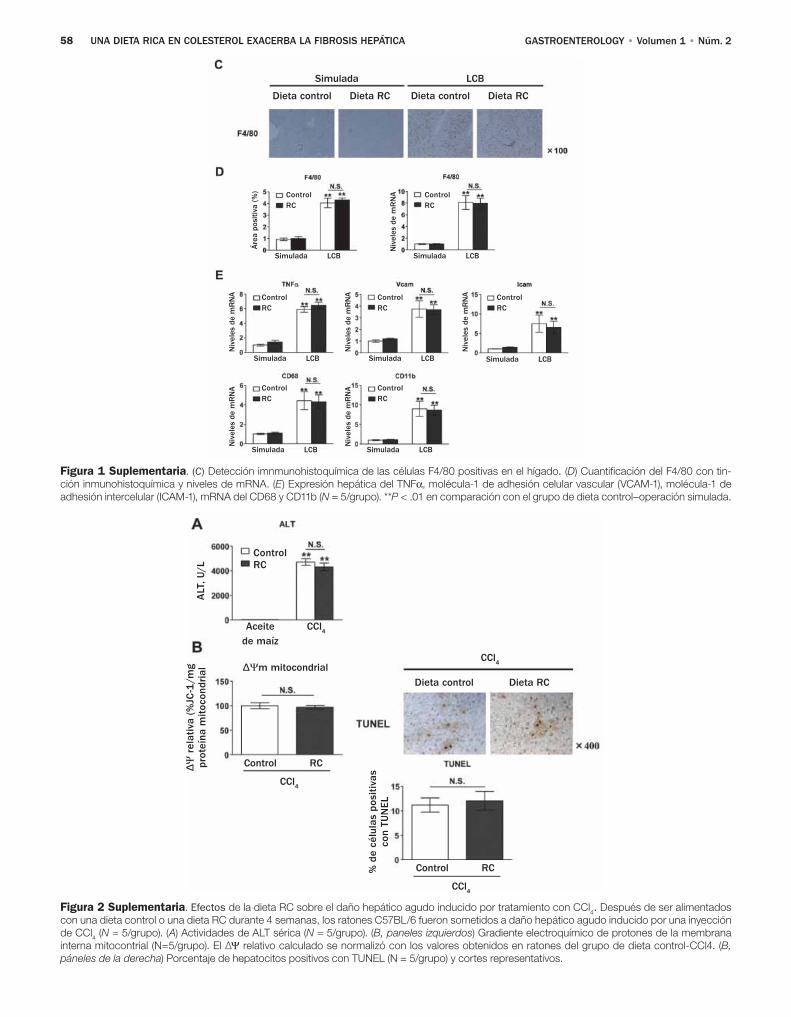
58 UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA GASTROENTEROLOGY • Volumen 1 • Núm. 2
Figura 1 Suplementaria. (C) Detección imnmunohistoquímica de las células F4/80 positivas en el hígado. (D) Cuantificación del F4/80 con tin-
ción inmunohistoquímica y niveles de mRNA. (E) Expresión hepática del TNFα, molécula-1 de adhesión celular vascular (VCAM-1), molécula-1 de
adhesión intercelular (ICAM-1), mRNA del CD68 y CD11b (N = 5/grupo). **P < .01 en comparación con el grupo de dieta control–operación simulada.
Figura 2 Suplementaria. Efectos de la dieta RC sobre el daño hepático agudo inducido por tratamiento con CCl4. Después de ser alimentados
con una dieta control o una dieta RC durante 4 semanas, los ratones C57BL/6 fueron sometidos a daño hepático agudo inducido por una inyección
de CCl4 (N = 5/grupo). (A) Actividades de ALT sérica (N = 5/grupo). (B, paneles izquierdos) Gradiente electroquímico de protones de la membrana
interna mitocontrial (N=5/grupo). El ΔΨ relativo calculado se normalizó con los valores obtenidos en ratones del grupo de dieta control-CCl4. (B,
páneles de la derecha) Porcentaje de hepatocitos positivos con TUNEL (N = 5/grupo) y cortes representativos.
