

Apresentação do PowerPoint - Americas Centro de...
25
Talassemias Márcio Hori
Transcript of Apresentação do PowerPoint - Americas Centro de...

Talassemias
Márcio Hori

Introdução
• Redução ou ausência de cadeias de globina na hemoglobina
• 1 par de genes de cadeias β (1 gene β no cromossomo 11) – β0 síntese de cadeias β ausente
– β+ síntese de cadeias β reduzida
• 2 pares de genes de cadeias α (2 genes α vizinhos e funcionantes no cromossomo 16) – Portador silencioso (3 genes ativos)
– Traço α-talassêmico (2 genes ativos)
– Doença da HbH (1 gene ativo)
– Hidropsia fetal (0 genes ativos – homozigose α0-talassemia)
• Morte intra-uterina no fim da gestação ou horas após o nascimento

• Talassemia desequilíbrio de síntese de cadeias de globina – Cadeias α em excesso em β talassemia
• Precipitação com formação de agregados em eritroblastos
– Lesão de membrana
– Interferência mecânica na célula
– Comprometimento da divisão celular
– Comprometimento do metabolismo celular
– Cadeia β em excesso em α talassemia • Cadeias β e γ mais solúveis tetrâmeros β4 (Hb H) e γ4 (Hb de Bart) mais
solúveis, mas instáveis, sem capacidade de oxigenação e precipitação na hemácia (redução de meia-vida da hemácia)
Morte do
eritroblasto
Eritropoiese ineficaz
Introdução

Manifestações clínicas
• Dependente da forma clínica – Talassemia maior
• Forma mais grave da doença, dependente de transfusão
• Manifestações aparecem no 1º ano de vida:
– Menor aumento de peso – Episódios de febre
– Diarreia – Apatia
– Irritabilidade – Palidez
• Manifestações desaparecem com o tratamento
– Talassemia menor
• Assintomáticos
• Anemia leve, piores em:
– Infância
– Infecções ou processos inflamatórios
– Gravidez

– Talassemia intermediária
• Sintomáticos com anemia estável (Hb 7-11 g/dL) sem necessidade regular de hemotransfusões
• Combinação de defeitos genéticos onde há redução de produção de cadeias β, mas de modo menos grave que na talassemia maior (ou defeito adicional que reduz produção de cadeias α)
• Manifestações clínicas predominantes:
– Volumosa esplenomegalia
– Redução de massa muscular
– Úlceras crônicas em pernas
– Alterações faciais
– Sintomas compressivos por grandes massas de tecido hematopoiético extramedular
– Piora da anemia com infecções ou deficiência de folato
Manifestações clínicas

• Anemia – Intensidade variável
– Mecanismos
• Sobrevida diminuída das hemácias
• Eritropoiese ineficaz
• Esplenomegalia aumento de hemólise e hemodiluição
• Deficiência de ácido fólico
• Menor crescimento pôndero-estatural – Hipóxia tecidual
– Grande consumo metabólico pela hiperplasia eritróide
Manifestações clínicas

– Redução de massa muscular
– Defeitos na maturidade sexual
• Menor crescimento somático (necessidade de massa corporal crítica para desencadear a puberdade)
• Aumento de ferro na hipófise e nas gônadas
• Hiperplasia eritroide – Desvio de grande fração do débito cardíaco expansão do volume
circulante e anemia dilucional
– Desvio de nutrientes para a medula óssea, em detrimento de outros tecidos
– Aumento de absorção intestinal de ferro
Manifestações clínicas

