
AP-1 TRANSCRIPTION FACTOR MEDIATES BOMBESIN … · adenomatous polyps and colorectal carcinomas of...
36
1 AP-1 TRANSCRIPTION FACTOR M EDIATES BOMBESIN-STIMULATED CYCLOOXYGENASE -2 EXPRESSION IN INTESTINAL EPITHELIAL CELLS Yan- Shi Guo * , Mark R. Hellmich *Ψ , Xiao Dong Wen * , and Courtney M. Townsend, Jr. * Departments of Surgery * and Physiology and Biophysics Ψ The University of Texas Medical Branch Galveston, Texas 77555 RUNNING TITLE: Mechanism of BBS-stimulated COX-2 expression Correspondence to: Courtney M. Townsend, Jr., M.D. Department of Surgery The University of Texas Medical Branch 301 University Boulevard Galveston, Texas 77555-0527 ph: (409) 772-1285 fax: (409) 772-5611 e-mail: [email protected] Supported by grants from the National Institutes of Health (PO1 DK35608, R01 DK48345) Copyright 2001 by The American Society for Biochemistry and Molecular Biology, Inc. JBC Papers in Press. Published on April 5, 2001 as Manuscript M101801200 by guest on March 20, 2019 http://www.jbc.org/ Downloaded from
-
Upload
truongngoc -
Category
Documents
-
view
217 -
download
0
Transcript of AP-1 TRANSCRIPTION FACTOR MEDIATES BOMBESIN … · adenomatous polyps and colorectal carcinomas of...

1
AP-1 TRANSCRIPTION FACTOR M EDIATES BOMBESIN-STIMULATED CYCLOOXYGENASE-2
EXPRESSION IN INTESTINAL EPITHELIAL CELLS
Yan-Shi Guo∗ , Mark R. Hellmich∗ Ψ, Xiao Dong Wen∗ , and Courtney M. Townsend, Jr.∗
Departments of Surgery* and Physiology and BiophysicsΨ
The University of Texas Medical Branch
Galveston, Texas 77555
RUNNING TITLE: Mechanism of BBS-stimulated COX-2 expression
Correspondence to: Courtney M. Townsend, Jr., M.D.
Department of Surgery
The University of Texas Medical Branch
301 University Boulevard
Galveston, Texas 77555-0527
ph: (409) 772-1285
fax: (409) 772-5611
e-mail: [email protected]
Supported by grants from the National Institutes of Health (PO1 DK35608, R01 DK48345)
Copyright 2001 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on April 5, 2001 as Manuscript M101801200 by guest on M
arch 20, 2019http://w
ww
.jbc.org/D
ownloaded from

2
ABBREVIATIONS
AP-1 activator protein-1
BBS bombesin
COX-2 cyclooxygenase-2
EMSA electrophoretic mobility shift assay
ERK extracellular signal-regulated kinase
GRP-R gastrin-releasing peptide receptor
IP immunoprecipitate
JNK c-Jun N-terminal kinase
LPS lipopolysaccharide
MAPK mitogen-activated protein kinase
MEK MAPK kinase
PG prostaglandin
SRE serum response element
TCF ternary complex factor
TRE TPA response element
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

3
SUMMARY
Colorectal carcinogenesis is a complex, multi-step process involving genetic alterations
and progressive changes in signaling pathways regulating intestinal epithelial cell proliferation,
differentiation and apoptosis. Although cyclooxgenase-2 (COX-2), gastrin-releasing peptide
(GRP) and its receptor, GRP-R, are not normally expressed by the epithelial cells lining the
human colon, the levels of all three proteins are aberrantly overexpressed in premalignant
adenomatous polyps and colorectal carcinomas of humans. Overexpression of these proteins is
associated with altered epithelial cell growth, adhesion and tumor cell invasiveness, both in vitro
and in vivo; however, a mechanistic link between GRP-R-mediated signaling pathways and
increased COX-2 overexpression has not been established. We report that bombesin (BBS), a
homolog of GRP, potently stimulates the expression of COX-2 mRNA and protein as well as the
release of PGE2 from a rat intestinal epithelial cell line engineered to express GRP-R. BBS
stimulation of COX-2 expression requires an increase in [Ca2+]i, activation of extracellular
signal-regulated kinase (ERK)-1 and -2 as well as p38MAPK and increased activation and
expression of the transcription factors, Elk-1, ATF-2, c-Fos and c-Jun. These data suggest that
the expression of GRP-R in intestinal epithelial cells may play a role in carcinogenesis by
stimulating COX-2 overexpression through an AP-1-dependent pathway.
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

4
Colorectal cancers are the third leading cause of cancer deaths in the United States (1).
One in twenty Americans is at risk of developing this disease during their lifetime. Considerable
experimental data have accumulated indicating an important role for cyclooxygenase-2 (COX-2)
in colorectal carcinogenesis. COX-2 is a key enzyme in the biosynthesis of prostaglandins (PGs)
from arachidonic acid and is overexpressed in 85-90% of human colon cancers and 40-50% of
premalignant adenomas (2). Several large epidemiological studies have shown that mortality
from colorectal cancers decreases (40-50%) in persons who regularly take aspirin or other
nonsteroidal antiinflammatory drugs (NSAIDs) (3), which inhibit COX activity. Additionally,
experiments with adenomatous polyposis coli (APC) gene-deficient mice (Min mice) revealed
that inhibition of COX activity with NSAIDs resulted in a reduction in the number and
multiplicity of spontaneously formed tumors (4-6) and, APC∆716 /COX-2 double-knockout mice
showed reduction in both the neoplastic growth and number of intestinal tumors (7). Although
mounting evidence supports an important role for COX-2 in colorectal carcinogenesis, the
molecular mechanisms leading to COX-2 overexpression in intestinal epithelial cells are not
completely understood.
Like COX-2, the mammalian homologue of bombesin (BBS), gastrin-releasing peptide
(GRP), and its cognate G-protein-coupled receptor, GRP receptor (GRP-R), are aberrantly
overexpressed in premalignant adenomatous polyps and colorectal cancers. Preston et al (8)
showed that 24% of colorectal cancers, but not the adjacent non-malignant mucosa, exhibited
high-affinity binding sites for GRP. Immunohistological analysis of archival tissue specimens
from colonic polyps and colon cancers revealed that 42% (n=5) of high-grade dysplastic polyps
and 62% (n=50) of colon cancers stained positively for both GRP and GRP-R protein (9). We
have found that 42% (n=5) of freshly resected adenomatous polyps and 67% (n=12) of colorectal
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

5
cancers contain cytokeratin-positive cells that exhibit an increase in the concentration of free
intracellular Ca2+ ([Ca2+]i) in response to BBS-stimulation, indicating the presence of functional
BBS-receptor (unpublished data).
Although the precise role of BBS-like peptides and GRP-R in colorectal carcinogenesis
has not been defined, recent observations that aspirin inhibits BBS-induced DNA synthesis in
Swiss 3T3 fibroblasts (10) and GRP stimulates expression of COX-2 in the same cell line (11),
have raised the possibility that GRP-R-mediated signaling pathways may contribute to the
upregulation of COX-2 expression during colorectal carcinogenesis. The aims of our study were
to examine whether the expression of GRP-R leads to BBS-dependent upregulation of COX-2 in
the rat intestinal epithelial cell line, RIE-1 and if so, to determine the molecular signaling
pathways linking GRP-R to the regulation of COX-2 expression.
We selected the RIE-1 cell line for these studies for several reasons: 1) They are a non-
tumorigenic intestinal epithelial cell line which, like the normal human colonic epithelium, do
not express endogenous GRP-R or other BBS receptor subtypes. 2) Unlike many epithelioid cell
lines derived from cancer cells, the endogenous level of COX-2 expression, under normal culture
conditions, is very low. 3) Constitutive overexpression of COX-2 in RIE-1 cells increases their
tumorigenic potential (12). 4) The aberrant overexpression of GRR-R and COX-2 in
premalignant adenomatous polyps suggests that these proteins may play a role in the early stages
of colon carcinogenesis.
To evaluate the potential role of GRP-R-mediated signaling pathways in COX-2 gene
expression, we developed RIE-1 cell lines expressing recombinant GRP-R called RIE/GRPR.
We found that the GRP-R agonist, BBS, markedly stimulates COX-2 mRNA and protein
expression as well as the release of PGE2 from these cells. The increase in COX-2 expression is
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