CCl4
Dieta control Dieta RC
Simulada
Dieta control Dieta RC
LCB
Dieta control Dieta RC
Áre
a p
osi
tiva
(%
)
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
AN
ivele
s de m
RN
A
Niv
ele
s de m
RN
AN
ivele
s de m
RN
A
Niv
ele
s de m
RN
A
ControlRC
Control
RC
Control
RC
Control
RC
Control
RC
Control
RC
Control
RC
Control
RCA
LT, U/L
ΔΨ
rela
tiva
(%
JC-1
/mg
pro
teín
a m
itocondri
al
% d
e c
élu
las
posi
tiva
s con T
UN
EL
Aceite CCl4
de maíz
Control RC
CCl4
Control RC
CCl4
ΔΨm mitocondrial
Simulada LCB Simulada LCB
Simulada LCB Simulada LCB Simulada LCB
Simulada LCB Simulada LCB
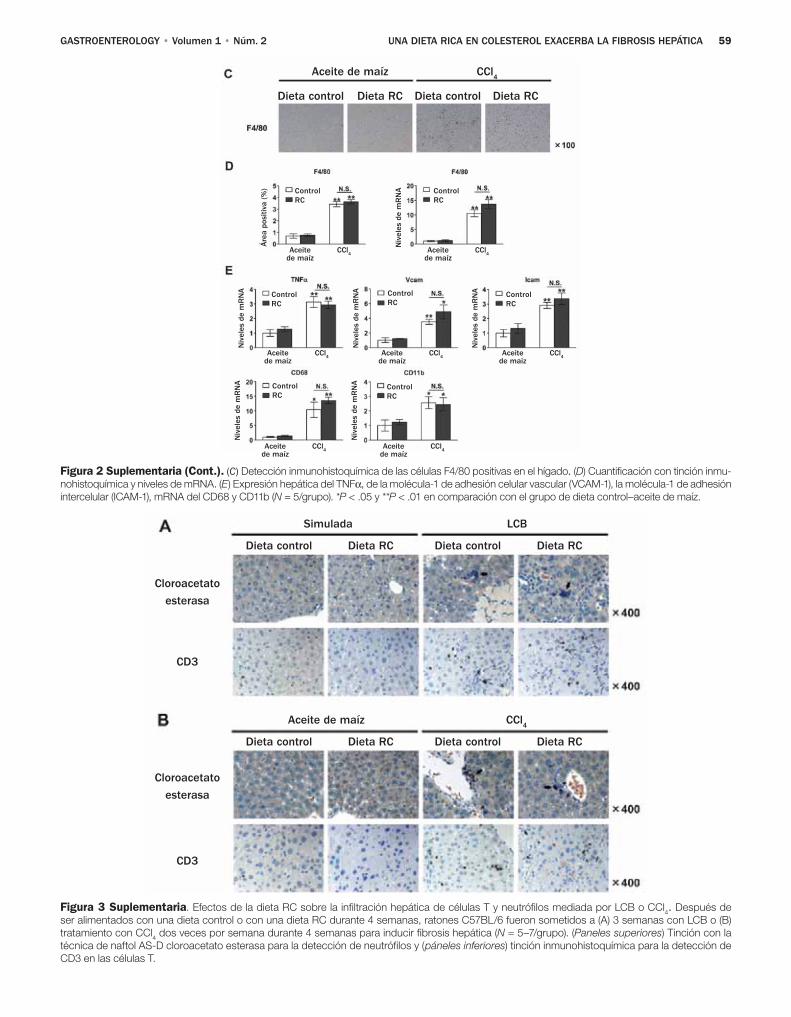
UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA 59GASTROENTEROLOGY • Volumen 1 • Núm. 2
Figura 2 Suplementaria (Cont.). (C) Detección inmunohistoquímica de las células F4/80 positivas en el hígado. (D) Cuantificación con tinción inmu-
nohistoquímica y niveles de mRNA. (E) Expresión hepática del TNFα, de la molécula-1 de adhesión celular vascular (VCAM-1), la molécula-1 de adhesión
intercelular (ICAM-1), mRNA del CD68 y CD11b (N = 5/grupo). *P < .05 y **P < .01 en comparación con el grupo de dieta control–aceite de maíz.