– Alterações ósseas
• Decorrentes de expansão da medula óssea devido à anemia
• Observam-se:
•Protuberância da região frontal e regiões malares •Depressão na ponta do nariz e horizontalização dos orifícios nasais •Hipertrofia dos maxilares maior exposição de dentes e gengivas superiores
Manifestações clínicas
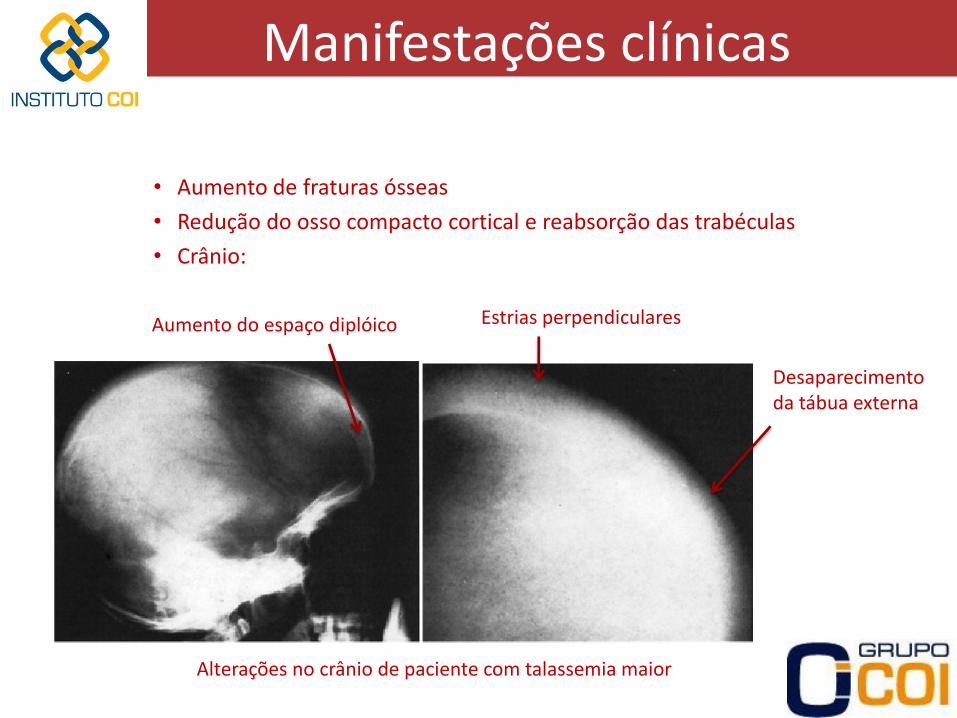
• Aumento de fraturas ósseas
• Redução do osso compacto cortical e reabsorção das trabéculas
• Crânio:
Aumento do espaço diplóico Estrias perpendiculares
Desaparecimento da tábua externa
Alterações no crânio de paciente com talassemia maior
Manifestações clínicas

• Ossos longos e ossos das mãos e pés com:
– Redução da cortical
– Rarefação óssea
– Aparecimento de cistos
• Aumento de ferro pode contribuir para a osteopatia pela talassemia
– Hipoparatireoidismo osteoporose
– Diminuição de vitamina C escorbuto
Manifestações clínicas

• Artralgia de grandes articulações
• Esplenomegalia – Por destruição aumentada das hemácias anormais
– Eritropoiese extramedular no baço
– Hiperesplenismo pancitopenia
• Aumento do ferro – Causas:
• Aumento de absorção intestinal do ferro
• Transfusões de repetição (principal)
– Alterações endócrinas
• Atraso de crescimento e puberdade
• Diabetes mellitus
• Hipoparatireoidismo
Manifestações clínicas

– Alterações cardíacas
• Pericardite
• Insuficiência cardíaca
• Arritmias
– Alterações hepáticas
• Cirrose hepática
Manifestações clínicas
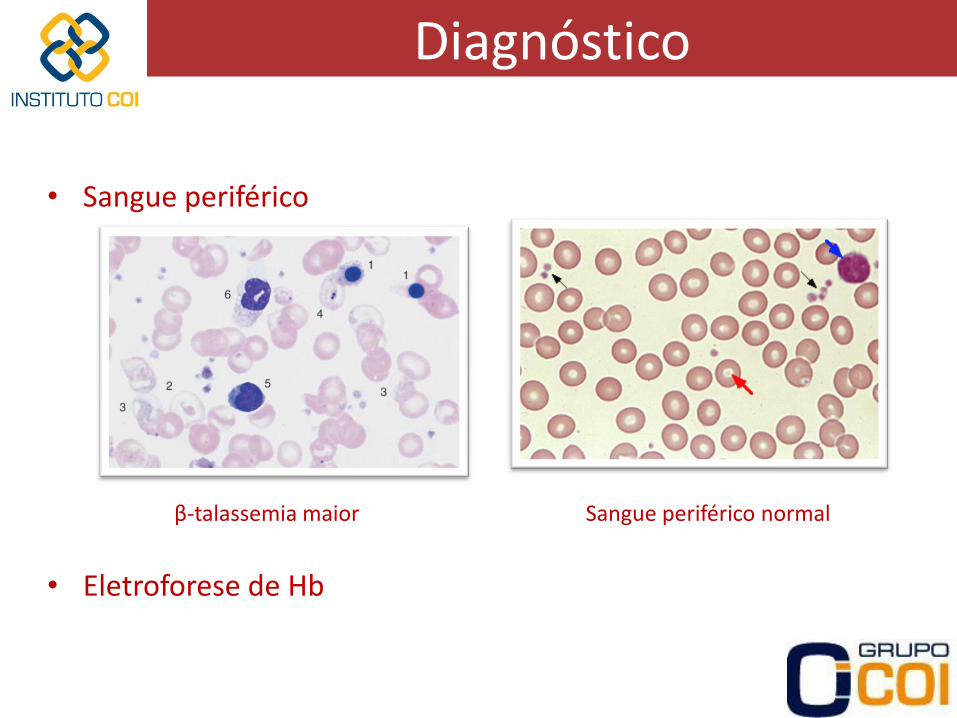
Diagnóstico
• Sangue periférico
Sangue periférico normal β-talassemia maior
• Eletroforese de Hb