6
largely due to BBS-enhanced transcription of the COX-2 gene and is dependent on an agonist-
stimulated increase in [Ca2+]i, activation of MAP kinase-dependent pathways and the increased
expression and activation of AP-1 transcription factor. These findings partially identify the
signaling pathways coupling GRP-R to the upregulation of COX-2 expression and identify the
regulation of COX-2 gene expression as a potential mechanism by which aberrantly expressed
GRP-R plays a role in colorectal carcinogenesis.
EXPERIMENTAL PROCEDURES
Plasmids
The mouse GRP-R expression vector was a gift from Dr. James F. Battey (National
Institutes of Health, Bethesda, MD). The mouse COX-2 promoter constructs, TIS10L-luc,
TIS10-80-luc and TIS10-40-luc, as well as mouse PGs-2 (COX-2) cDNA probe were kindly
provided by Dr. Harvey R. Herschman (Los Angeles, CA). The reporter constructs Gal4-
Elk(307-408), Gal-4-Sap(268-431), Gal4-luc were gifts from Dr. Ralf Janknecht (La Jolla, CA).
c-fos-luc and 3XTRE-luc were provided by Dr. Johannes L. Bos (Utrecht, The Netherlands) and
Dr. Joan Massagué (New York, NY), respectively. Mouse c-Fos and c-Jun cDNA probes were
purchased from the American Type Culture Collection (Rockville, MD).
Antibodies
The anti-COX-2 antibodies were obtained from Cayman Chemical (Ann Arbor, MI). The
anti-active ERK-1 and –2 antibody (pTEpY) was purchased from Promega (Madison, WI).
Anti-phospho-p38MAPK, anti-phospho-ELK-1 and antiphospho-ATF-2 antibodies were obtained
from New England Biolabs, Inc. (Beverly, MA).
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

7
RIE/GRPR cell lines
RIE-1 cells were a gift from Dr. Kenneth D. Brown (Cambridge Research Station,
Babraham, Cambridge, UK). RIE-1 cells were transfected with mouse GRP receptor using
lipofectamine (Gibco/BRL, Grand Island, NY) according to the manufacturer’s
recommendations. G418-resistant colonies were selected as described previously (13). The
number of binding sites (Bmax) and their binding affinities (Kd) were determined using [125I]BBS
binding assays as described (14). Agonist-induced changes in [Ca2+]i were detected using the
Ca2+-sensitive dye AM-fura-2 as previously described (15). Cells were cultured at 37oC in a
humidified atmosphere of 95% air and 5% CO2 in DMEM medium supplemented with 5%
heated-inactivated fetal bovine serum (FBS, Hyclone, Logan, UT) and Geneticin (G418,
400 µg/ml, GIBCO/BRL).
RNA isolation and Northern blot analysis
Total cellular RNA was extracted by the method of Chomczynski and Sacchi (16). RNA
samples (30 µg/lane) were separated on 1.2% agarose-formaldehyde gels and blotted onto
Nytran plus filters (Schleicher and Shuell, Inc., Keene, NH). The blots were hybridized with
cDNA probes labeled with [α32P]dATP by random primer extension. Specific hybridization was
visualized by autoradiography. To ensure RNA integrity and to confirm equal loading between
lanes, the filters were stripped and rehybridized with a probe for 18S rRNA.
Western blot analysis
Immunoblot analysis was performed as described previously (13). The cells were lysed
for 30 min in a solution consisting of 1 X PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate,
0.1% SDS, 1 mM phenylmethylsulfonyl fluoride, 10 µg/ml aprotinin, and 1 mM sodium
orthovanidate. Cellular proteins were denatured by heating, resolved by SDS-polyacrylamide
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

8
gel electrophoresis and transferred to nitrocellulose membranes and probed with the indicated
antibodies and then with a peroxide-coupled second antibody (Promega). Proteins were detected
using the enhanced chemiluminescence system (ECL, Amersham Pharmacia Biotech).
PGE2 assay
RIE/GRPR cells were plated in 24-well plates. Thirty-six hours later, the medium was
replaced with serum-free DMEM overnight. The cells were then incubated with or without BBS
for the indicated times. Media were collected from each well and analyzed for PGE2 by ELISA
(Cayman Chemical, Ann Arbor, MI).
Immunofluorescence microscopy
RIE/GRPR cells were cultured in Lab-TekII chamber slides (Nalge Nunc International,
Naperville, IL). Prior to immunostaining with anti-COX-2 antiserum, the cells were incubated
with or without BBS (100 nM) for 6 h at 37oC, fixed with a 4% paraformaldehyde (15 min),
permeabilized with 0.3% Triton X-100 (10 min) and incubated in blocking solution (1% BSA in
PBS, 20 min). After incubating the cells with anti-COX-2 antiserum (1:400) for 90 min at room
temperature, the cells were washed three times with PBS and incubated with a goat anti-rabbit
IgG antibody labeled with Alexa 488 (Molecular Probes, Eugene, OR) (1:2000, 30 min).
Specific immunostaining was visualized with a Nikon Eclipse fluorescence microscope.
Luciferase assay
RIE/GRPR cells were seeded in 6-well plates at a density of 2x105 cells/well. After
overnight adhesion, cells were transfected with 2 µg of promoter/luciferase reporter gene DNA
using Fugene 6 (Roche Molecular Biochemicals, Indianapolis, IN). Prior to assaying for
luciferase activity, cells were incubated in DMEM without serum (24 h) and treated with BBS
for 6 h. Luciferase activity in 20 µl of cell extract was assayed using the Luciferase Assay
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

9
System (Promega, Madison WI). Transfection efficiency was assessed using a β-galactosidase
expression plasmid as described previously (17).
Electrophoretic mobility shift assay
Nuclear extracts were prepared as previously described (18). An oligonucleotide
(Stratagene, La Jolla, CA) whose sequence corresponded to the AP-1 binding site consensus
sequence was end-labeled with [γ-32P]ATP and T4 polynucleotide kinase. Electrophoretic
mobility shift assay reaction mixtures contained 50,000 cpm of 32P-end-labeled oligonucleotide,
20 µg of nuclear protein extract and 1.0 µg of poly (dl • dc; Pharmacia Biotech, Inc.) in a final
volume of 20 µl. Reaction mixtures were resolved on 4% non-denaturing polyacrylamide gel
electrophoresis at 200 V for 2 h. Gels were dried and visualized by autoradiography (17).
Statistical analysis
All experiments were repeated on at least two separate occasions. Results from Northern
and Western blots were quantified by densitometry. Values are expressed as mean ± SEM.
Differences between means were compared using the analysis of variance (ANOVA) test and
were considered significantly different at the level of p <0.05.
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