Figura 3 Suplementaria. Efectos de la dieta RC sobre la infiltración hepática de células T y neutrófilos mediada por LCB o CCl4. Después de
ser alimentados con una dieta control o con una dieta RC durante 4 semanas, ratones C57BL/6 fueron sometidos a (A) 3 semanas con LCB o (B)
tratamiento con CCl4 dos veces por semana durante 4 semanas para inducir fibrosis hepática (N = 5–7/grupo). (Paneles superiores) Tinción con la
técnica de naftol AS-D cloroacetato esterasa para la detección de neutrófilos y (páneles inferiores) tinción inmunohistoquímica para la detección de
CD3 en las células T.
Cloroacetato
esterasa
Dieta control Dieta RC Dieta control Dieta RC
Dieta control Dieta RC Dieta control Dieta RC
Simulada LCB
Aceite de maíz CCl4
Aceite de maíz CCl4
Aceite CCl4
de maíz
Aceite CCl4
de maíz
Aceite CCl4
de maíz Aceite CCl
4
de maíz Aceite CCl
4
de maíz
Aceite CCl4
de maíz
Aceite CCl4
de maíz
Áre
a p
osi
tiva
(%
)
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
ControlRC
ControlRC
ControlRC
ControlRC
ControlRC
ControlRC
ControlRC
Dieta control Dieta RC Dieta control Dieta RC
CD3
CD3
Cloroacetato
esterasa
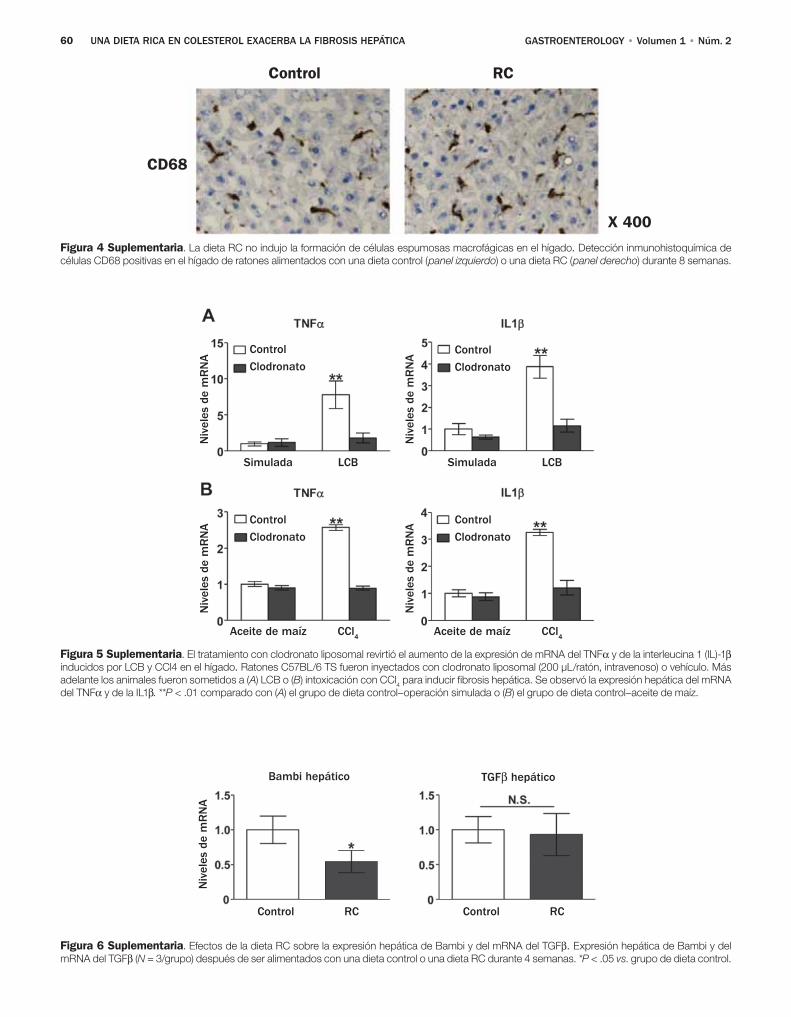
60 UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA GASTROENTEROLOGY • Volumen 1 • Núm. 2
Figura 4 Suplementaria. La dieta RC no indujo la formación de células espumosas macrofágicas en el hígado. Detección inmunohistoquímica de
células CD68 positivas en el hígado de ratones alimentados con una dieta control (panel izquierdo) o una dieta RC (panel derecho) durante 8 semanas.