• β-talassemia maior – Achados clínicos e história familiar nos pais
– Anemia microcítica (Hb < 9 g/dL) e hipocrômica
– Anisopoiquilocitose intensa, esquizócitos, hemácias e eritroblastos com granulações basofílicas, hemácias em alvo, eritroblastos, desvio à esquerda dos granulócitos
– Se hiperesplenismo leucopenia e trombocitopenia
– Eletroforese de Hb • Ausência de HbA
• Aumento de HbF
• HbA2 variável
Diagnóstico

• β-talassemia menor – Casos clássicos
• Anemia leve (10,5-13g/dL), mas que pode ser mais intensa na infância
• Microcitose e hipocromia com ferro sérico normal
• Eletroforese de Hb
– Aumento de HbA2 (3,5 a 6%)
– HbF normais ou ligeiramente elevados (<5%)
• Marcadores de hemólise – LDH elevada
– Haptoglobina reduzida
– Bilirrubina elevada às custas de indireta
– Reticulocitose
Diagnóstico

α-talassemia – Clínica e Diagnóstico
• Hidropsia fetal (nenhum gene) – Morte intrauterina ou horas após o nascimento
– Grande hepatoesplenomegalia, edema
• Doença por HbH – 1 gene α globina ativo
– Período neonatal predomina HbF, com 10-20% Hb de Bart (γ4) e pouca HbH
– Vida adulta Predomínio de HbA, com 5-30% HbH (β4) – Quadro clínico de talassemia maior ou intermediária
• Anemia hemolítica crônica de intensidade variável
• Esplenomegalia
• Alterações ósseas
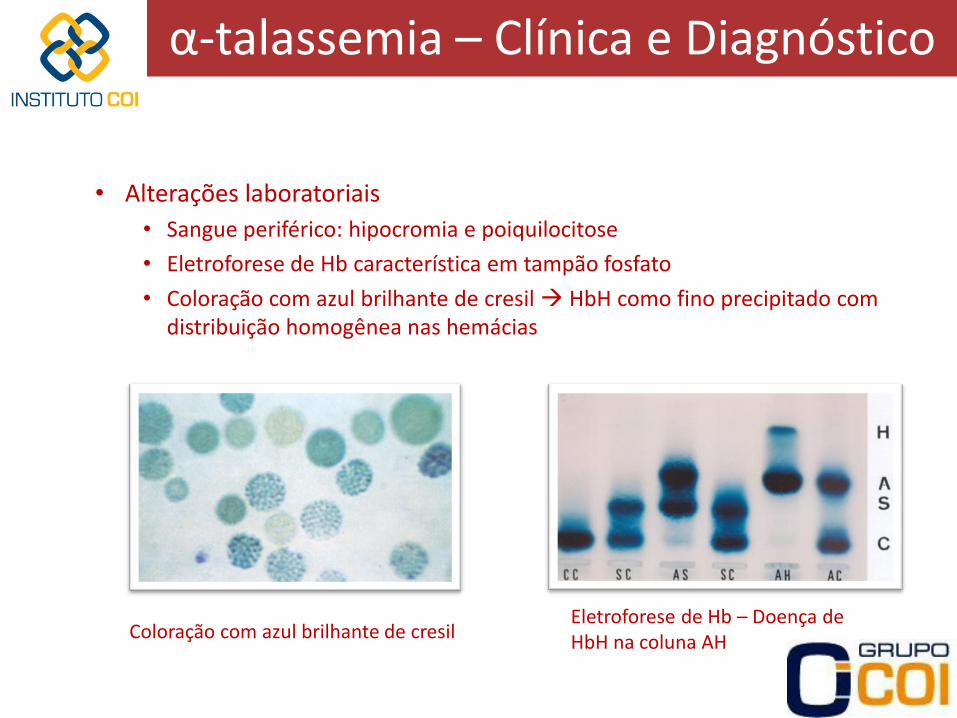
• Alterações laboratoriais
• Sangue periférico: hipocromia e poiquilocitose
• Eletroforese de Hb característica em tampão fosfato
• Coloração com azul brilhante de cresil HbH como fino precipitado com distribuição homogênea nas hemácias
Coloração com azul brilhante de cresil Eletroforese de Hb – Doença de HbH na coluna AH
α-talassemia – Clínica e Diagnóstico