10
RESULTS
RIE/GRPR cells
The RIE-1 cell line, isolated originally from the rat small intestine, exhibits epithelioid
morphology, the normal rat diploid number of chromosomes and does not form colonies in soft
agar (19). These properties have made them one of the preferred cell models for examining non-
tumorigenic intestinal epithelial cell physiology and biochemistry in vitro. Constitutive
overexpression of recombinant COX-2 in RIE-1 cells is associated with altered cell adhesion to
extracellular matrix, decreased expression of E-cadherin, increased expression of the anti-
apoptotic gene product, BCL-2, and decreased apoptosis (12). To determine whether BBS and
GRP-R could regulate endogenous COX-2 expression in RIE-1 cells, cells were stably
transfected with an expression plasmid containing a mouse GRP-R cDNA downstream of the
constitutively active CMV promoter. After selection with G418, surviving cell clones were
evaluated for the level of receptor expression and activity using radiolabeled ligand binding and
Ca2+-imaging with the calcium indicator dye AM-fura-2, respectively. Five clones were isolated
with Bmax values ranging from approximately 3000 to 8900 binding sites per cell. The calculated
affinity constants exhibited a range of values from 0.33 nM to 1.3 nM. For the remainder of the
studies, we used the RIE/GRPR cell line with an affinity constant for BBS of 0.54 nM and Bmax
values of approximate 3000 receptors per cell. Fura-2 imaging experiments revealed that greater
than 99% of these cells exhibited an increase in [Ca2+]i upon stimulation with BBS (1 nM).
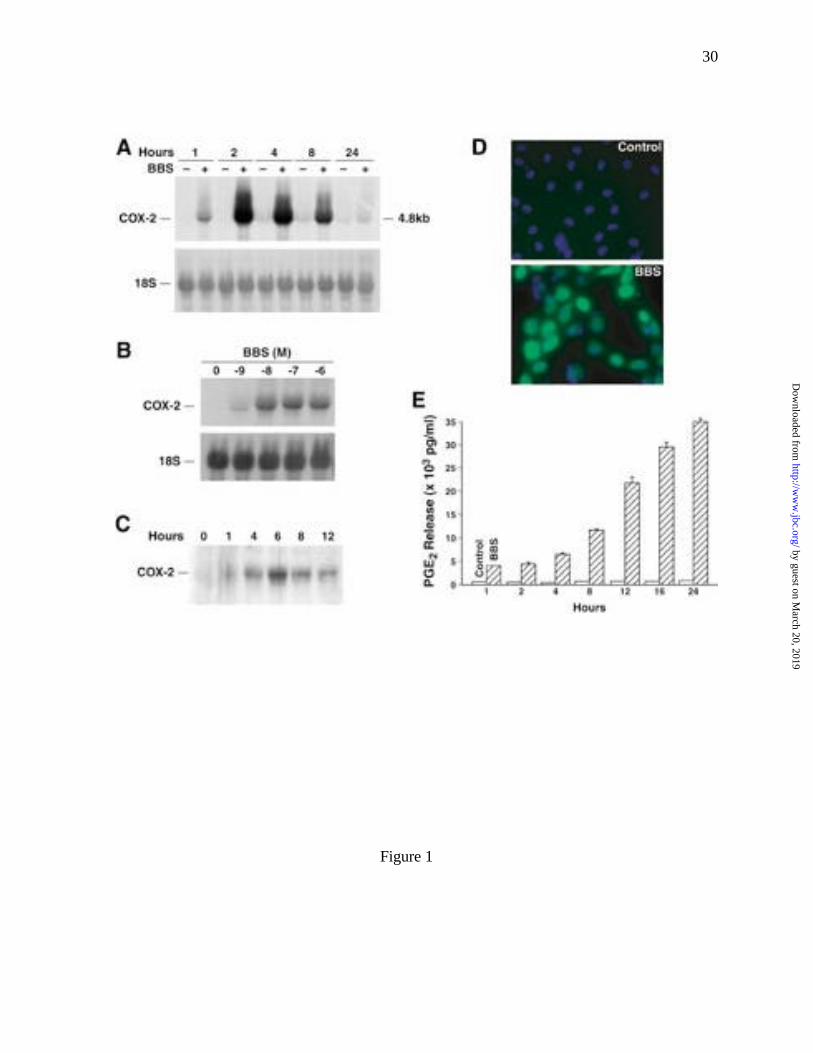
BBS stimulates COX-2 expression and activity
BBS stimulated time- and dose-dependent increases in the expression of COX-2 mRNA
and protein in RIE/GRPR cells. Compared to untreated cells, the level of COX-2 mRNA
increased in cells treated with BBS (100 nM) by 3-, 20-, 15-, 8- and 0.5-fold at 1, 2, 4, 8 and
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

11
24 h, respectively (Fig. 1A). The increase in COX-2 mRNA at 2 h was dependent on the
concentration of BBS used to stimulate the cells. Maximum increases in COX-2 mRNA levels
were detected in cells stimulated with 10, 100 and 1000 nM BBS (Fig. 1B). BBS treatment also
stimulated a time-dependent increase in the level of COX-2 protein. Western blots revealed an
increase in COX-2 protein by 1 h following BBS (100 nM) stimulation and a peak in expression
at 6 h (Fig. 1C). Untreated RIE/GRPR cells showed no detectable expression of COX-2 protein.
Consistent with the Western blot data, immunofluorescence staining showed an increase in
COX-2 immunoreactivity in RIE/GRPR cells following stimulation with BBS (Fig. 1D, BBS),
whereas untreated cells did not exhibit COX-2 immunoreactivity (Fig. 1D, Control).
COX converts arachidonic acid, released from phospholipid stores by the action of
phospholipase A2, to prostaglandin H2, the common precursor of all prostaglandins. To assess
whether increased COX-2 expression was associated with an increased prostaglandin synthesis,
the levels of Prostaglandin E2 (PGE2) released from RIE/GRPR cells were measured using an
ELISA assay. Compared with untreated control cultures, PGE2 levels in the media of RIE/GRPR
cells treated with BBS increased by 6.8-fold at 1 h and continued to increase to 45-fold at 24 h
(Fig. 1E).
Increases in [Ca2+]i and MAPK activity mediate BBS regulation of COX-2 expression
Agonist binding to GRP-R initiates the activation of intracellular signaling pathways
(20,21) involving specific heterotrimeric G-proteins (22,23), generation of the second
messengers, inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG), release of Ca2+ from
IP3-sensitive stores (15) and activation of various protein kinases including protein kinase C
(PKC) (15), protein kinase D (PKD) (24), the Src-family of non-receptor tyrosine kinases (25)
and the mitogen-activated protein kinase (MAPK) cascades (26).
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

12
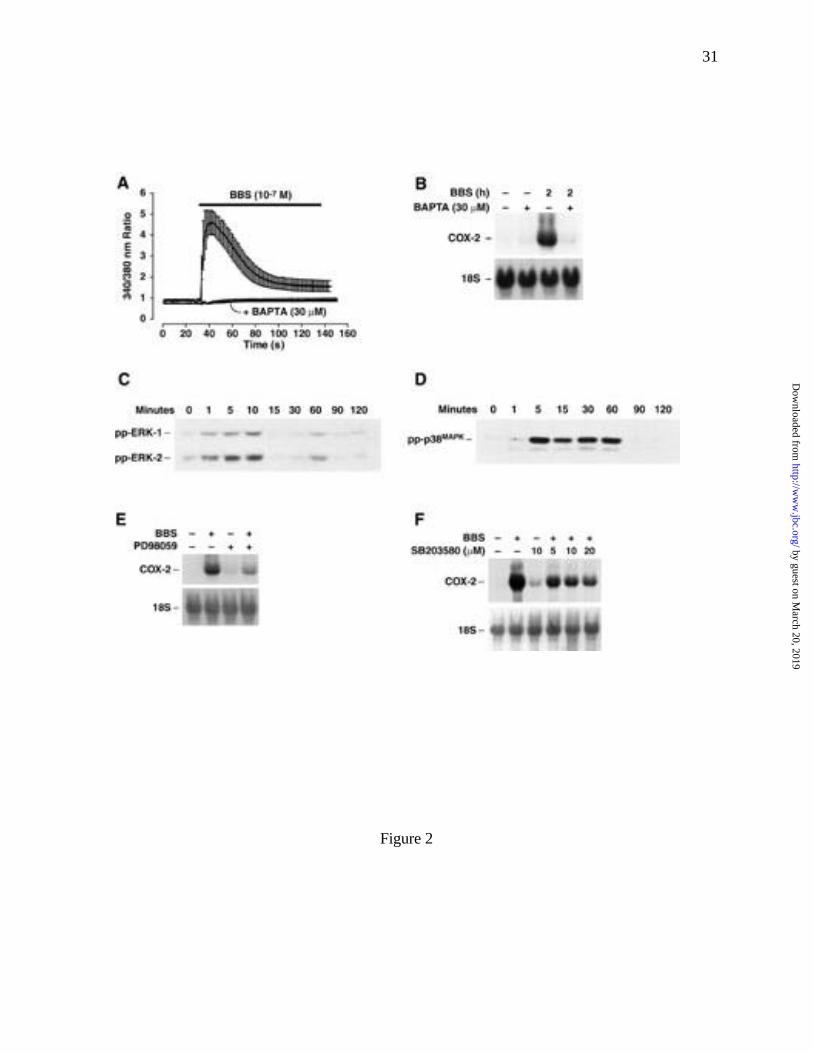
BBS stimulation of RIE/GRPR cells induced a rapid increase in [Ca2+]i (Fig. 2A). To
assess the role of [Ca2+]i in BBS regulation of COX-2 mRNA expression, cells were pretreated
for 1 h with the membrane-permeable chelating agent, AM-BAPTA (30 µM). Treatment with
the chelator was sufficient to inhibit the BBS-induced increase in [Ca2+]i (Fig. 2A) and COX-2
mRNA expression (Fig. 2B), but did not affect either cell viability, measured by trypan blue
exclusion assay (% blue cells; vehicle [0.1% DMSO] 3.56 ± 0.79 vs. AM-BAPTA 3.23 ± 0.67),
or alter the expression of 18S ribosomal RNA. Together these data indicate that an agonist-
induced increase in [Ca2+]i is required for BBS-stimulated increases in COX-2 mRNA levels in
RIE/GRPR cells.
Mitogen-activated protein kinase pathways mediate the regulation of COX-2 expression
to a variety of extracellular stimuli (27-29). Three related MAPK cascades have been described
(30,31), they are referred to as the extracellular signal-regulated protein kinase (ERK) pathway,
the c-Jun N-terminal kinase (JNK) pathway and the p38 MAP kinase (p38MAPK) pathway. The
activities of MAPKs are regulated by upstream dual-specificity MAPK kinases (MEKs). MEKs
activate MAPKs, such as ERKs, JNK and p38MAPK, by phosphorylation on both threonine and
tyrosine resides. To determine whether BBS activated MAPK pathways in RIE/GRPR cells, the
levels of phosphorylated ERK, p38MAPK and JNK proteins were determined by immunoblotting.
Bombesin treatment stimulated the activation of the two ERK isozymes, ERK-1 and -2,
as well as p38MAPK (Fig. 2C, D), but did not activate JNK (data not shown). Western blots of
RIE/GRPR cell extracts, probed with antibodies selective for the phosphorylated (activated)
forms of ERK-1 and -2, showed that BBS induced a time-dependent increase in phosphorylated
ERK-1 and -2 (Fig. 2C). ERK-1 and -2 phosphorylation was increased 1 min after BBS
treatment and reached a peak at 10 min before returning to baseline at 15 and 30 min. A second,
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