Figura 5 Suplementaria. El tratamiento con clodronato liposomal revirtió el aumento de la expresión de mRNA del TNFα y de la interleucina 1 (IL)-1β
inducidos por LCB y CCl4 en el hígado. Ratones C57BL/6 TS fueron inyectados con clodronato liposomal (200 μL/ratón, intravenoso) o vehículo. Más
adelante los animales fueron sometidos a (A) LCB o (B) intoxicación con CCl4 para inducir fibrosis hepática. Se observó la expresión hepática del mRNA
del TNFα y de la IL1β. **P < .01 comparado con (A) el grupo de dieta control–operación simulada o (B) el grupo de dieta control–aceite de maíz.
Figura 6 Suplementaria. Efectos de la dieta RC sobre la expresión hepática de Bambi y del mRNA del TGFβ. Expresión hepática de Bambi y del
mRNA del TGFβ (N = 3/grupo) después de ser alimentados con una dieta control o una dieta RC durante 4 semanas. *P < .05 vs. grupo de dieta control.
Control RC Control RC
Control
Clodronato
Simulada LCB
Aceite de maíz CCl4
Simulada LCB
Aceite de maíz CCl4
Control
Clodronato
Control
Clodronato
Control
Clodronato
Bambi hepático TGFβ hepático
Control
CD68
X 400
RC
Niv
ele
s de m
RN
AN
ivele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
Niv
ele
s de m
RN
A
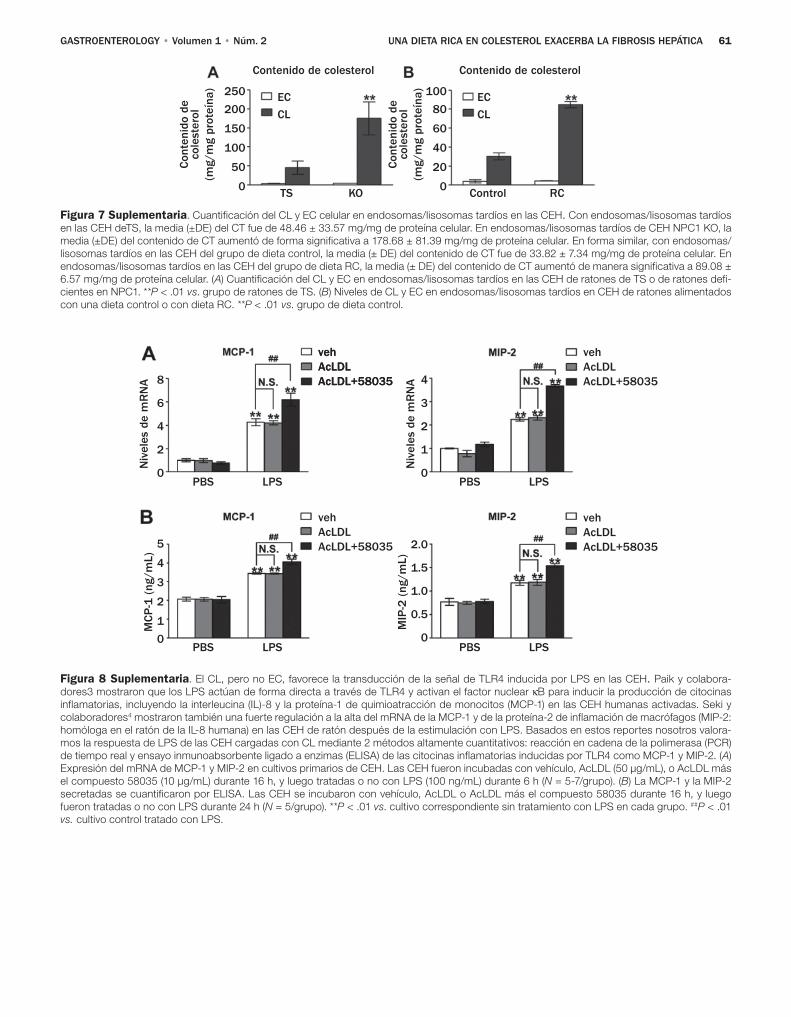
UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA 61GASTROENTEROLOGY • Volumen 1 • Núm. 2
Figura 7 Suplementaria. Cuantificación del CL y EC celular en endosomas/lisosomas tardíos en las CEH. Con endosomas/lisosomas tardíos
en las CEH deTS, la media (±DE) del CT fue de 48.46 ± 33.57 mg/mg de proteína celular. En endosomas/lisosomas tardíos de CEH NPC1 KO, la
media (±DE) del contenido de CT aumentó de forma significativa a 178.68 ± 81.39 mg/mg de proteína celular. En forma similar, con endosomas/
lisosomas tardíos en las CEH del grupo de dieta control, la media (± DE) del contenido de CT fue de 33.82 ± 7.34 mg/mg de proteína celular. En
endosomas/lisosomas tardíos en las CEH del grupo de dieta RC, la media (± DE) del contenido de CT aumentó de manera significativa a 89.08 ±
6.57 mg/mg de proteína celular. (A) Cuantificación del CL y EC en endosomas/lisosomas tardíos en las CEH de ratones de TS o de ratones defi-
cientes en NPC1. **P < .01 vs. grupo de ratones de TS. (B) Niveles de CL y EC en endosomas/lisosomas tardíos en CEH de ratones alimentados
con una dieta control o con dieta RC. **P < .01 vs. grupo de dieta control.