• Traço α-talassêmico – 2 genes de cadeias α ativos
– Assintomáticos
– Sangue periférico: microcitose e hipocromia
– Período neonatal 5-10% Hb de Bart
– Vida adulta hipocromia, ferro sérico normal, detecção por métodos moleculares (DNA)
• Portador silencioso – 3 genes de cadeia α ativos
– Período neonatal 1-2% Hb de Bart
– Vida adulta ligeira hipocromia de detecção difícil ou sangue periférico normal
– Detecção por métodos moleculares (DNA)
α-talassemia – Clínica e Diagnóstico

• Associação com outras hemoglobinopatias – Associação com HbS doença de menor gravidade
– Associação com homozigotos para β talassemia doença menos grave talassemia intermediária
α-talassemia – Clínica e Diagnóstico

• Transfusional • Efeitos favoráveis sobre:
– Crescimento e atividade física
– Redução ou ausência de deformações ósseas
– Esplenomegalia
• Benefícios mais evidentes quando iniciado precocemente
• Tx de hemácias 20 mL/kg a cada 3-4 semanas manter Hb pré-tx 10 a 12g/dL
• Se sobrecarga cardíaca ou Hb <5g/dL Tx de hemácias 5-10 mL/Kg a cada 2-3 dias
Tratamento

– Programa regular também para talassemia intermediária, apesar de Hb>7g/dL, se houver:
• Deformações ósseas graves
• Retardo do crescimento
• Aumento progressivo do baço
• Úlceras persistentes nas pernas
• Terapia de quelação de ferro – Iniciar 1 ano após o começo do programa regular de transfusão
– Única forma de reduzir os danos decorrentes da hemocromatose secundária
– Desferoxamina (parenteral) ou deferasirox (oral)
• Reposição de ácido fólico – Evitar deficiência de folato em decorrência do elevado turnover
eritrocitário
Tratamento

• Esplenectomia – Avaliar risco x benefício
– Indicações:
• Trombocitopenia importante
• Consumo transfusional de sangue elevado (>240 mL/Kg/ano para manter Hb>10g/dL)
– Complicação sepse por germes encapsulados
• Prevenção:
– Adiar a cirurgia em paciente < 5 anos, se possível
– Vacinação para pneumococo, Haemophyllus influenzae e meningococo
– Antibioticoprofilaxia com penicilina V oral ou benzatina após a esplenectomia nos primeiros anos após a cirurgia ou até a adolescência
– Atendimento médico imediato em caso de febre
Tratamento

• Tratamento das complicações – Atraso puberal reposição hormonal pode ser necessária
– Insuficiência cardíaca iECA, espirolonolactona, β-bloqueadores
– Diabetes mellitus hipoglicemiantes orais, insulina
• Acompanhamento psicológico – Profundos impactos de uma doença crônica desde a infância
– Necessidade de acompanhamento médico regular
– Necessidade de uso crônico de quelantes de ferro e hemotransfusões
Tratamento

• Transplante alogênico de medula óssea – Apenas em casos muito selecionados
– Apenas se houver doador aparentado
– Melhores resultados se precocemente realizado
• Pacientes com menos complicações decorrentes da talassemia
• Menor grau de sobrecarga de ferro
Tratamento


