13
smaller increase in ERK-1 and -2 activation was detected at 60 min after BBS stimulation (Fig.
2C). The active status of ERK-1 and -2 was confirmed by in vitro phosphorylation experiments
using immunoprecipitated (IP) ERK-1 and -2 and the substrate, myolin basic protein (MBP). A
5-fold increase in MBP phosphorylation was observed when using IP proteins from RIE/GRPR
cells treated for 10 min with BBS vs. IP proteins from untreated cultures (data not shown). In
addition to ERK activation, BBS stimulated a time-dependent activation of p38MAPK. An
increase in the phosphorylated form of p38MAPK was detected 5 min following BBS treatment
(Fig. 2D). In contrast to the transient activation of ERK-1 and -2, p38MAPK phosphorylation
reached a maximum by 5 min and remained elevated up to 60 min after agonist stimulation (Fig.
2D).
To determine whether MAPK activation was required for BBS-induced increases in
COX-2 mRNA levels, cells were pretreated with selective inhibitors of MEK (PD98059) and
p38MAPK (SB203580). PD98059 (10 µM) and SB203580 (5-20 µM) significantly, but
incompletely, inhibited the BBS-stimulated increases in COX-2 mRNA levels (Fig. 2E, F).
Although the indicated concentrations of PD98059 and SB203580 were sufficient to completely
inhibit agonist-dependent kinase activation, the inhibition by either compound alone was
insufficient to completely block the BBS-stimulated increases in COX-2 mRNA levels,
suggesting that both MEK/ERK and p38MAPK-dependent pathways are partially involved in
GRP-R-mediated regulation of COX-2 mRNA levels.
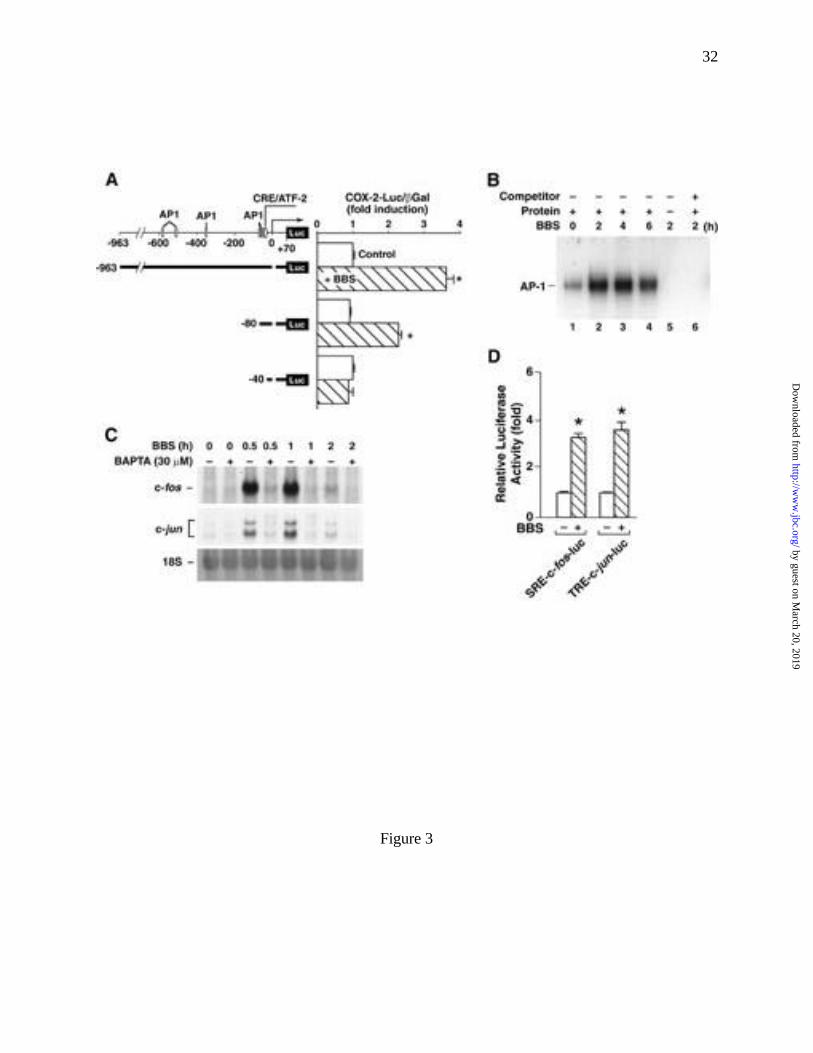
BBS stimulated COX-2 promoter activity
COX-2 expression is regulated by both transcriptional and posttranscriptional
mechanisms (32-35). To determine whether BBS regulated COX-2 promoter activity,
RIE/GRPR cells were transfected with different size fragments of the mouse COX-2 promoter
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

14
coupled to a luciferase reporter gene. BBS (100 nM) induced a 3.6-fold increase in luciferase
activity in cells transiently expressing TIS10L-luc (-963 to +30) compared with untreated control
cultures. A 2.3-fold induction was observed when using a shorter fragment of the COX-2
promoter (TIS10-80luc -80 to +30). BBS did not stimulate an increase in luciferase activity in
cells containing the shortest COX-2 promoter construct (TIS10-40luc [-40 to +30]) (Fig. 3A).
We also assessed the effects of BBS treatment on RIE/GRPR cells transfected with rat and
human COX-2 promoter/luciferase reporter constructs because differences exist in the sequences
of mouse, rat and human COX-2 promoters. Similar to the mouse promoter, BBS-induced
increases in luciferase activity were detected in cells expressing both the rat and human COX-2
reporter constructs (data not shown). Together these data demonstrated that GRP-R-mediated
signaling pathways are linked to regulation of COX-2 promoter activity in RIE/GRPR cells.
BBS activates AP-1 transcription factor regulating COX-2 expression
The COX-2 promoter contains multiple potential cis-activating regulator elements. To
date, CRE, E-box, NF-IL6 (C/EBPβ) and NF-κB transcriptional elements have been identified as
being involved in receptor-mediated COX-2 expression (32,34,36-40). Additionally, numerous
potential cis-activating consensus sequences have been identified within the COX-2 promoter,
including AP-1, AP-2, SP-1, MEF-2, STAT1 and STAT3 sites (35,41,42). The identities of the
cis-elements regulated by BBS- and GRP-R-mediated signaling pathways are unknown. In
several cell models, BBS is a potent stimulator of the activator protein-1 (AP-1) transcription
factor complex (43-45). The AP-1 complex is composed of hetero- and homodimers of the Jun
and Fos families of transcription factors, which bind to a specific DNA consensus sequence
(TGA(C/G)TCA) (46). Electrophoretic mobility shift assay (EMSA), using an end-labeled
oligonucleotide probe containing the AP-1 consensus binding sequence, showed an increase in
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