Figura 8 Suplementaria. El CL, pero no EC, favorece la transducción de la señal de TLR4 inducida por LPS en las CEH. Paik y colabora-
dores3 mostraron que los LPS actúan de forma directa a través de TLR4 y activan el factor nuclear κB para inducir la producción de citocinas
inflamatorias, incluyendo la interleucina (IL)-8 y la proteína-1 de quimioatracción de monocitos (MCP-1) en las CEH humanas activadas. Seki y
colaboradores4 mostraron también una fuerte regulación a la alta del mRNA de la MCP-1 y de la proteína-2 de inflamación de macrófagos (MIP-2:
homóloga en el ratón de la IL-8 humana) en las CEH de ratón después de la estimulación con LPS. Basados en estos reportes nosotros valora-
mos la respuesta de LPS de las CEH cargadas con CL mediante 2 métodos altamente cuantitativos: reacción en cadena de la polimerasa (PCR)
de tiempo real y ensayo inmunoabsorbente ligado a enzimas (ELISA) de las citocinas inflamatorias inducidas por TLR4 como MCP-1 y MIP-2. (A)
Expresión del mRNA de MCP-1 y MIP-2 en cultivos primarios de CEH. Las CEH fueron incubadas con vehículo, AcLDL (50 μg/mL), o AcLDL más
el compuesto 58035 (10 μg/mL) durante 16 h, y luego tratadas o no con LPS (100 ng/mL) durante 6 h (N = 5-7/grupo). (B) La MCP-1 y la MIP-2
secretadas se cuantificaron por ELISA. Las CEH se incubaron con vehículo, AcLDL o AcLDL más el compuesto 58035 durante 16 h, y luego
fueron tratadas o no con LPS durante 24 h (N = 5/grupo). **P < .01 vs. cultivo correspondiente sin tratamiento con LPS en cada grupo. ##P < .01
vs. cultivo control tratado con LPS.
PBS LPS
TS KO Control RC
PBS LPS
PBS LPS