15
AP-1 binding activity in nuclear proteins extracts of RIE/GRPR cells following treatment with
BBS (Fig. 3B). The BBS-stimulated increases in AP-1 binding activity were detected by 2 h,
reached a maximum at 4 h and decreased thereafter (Fig. 3B, lanes 2-4). Preceding the increase
in AP-1 binding activity was a BBS-stimulated increase in both c-fos and c-jun mRNA
expression. BBS induces a transient (time- and Ca2+-dependent) increase in c-Fos and c-Jun
mRNA levels (Fig. 3C). Steady-state mRNA levels of both transcription factors were increased
by 0.5 h, peaked by 1 h and then returned to near baseline levels by 2 h. Like BBS-regulation of
COX-2 mRNA levels, the agonist-stimulated increase in c-Fos and c-Jun mRNA were inhibited
by cells pretreated with AM-BAPTA (30 µM) (Fig. 3C). Additionally, RIE/GRPR cells
transfected with either 5�-promoter sequences of c-fos or c-jun coupled to the luciferase reporter
gene showed a 3.3-fold and 3.5-fold increase in luciferase activity compared to untreated control
cells, respectively (Fig. 3D). Together these data indicate that GRP-R activation stimulates Fos
and Jun expression in RIE/GRPR cells through the activation of their respective promoters.
Expression of the c-fos and c-jun genes are regulated, in part, by ternary complex factors
(TCFs) and ATF-2, respectively. TCFs belong to the ets-domain family of DNA binding
proteins which includes Elk-1, Sap1 and Sap2. Phosphorylation of Elk-1 by MAPKs increases
its ability to form complexes with serum response factor (SRF) and results in serum response
element (SRE)-dependent activation of the c-fos promoter (47,48). The phosphorylation of
ATF-2 by p38MAPK increases TPA-response element (TRE)-dependent transcriptional activity of
c-jun (48). To determine whether these transcriptional factors were involved in BBS signaling,
we examined the effect of BBS on their phosphorylation state using antibodies directed against
the phosphorylated (activated) forms of Elk-1 and ATF-2. An increase in phosphorylated Elk-1
and ATF-2 was detected 5 min and 15 min after BBS treatment and continued for 60 min and 30
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

16
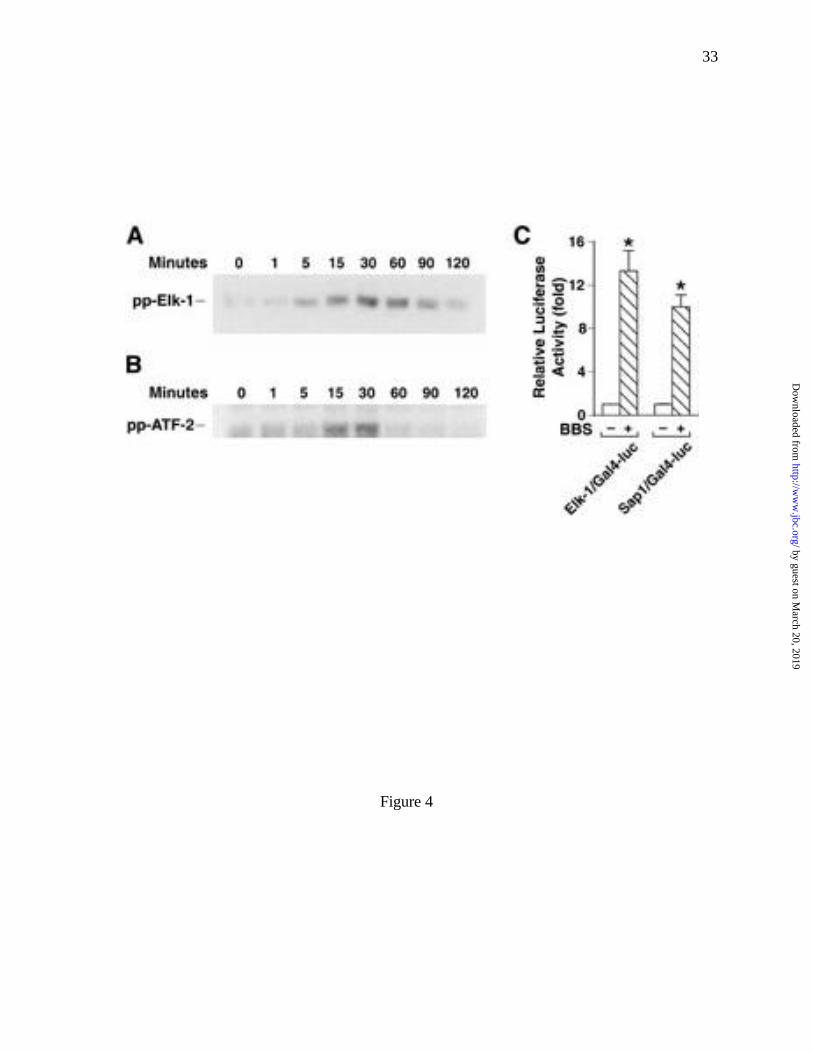
min, respectively (Fig. 4A, B). Additionally, BBS increased the promoter activities of Elk-1 and
Sap-1 by 12-fold and 9-fold, respectively (Fig. 4C).
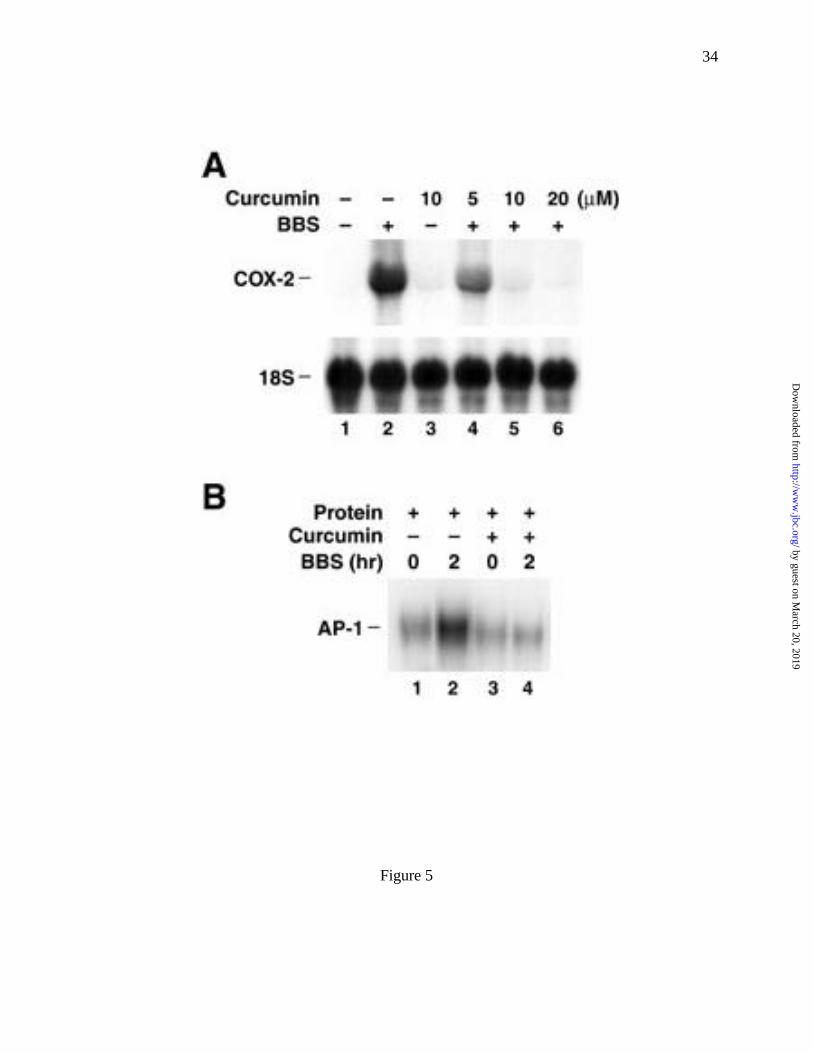
To assess the role of AP-1 activation in agonist-stimulated COX-2 expression, we treated
RIE/GRPR cells with diferulolymethane (curcumin). Curcumin is an inhibitor of AP-1 binding
(49-51). RIE/GRPR cells were preincubated with or without curcumin for 1 h, and then
stimulated with BBS (100 nM) for an additional 2 h. BBS-induced increases in COX-2 mRNA
levels were inhibited in a dose-dependent manner by curcumin (Fig. 5A), with complete
inhibition at 10 µM. In addition, we found that 10 µM curcumin completely inhibited BBS-
stimulated increases in AP-1 binding activity (Fig. 5B). Together these data demonstrate that
BBS stimulation of AP-1 binding is an important intermediate in its regulation of COX-2 gene
expression in the intestinal epithelial cell line, RIE/GRPR.
DISCUSSION
The aberrant overexpression of COX-2, BBS-like peptides and GRP-R have been
demonstrated in various carcinomas, including lung, pancreatic, gastric, breast, prostate and
colorectal carcinomas (2,36,37,52-61). While a growing body of experimental evidence suggests
COX-2 plays an important role in the development of colorectal carcinogenesis, little is known
about the molecular mechanisms leading to its upregulation. Recent data from mouse Swiss 3T3
fibroblasts showing GRP-R activation results in increased COX-2 (11) and aspirin inhibits BBS-
stimulated DNA synthesis (10) expression suggest that the aberrant overexpression of GRP-R
and COX-2 in some adenomatous polyps and colorectal cancers may be more than coincidental.
To evaluate potential mechanistic links between GRP-R-mediated signaling pathways and the
regulation of COX-2 expression in an intestinal epithelial cell line, we developed the RIE/GRPR
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

17
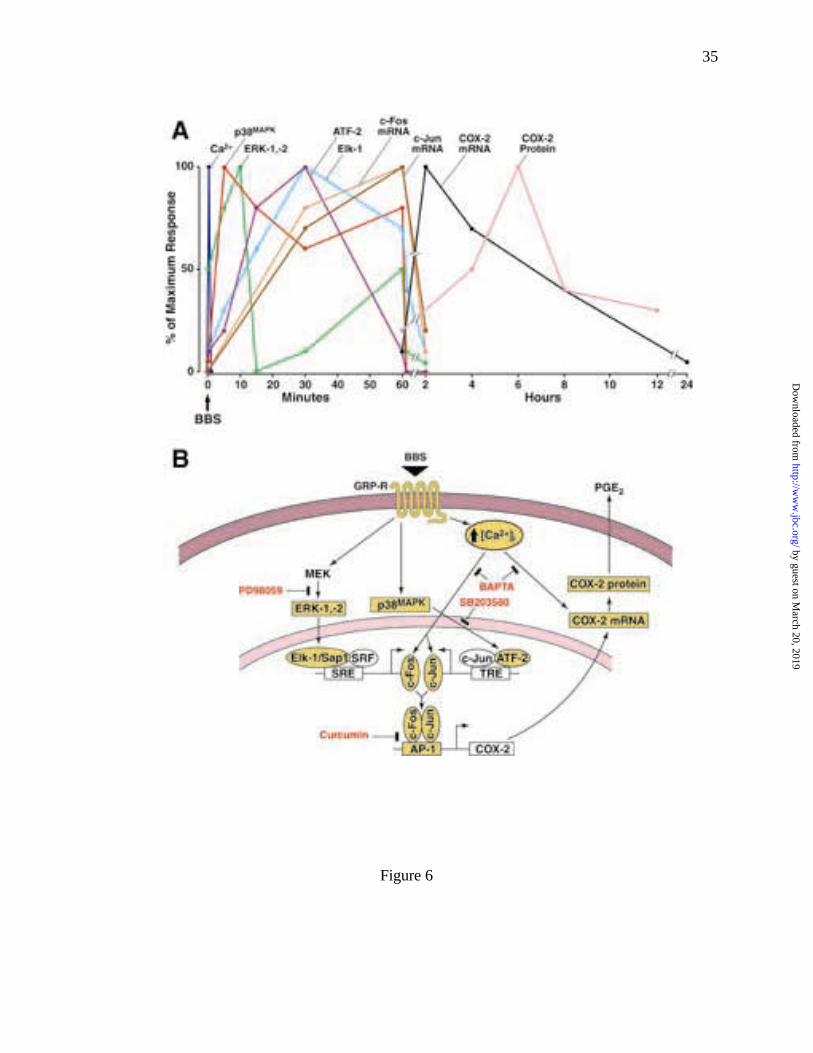
cell lines. In this cell model, we found that the GRP-R agonist, BBS, is a potent stimulator of
COX-2 expression. The data presented in this report allow us to partially define the temporal
sequence of molecular events involved in BBS stimulation of COX-2 expression in RIE/GRPR
cells (Fig. 6A). Agonist binding to GRP-R stimulates a rapid and transient increase in [Ca2+]i,
followed by the slower, transient activation of the MAPKs: ERK-1, -2 and p38MAPK. The
activation of both ERKs and p38MAPK occurred within 1 min of BBS stimulation. The levels of
phosphorylated ERK-1 and -2 returned to baseline by 15 min, whereas p38MAPK remained
activated for up to 60 min. A second smaller increase in ERK activity was observed at 60 min.
Subsequent to ERK activation, but within the period of elevated p38MAPK, the levels of c-Fos and
c-Jun mRNA increased. The increases in c-Fos and c-Jun mRNA were preceded by increased
activation (phosphorylation) of the transcription factors Elk-1 and ATF-2, regulators of the SRE
and TRE transcriptional elements, respectively. The observed temporal sequence of changing
gene expression and protein activation, coupled with the effects of various selective inhibitors,
suggests the model of GRP-R-mediated regulation of COX-2 gene expression depicted in Figure
6B.
Mitogen-activated protein kinase pathways mediate the stimulatory effects of different
extracellular stimuli on COX-2 expression in a stimulus- and cell-type specific manner. We
have shown that BBS stimulation of COX-2 expression in RIE/GRPR cells involves both ERK
and p38MAPK pathways, but not the JNK pathway. Similarly, ERK and p38MAPK pathways
mediate the induction of COX-2 expression by transforming growth factor (TGF)-α and
interferon-γ in human epidermal keratinocytes and squamous carcinoma cells (62). Whereas
JNK and p38MAPK pathways regulate interleukin (IL)-1β-stimulated COX-2 expression in renal
mesangial cells (63), all three MAPK cascades (ERK, JNK and p38MAPK) are involved in the
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

18
induction of COX-2 expression by physiologic hypertonicity in renal medullary collecting duct
cells (29). Regulation of COX-2 by PDGF is mediated through ERK and JNK pathways in NIH
3T3 cells (42), but ERK-2 is required for oxytocin-stimulated PGE2 synthesis in uterine
endometrial and amnion cells (64). Together these studies demonstrate the central role MAPK
cascades play in regulation of COX-2 expression to a variety of extracellular stimuli.
We have found that the increase of the steady-state level of COX-2 by BBS is regulated
by a transcriptional mechanism (Fig. 3A). Furthermore, BBS-mediated COX-2 expression
occurs predominantly through the Ca2+/MAPK/AP-1-dependent signaling pathways. Activated
ERKs can directly phosphorylate transcription factors, such as Elk-1 and Sap-1, which then bind
to the serum-responsive element (SRE) of c-fos promoter (47,48), and p38MAPK phosphorylates
ATF-2, which binds to TRE within the c-Jun promoter. We show that BBS stimulated an
increase in the phosphorylation of Elk-1 and ATF-2, which precedes an increase in the steady-
state levels of c-Fos and c-Jun mRNA and AP-1 binding activity. Inhibition of BBS-induced
increases in [Ca2+]i blocked increases in both COX-2 mRNA levels and levels of c-Fos and c-Jun
mRNA (Figs. 2B, 3C), as well as AP-1 binding (data not shown). Furthermore, inhibition of
ERK and p38MAPK activation suppressed AP-1 binding and COX-2 expression. Finally,
inhibition of AP-1 binding with curcumin blocked BBS-stimulated increases in COX-2 mRNA
levels. These findings indicate that the stimulation of COX-2 expression by BBS is mediated, in
large part, by an AP-1 transcription factor. Our findings are supported by studies showing AP-1
activation mediates the induction of COX-2 in response to stimulation with superoxide,
lipopolysaccharide (LPS), IL-1β and nitric oxide in RAW264.7 mouse macrophages and human
pulmonary type II A549 epithelial cells, and to platelet-activating factor and IL-1 in human
epidermal keratinocytes (65-68).
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