PBS LPS
veh
AcLDL
AcLDL+58035
veh
AcLDL
AcLDL+58035
EC
CL
EC
CL
Contenido de colesterol Contenido de colesterol
veh
AcLDL
AcLDL+58035
veh
AcLDL
AcLDL+58035
veh
AcLDL
AcLDL+58035
Niv
ele
s de m
RN
A
Conte
nid
o d
e
cole
stero
l
(mg/m
g p
rote
ína)
Conte
nid
o d
e
cole
stero
l
(mg/m
g p
rote
ína)
Niv
ele
s de m
RN
A
MCP
-1 (
ng/m
L)
MIP
-2 (
ng/m
L)
8
6
4
2
0
250
200
150
100
50
0
100
80
60
40
20
0
5
4
3
2
1
0
4
3
2
1
0
2.0
1.5
1.0
0.5
0
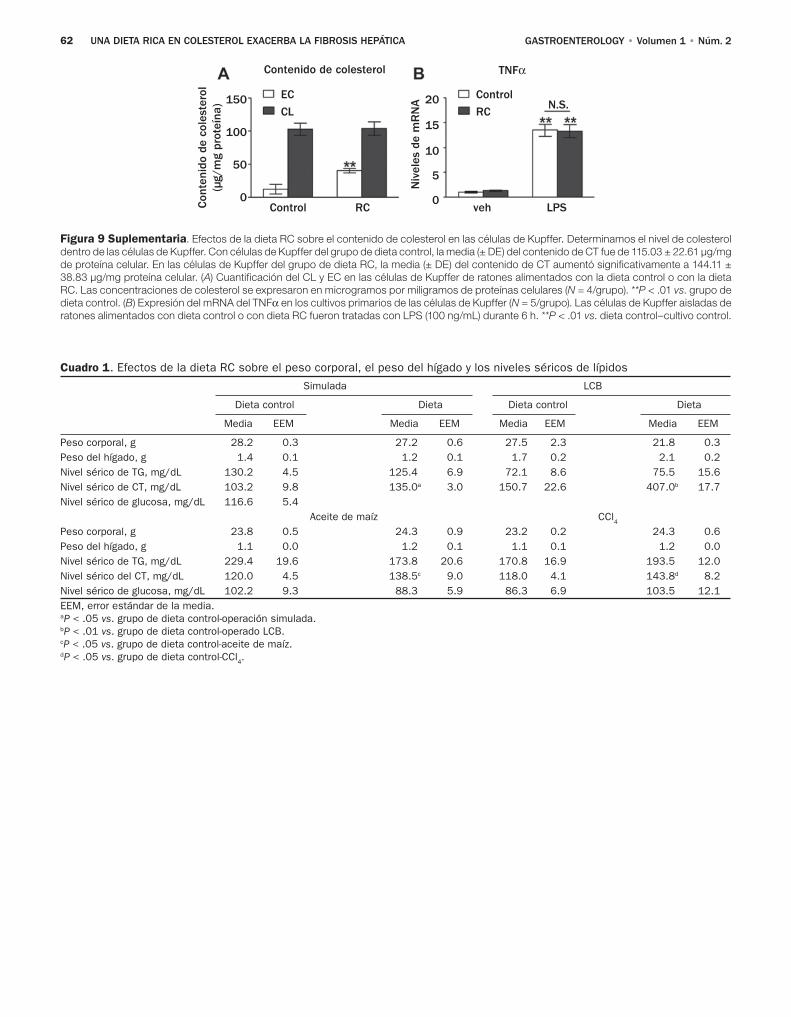
62 UNA DIETA RICA EN COLESTEROL EXACERBA LA FIBROSIS HEPÁTICA GASTROENTEROLOGY • Volumen 1 • Núm. 2
Figura 9 Suplementaria. Efectos de la dieta RC sobre el contenido de colesterol en las células de Kupffer. Determinamos el nivel de colesterol
dentro de las células de Kupffer. Con células de Kupffer del grupo de dieta control, la media (± DE) del contenido de CT fue de 115.03 ± 22.61 μg/mg
de proteína celular. En las células de Kupffer del grupo de dieta RC, la media (± DE) del contenido de CT aumentó significativamente a 144.11 ±
38.83 μg/mg proteína celular. (A) Cuantificación del CL y EC en las células de Kupffer de ratones alimentados con la dieta control o con la dieta
RC. Las concentraciones de colesterol se expresaron en microgramos por miligramos de proteínas celulares (N = 4/grupo). **P < .01 vs. grupo de
dieta control. (B) Expresión del mRNA del TNFα en los cultivos primarios de las células de Kupffer (N = 5/grupo). Las células de Kupffer aisladas de
ratones alimentados con dieta control o con dieta RC fueron tratadas con LPS (100 ng/mL) durante 6 h. **P < .01 vs. dieta control–cultivo control.