19
Posttranscriptional mechanisms can also play a role in the regulation of COX-2
expression (34,35). The stability of COX-2 mRNA is mediated by sequences within its 3′-
untranslated regions (3′-UTR) (41). In RIE-1 cells, TGF-β1 enhanced Ha-ras-induced COX-2
expression via stabilization of COX-2 3′-UTR (35). In human monocytes, the increase of
COX-2 induction by LPS was due to p38MAPK stabilizing COX-2 mRNA (34). Lasa et al (41)
recently reported that only 123 nucleotides immediately 3′ to the translation termination condon
are required for the regulation of mRNA stability by p38MAPK. Although the mechanism of
p38MAPK-mediated COX-2 mRNA stabilization is unclear, the p38MAPK inhibitor (SB203580) has
been shown to decrease the cellular half-life of COX-2 mRNA (34,41). We found in RIE/GRPR
cells that BBS induced a significant increase in p38MAPK activation; however, SB203580
treatment did not decrease the half-life of the agonist-induced increases in COX-2 mRNA levels
(data not shown), suggesting that BBS-induced p38MAPK activation regulates COX-2
transcription rather than mRNA stability. Which pathway is favored, transcription vs. message
stability, may be dependent on the precise kinetics of the p38MAPK activation. The extent and
duration of p38MAPK activation in response to a specific stimuli may dictate whether pathways
involved in the transcriptional regulation of COX-2 expression are favored over pathways
regulating the stability of COX-2 mRNA.
In summary, our results demonstrated that BBS is a potent inducer of COX-2 expression
in intestinal epithelial cells, and this action is mediated, at least in part, by Ca2+/MAPK/AP-1-
dependent signaling pathways. These data suggest regulation of COX-2 gene expression has a
potential mechanism by which aberrantly expressed GRP-R plays a role in colorectal
carcinogenesis.
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

20
ACKNOWLEDGMENTS
We thank Dr. Kenneth D. Brown for the gift of the RIE-1 cell line, and Drs. James F.
Battey, Harvey R. Herschman, Ralf Janknecht, Johannes L. Bos and Joan Massagué for
providing plasmid constructs. We also wish to thank Jell H. Hsieh and Kirk L. Ives for their
technical support and Eileen Figueroa, Karen Martin, and Steve Schuenke for the preparation of
this manuscript.
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

21
REFERENCES
1. Greenlee, R. T., Murray, T., Bolden, S., and Wingo, P. A. (2000) CA Cancer J Clin
50(1), 7-33
2. Eberhart, C. E., Coffey, R. J., Radhika, A., Giardiello, F. M., Ferrenbach, S., and DuBois,
R. N. (1994) Gastroenterology 107(4), 1183-8
3. Moghimzadeh, E., Ekman, R., Hakanson, R., Yanaihara, N., and Sundler, F. (1983)
Neuroscience 10(2), 553-63
4. Jacoby, R. F., Marshall, D. J., Newton, M. A., Novakovic, K., Tutsch, K., Cole, C. E.,
Lubet, R. A., Kelloff, G. J., Verma, A., Moser, A. R., and Dove, W. F. (1996) Cancer
Res 56(4), 710-4
5. Boolbol, S. K., Dannenberg, A. J., Chadburn, A., Martucci, C., Guo, X. J., Ramonetti, J.
T., Abreu-Goris, M., Newmark, H. L., Lipkin, M. L., DeCosse, J. J., and Bertagnolli, M.
M. (1996) Cancer Res 56(11), 2556-60
6. Beazer-Barclay, Y., Levy, D. B., Moser, A. R., Dove, W. F., Hamilton, S. R., Vogelstein,
B., and Kinzler, K. W. (1996) Carcinogenesis 17(8), 1757-60.
7. Oshima, M., Dinchuk, J. E., Kargman, S. L., Oshima, H., Hancock, B., Kwong, E.,
Trzaskos, J. M., Evans, J. F., and Taketo, M. M. (1996) Cell 87(5), 803-9
8. Preston, S. R., Miller, G. V., and Primrose, J. N. (1996) Crit Rev Oncol Hematol 23(3),
225-38
9. Carroll, R. E., Matkowskyj, K. A., Chakrabarti, S., McDonald, T. J., and Benya, R. V.
(1999) Am J Physiol 276(3 Pt 1), G655-65
10. Castano, E., Dalmau, M., Marti, M., Berrocal, F., Bartrons, R., and Gil, J. (1997) J
Pharmacol Exp Ther 280(1), 366-72
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

22
11. Hecht, J. R., Duque, J., Reddy, S. T., Herschman, H. R., Walsh, J. H., and Slice, L. W.
(1997) Prostaglandins 54(5), 757-68
12. Tsujii, M., and DuBois, R. N. (1995) Cell 83(3), 493-501
13. Guo, Y. S., Jin, G. F., Houston, C. W., Thompson, J. C., and Townsend, C. M., Jr. (1998)
J Cell Physiol 175(2), 141-8
14. Guo, Y. S., Jin, G. F., Townsend, C. M., Zhang, T., Sheng, H. M., Beauchamp, R. D., and
Thompson, J. C. (1995) J Am Coll Surg 181(2), 145-54.
15. Hellmich, M. R., Ives, K. L., Udupi, V., Soloff, M. S., Greeley, G. H., Christensen, B. N.,
and Townsend, C. M. (1999) J Biol Chem 274(34), 23901-9.
16. Chomczynski, P., and Sacchi, N. (1987) Anal Biochem 162(1), 156-9
17. Evers, B. M., Wang, X., Zhou, Z., Townsend, C. M., Jr., McNeil, G. P., and Dobner, P.
R. (1995) Mol Cell Biol 15(7), 3870-81
18. Shapiro, D. J., Sharp, P. A., Wahli, W. W., and Keller, M. J. (1988) DNA 7(1), 47-55
19. Blay, J., and Brown, K. D. (1984) Cell Biol Int Rep 8(7), 551-60
20. Kroog, G. S., Jensen, R. T., and Battey, J. F. (1995) Med Res Rev 15(5), 389-417
21. Rozengurt, E. (1998) J Cell Physiol 177(4), 507-17
22. Hellmich, M. R., Battey, J. F., and Northup, J. K. (1997) Proc Natl Acad Sci U S A 94(2),
751-6.
23. Jian, X., Sainz, E., Clark, W. A., Jensen, R. T., Battey, J. F., and Northup, J. K. (1999) J
Biol Chem 274(17), 11573-81
24. Zugaza, J. L., Waldron, R. T., Sinnett-Smith, J., and Rozengurt, E. (1997) J Biol Chem
272(38), 23952-60.
25. Rodriguez-Fernandez, J. L., and Rozengurt, E. (1996) J Biol Chem 271(44), 27895-901
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

23
26. Pang, L., Decker, S. J., and Saltiel, A. R. (1993) Biochem J 289(Pt 1), 283-7
27. Cheng, H. F., Wang, J. L., Zhang, M. Z., McKanna, J. A., and Harris, R. C. (2000) J Clin
Invest 106(5), 681-8
28. Molina-Holgado, E., Ortiz, S., Molina-Holgado, F., and Guaza, C. (2000) Br J
Pharmacol 131(1), 152-9
29. Yang, T., Huang, Y., Heasley, L. E., Berl, T., Schnermann, J. B., and Briggs, J. P. (2000)
J Biol Chem 275(30), 23281-6
30. Seger, R., and Krebs, E. G. (1995) Faseb J 9(9), 726-35.
31. Davis, R. J. (1994) Trends Biochem Sci 19(11), 470-3
32. Xie, W., Fletcher, B. S., Andersen, R. D., and Herschman, H. R. (1994) Mol Cell Biol
14(10), 6531-9
33. Yamamoto, K., Arakawa, T., Ueda, N., and Yamamoto, S. (1995) J Biol Chem 270(52),
31315-20
34. Dean, J. L., Brook, M., Clark, A. R., and Saklatvala, J. (1999) J Biol Chem 274(1), 264-9
35. Sheng, H., Shao, J., Dixon, D. A., Williams, C. S., Prescott, S. M., DuBois, R. N., and
Beauchamp, R. D. (2000) J Biol Chem 275(9), 6628-35
36. Wolff, H., Saukkonen, K., Anttila, S., Karjalainen, A., Vainio, H., and Ristimaki, A.
(1998) Cancer Res 58(22), 4997-5001
37. Chan, G., Boyle, J. O., Yang, E. K., Zhang, F., Sacks, P. G., Shah, J. P., Edelstein, D.,
Soslow, R. A., Koki, A. T., Woerner, B. M., Masferrer, J. L., and Dannenberg, A. J.
(1999) Cancer Res 59(5), 991-4
38. Tomozawa, S., Tsuno, N. H., Sunami, E., Hatano, K., Kitayama, J., Osada, T., Saito, S.,
Tsuruo, T., Shibata, Y., and Nagawa, H. (2000) Br J Cancer 83(3), 324-8
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