Cuadro 1. Efectos de la dieta RC sobre el peso corporal, el peso del hígado y los niveles séricos de lípidos
Simulada LCB
Dieta control Dieta Dieta control Dieta
Media EEM Media EEM Media EEM Media EEM
Peso corporal, g 28.2 0.3 27.2 0.6 27.5 2.3 21.8 0.3
Peso del hígado, g 1.4 0.1 1.2 0.1 1.7 0.2 2.1 0.2
Nivel sérico de TG, mg/dL 130.2 4.5 125.4 6.9 72.1 8.6 75.5 15.6
Nivel sérico de CT, mg/dL 103.2 9.8 135.0a 3.0 150.7 22.6 407.0b 17.7
Nivel sérico de glucosa, mg/dL 116.6 5.4
Aceite de maíz CCl4
Peso corporal, g 23.8 0.5 24.3 0.9 23.2 0.2 24.3 0.6
Peso del hígado, g 1.1 0.0 1.2 0.1 1.1 0.1 1.2 0.0
Nivel sérico de TG, mg/dL 229.4 19.6 173.8 20.6 170.8 16.9 193.5 12.0
Nivel sérico del CT, mg/dL 120.0 4.5 138.5c 9.0 118.0 4.1 143.8d 8.2
Nivel sérico de glucosa, mg/dL 102.2 9.3 88.3 5.9 86.3 6.9 103.5 12.1
EEM, error estándar de la media.aP < .05 vs. grupo de dieta control-operación simulada.bP < .01 vs. grupo de dieta control-operado LCB.cP < .05 vs. grupo de dieta control-aceite de maíz.dP < .05 vs. grupo de dieta control-CCl
4.
Contenido de colesterol
veh LPS Control RC
EC
CL
Control
RCN.S.
TNFα
150
100
50
0
20
15
10
5
0Conte
nid
o d
e c
ole
stero
l
(μg/m
g p
rote
ína)
Niv
ele
s de m
RN
A

63
AUTOEVALUACIÓN
Examen para obtener el puntaje para recertifi cación
INSTRUCCIONES GENERALES
¿Cómo contestar el examen?
Estimado doctor, en cada número de la revista usted encontrará al fi nal de cada publicación las preguntas
que contiene el examen, mismo que deberá ser contestado vía Internet.
¿Cómo presentar el examen?
En el examen correspondiente a Gastroenterology®, usted cuenta con tres oportunidades para obtener
el mínimo de aciertos requerido (80% de respuestas correctas) para acreditarlo y así obtener el puntaje
para recertifi cación.
Para tener acceso a la versión electrónica de este examen usted deberá:
1. Ingresar en Internet al sitio Sistema Interactivo de Evaluación Médica (SIEM®) en la siguiente
dirección:
www.evaluacionmedica.com.mx
2. Acceder a la evaluación de Gastroenterology® haciendo doble clic sobre la imagen de la portada de
la revista.
3. Ingresar la siguiente clave: GSV1N2
4. Seguir las instrucciones subsecuentes. Este examen está integrado por reactivos de opción múltiple.
Lea cuidadosamente cada enunciado, seleccione la respuesta que considere correcta y márquela.
Con el fi n de mantener un control sobre el examen, la clave de acceso se ligará con su cédula profesional
por lo que es muy importante que escriba ambas correctamente a fi n de evitar problemas posteriores.
Por ningún motivo se activarán claves nuevas.
PUNTAJE OTORGADO
La Asociación Mexicana de Gastroenterología, avala la edición mexicana de
Gastroenterology® como material de educación y actualización médica.
El Consejo Mexicano de Gastroenterología A.C. otorgará 6 puntos de recertifi cación
(1 punto por cada número de Gastroenterology® ) a los médicos que aprueben con un
mínimo de 80% la evaluación de cada ejemplar.
AUTOEVALUACION 63 3/14/12 11:41 AM

GASTROENTEROLOGY • Volumen 1 • Núm. 264
1. En la fibrosis hepática ¿Qué células se encuentran involucradas en su fisiopatología?a) Células de Kupfferb) Células estelares hepáticasc) Apoptosis del hepatocitod) Todas las anteriores e) Sólo a y c
2. ¿Cuál es el porcentaje de pacientes que no alcanza una respuesta virológica sostenida con el tratamiento estándar actual para los pacientes infectados con el genotipo 1 del VHC consistente en peginterferón alfa (PegIFN) y ribavirina (RBV) durante 48 semanas?f) Cerca de 20%g) Cerca de 30%h) Cerca de 40%i) Cerca de 45%j) Cerca de 60%
3. Los siguientes enunciados son verdaderos en relación al esófago de Barrett, excepto:a) Tiene una prevalencia estimada de 1 a 2% en los países
occidentales.b) La enfermedad crónica por reflujo gastroesofágico es
importante en la fisiopatologíac) Cerca de 10% de los pacientes con enfermedad por
reflujo gastroesofágico desarrollará EB.d) Cerca de 15% de los pacientes con reflujo
gastroesofágico desarrollará EB. e) Los pacientes con Barrett tienen un riesgo aumentado 30
a 125 veces de desarrollar adenocarcinoma esofágico, con una incidencia anual aproximada de 0.5%.
4. ¿Cuál sería el papel de las estatinas en la quimioprevención para el desarrollo de adenocarcinoma en el esófago de Barrett?a) Las estatinas inhiben la 3-hidroxi-3-metilglutaril
coenzima A reductasa, la enzima limitante de la biosíntesis del colesterol.
b) Se inhibe la formación de farnesil y geranilgeranil, esenciales para la activación de las proteínas intracelulares a través de prenilación, como es la proteína Ras, la cual es el activador de la cinasa regulada por señales extracelulares, la vía activada por mitógenos.
c) Como resultado, las estatinas inhiben la proliferación e inducir apoptosis en el epitelio de Barrett, lo que lleva a disminución del riesgo de progresión neoplásica.
d) Sólo a y be) Sólo a, b y c
5. ¿Qué son los inflamasomas?a) Son complejos carboproteicos relacionados con la
respuesta inflamatoria inmediata a la infección por bacterias en el intestino.
b) Los inflamasomas son complejos de multiproteínas mediadoras de la activación de la caspasa-1, que favorecen la secreción de las citocinas proinflamatorias ineterleucina-1ß e interleucina-18 y la piroptosis.
c) Responde a la presencia de complejos celulares por linfocitos T
d) Son receptores de patrones de bacterias patógenase) Ninguna de las anteriores
6. Se sabe que los inflamasomas son importantes en la defensa de organismos patógenos que pueden invadir el tracto GI. ¿Cuáles son otras funciones que presentan? a) Para enviar señales al sistema nervioso entérico y
favorecer la secreción intestinalb) Inician y perpetúan una respuesta humoral mediada por
inmunoglobulina Mc) Mantienen la integridad del epitelio intestinal y favorecen
la reparación, lo que hace que sean factores importantes en la patogenia de las EII y del cáncer.
d) Destruyen a la flora intestinale) Favorecen a la patogenia del síndrome de intestino irritable.
7. ¿En cuál de los genotipos del virus de la hepatitis C la respuesta al tratamiento combinado con interferón pegilado (IFN-PEG) y ribavirina (RBV) es más efectivo?a) Genotipo 1Ab) Genotipo 1Bc) Genotipo 2 y 3 d) Genotipo 4e) Genotipo 6
8. Los avances en la comprensión del ciclo de vida del virus de la hepatitis C llevaron al desarrollo de muchos agentes antivirales de acción directa (AAD). ¿Cuáles de los siguientes antivirales se encuentran dentro de este grupo y que se proponen para el tratamiento futuro de la hepatitis C?a) Aciclovirb) Telaprevir y boceprevir—inhibidores de la serina proteasa
NS3/4ª c) Zanamivird) Amantadinae) Tenofovir
9. ¿Cuál de los siguientes mecanismo participa en la fisiopatología del daño hepático inducido por el alcohol?a) Células de Kupfferb) Hepatocitoc) Células estelares hepáticas, llamadas anteriormente
lipocitos d) Sólo a y ce) Todas las anteriores
10. De acuerdo con el trabajo de la Dra. Florine Kastelein, relacionado al aumento de la incidencia de esófago de Barrett y adenocarcinoma esofágica, el uso de estatinas y antiinflamatorios no esteroideos podían tener una función ¿qué es lo que encontró?a) El uso de AINE, pero no de estatinas disminuye el riesgo
de progresión neoplásica en pacientes con esófago de Barrett.
b) El uso de AINE y estatinas disminuye el riesgo de progresión neoplásica en pacientes con esófago de Barrett.
c) El uso de estatinas, pero no de AINE disminuye el riesgo de progresión neoplásica en pacientes con esófago de Barrett.
d) El uso de AINE y estatinas aumentan el riesgo de progresión neoplásica en pacientes con esófago de Barrett.
e) El uso de AINE y estatinas no tienen efecto en el riesgo de progresión neoplásica en pacientes con esófago de Barrett.
AUTOEVALUACION 64 3/14/12 11:41 AM


Cód
igo
de a
lmac
én: 107241
Portada Gastr_astra.pdf 1 3/14/12 12:14 PM