24
39. Fujita, T., Matsui, M., Takaku, K., Uetake, H., Ichikawa, W., Taketo, M. M., and
Sugihara, K. (1998) Cancer Res 58(21), 4823-6
40. Sawaoka, H., Tsuji, S., Tsujii, M., Gunawan, E. S., Sasaki, Y., Kawano, S., and Hori, M.
(1999) Lab Invest 79(12), 1469-77
41. Lasa, M., Mahtani, K. R., Finch, A., Brewer, G., Saklatvala, J., and Clark, A. R. (2000)
Mol Cell Biol 20(12), 4265-74
42. Xie, W., and Herschman, H. R. (1996) J Biol Chem 271(49), 31742-8
43. Reddy, S. T., Wadleigh, D. J., and Herschman, H. R. (2000) J Biol Chem 275(5), 3107-
13
44. Morris, J. K., and Richards, J. S. (1996) J Biol Chem 271(28), 16633-43
45. Murakami, M., Kambe, T., Shimbara, S., and Kudo, I. (1999) J Biol Chem 274(5), 3103-
15
46. Pennypacker, K. R., Hong, J. S., and McMillian, M. K. (1994) FASEB J 8(8), 475-8
47. Karin, M. (1995) J Biol Chem 270(28), 16483-6
48. Whitmarsh, A. J., and Davis, R. J. (1996) J Mol Med 74(10), 589-607
49. Mohan, R., Sivak, J., Ashton, P., Russo, L. A., Pham, B. Q., Kasahara, N., Raizman, M.
B., and Fini, M. E. (2000) J Biol Chem 275(14), 10405-12
50. Seol, D. W., Chen, Q., and Zarnegar, R. (2000) Oncogene 19(9), 1132-7
51. Hanazawa, S., Takeshita, A., Amano, S., Semba, T., Nirazuka, T., Katoh, H., and Kitano,
S. (1993) J Biol Chem 268(13), 9526-32
52. Williams, C. S., Mann, M., and DuBois, R. N. (1999) Oncogene 18(55), 7908-16
53. Dubois, R. N., Abramson, S. B., Crofford, L., Gupta, R. A., Simon, L. S., Van De Putte,
L. B., and Lipsky, P. E. (1998) Faseb J 12(12), 1063-73
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

25
54. Murata, H., Kawano, S., Tsuji, S., Tsuji, M., Sawaoka, H., Kimura, Y., Shiozaki, H., and
Hori, M. (1999) Am J Gastroenterol 94(2), 451-5
55. Yip-Schneider, M. T., Barnard, D. S., Billings, S. D., Cheng, L., Heilman, D. K., Lin, A.,
Marshall, S. J., Crowell, P. L., Marshall, M. S., and Sweeney, C. J. (2000)
Carcinogenesis 21(2), 139-46
56. Rolland, P. H., Martin, P. M., Jacquemier, J., Rolland, A. M., and Toga, M. (1980) J Natl
Cancer Inst 64(5), 1061-70
57. Gupta, S., Srivastava, M., Ahmad, N., Bostwick, D. G., and Mukhtar, H. (2000) Prostate
42(1), 73-8
58. Cuttitta, F., Carney, D. N., Mulshine, J., Moody, T. W., Fedorko, J., Fischler, A., and
Minna, J. D. (1985) Nature 316(6031), 823-6.
59. Giacchetti, S., Gauville, C., de Cremoux, P., Bertin, L., Berthon, P., Abita, J. P., Cuttitta,
F., and Calvo, F. (1990) Int J Cancer 46(2), 293-8.
60. Sun, B., Halmos, G., Schally, A. V., Wang, X., and Martinez, M. (2000) Prostate 42(4),
295-303.
61. Frucht, H., Gazdar, A. F., Park, J. A., Oie, H., and Jensen, R. T. (1992) Cancer Res 52(5),
1114-22.
62. Matsuura, H., Sakaue, M., Subbaramaiah, K., Kamitani, H., Eling, T. E., Dannenberg, A.
J., Tanabe, T., Inoue, H., Arata, J., and Jetten, A. M. (1999) J Biol Chem 274(41), 29138-
48
63. Guan, Z., Buckman, S. Y., Miller, B. W., Springer, L. D., and Morrison, A. R. (1998) J
Biol Chem 273(44), 28670-6
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

26
64. Strakova, Z., Copland, J. A., Lolait, S. J., and Soloff, M. S. (1998) Am J Physiol 274(4 Pt
1), E634-41
65. Lukiw, W. J., Pelaez, R. P., Martinez, J., and Bazan, N. G. (1998) Proc Natl Acad Sci U S
A 95(7), 3914-9
66. Miller, C., Zhang, M., He, Y., Zhao, J., Pelletier, J. P., Martel-Pelletier, J., and Di
Battista, J. A. (1998) J Cell Biochem 69(4), 392-413
67. von Knethen, A., Callsen, D., and Brune, B. (1999) J Immunol 163(5), 2858-66
68. von Knethen, A., and Brune, B. (2000) Biochemistry 39(6), 1532-40
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

27
FIGURE LEGENDS
Fig. 1 Bombesin (BBS) stimulated an increase in COX-2 expression and activity in
RIE/GRPR cells. (A) Time course of COX-2 mRNA expression. Sub-confluent,
serum-starved RIE/GRPR cells were treated with BBS (+)(100 nM) for the
indicated time and the steady-state levels of COX-2 mRNA expression were
determined by Northern blot. (B) Dose effect. RIE/GRPR cells were treated with
the indicated concentration of BBS for 2 h and the steady-state levels of COX-2
mRNA were assessed by Northern blot. (C) Time course of COX-2 protein
expression. RIE/GRPR cells were incubated with BBS (100 nM), lysed in RIPA
buffer at the indicated times and analyzed by Western blot as described in
Experimental Procedures. (D) Immunofluorescence staining for COX-2.
RIE/GRPR cells were cultured on glass-covered slides. After serum starving for
24 h, cells were stimulated with BBS (lower panel, BBS) for 6 h, fixed and
immunofluorescence stained as described under Experimental Procedures.
Untreated cultures are shown in the upper panel (Control). (E) BBS stimulated
the release of PGE2 from RIE/GRPR cells. The cells were plated in 24-well
plates for 36 h, serum starved for 24 h and incubated with or without BBS (100
nM) for 1 h to 24 h. Medium in each well was collected and analyzed for PGE2
levels by ELISA.
Fig. 2 Bombesin stimulation of COX-2 expression requires an increase in [Ca2+]i and the
activation of mitogen-activated protein kinases. (A) BBS (100 nM) stimulated an
increase in [Ca2+]i, which was blocked by a 1-h pretreatment of the RIE/GRPR
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

28
cells with the chelating agent AM-BAPTA (30 µM). (B) Effects of AM-BAPTA
treatment on BBS-stimulated COX-2 mRNA abundance. (C) BBS stimulated a
time-dependent increase in the levels of activated (phosphorylated) ERK-1
(ppERK-1) and ERK-2 (ppERK-2) proteins. (D) BBS stimulated a time-
dependent increase in the level of activated p38MAPK (pp-p38MAPK) protein. (E)
Inhibition of MEK with the selective inhibitor, PD988059 (10 µM), blocked the
BBS-stimulated accumulation of COX-2 mRNA. (F) Inhibition of the BBS-
induced increases in COX-2 mRNA by the selective p38MAPK inhibitor,
SB203580 (5-20 µM).
Fig. 3 BBS increased COX-2 gene transcriptional activity. (A) BBS stimulated the
COX-2 promoter activity. The left panel shows the cis-acting transcriptional
response elements of mouse COX-2 promoter and the constructs of three COX-2
promoter plasmids. The right panel shows the effect of BBS on the induction of
COX-2 promoter coupled to a luciferase reporter gene when transiently
transfected to RIE/GRPR cells. Luciferase activity (mean ± SD) from four
independent experiments is expressed relative to Control after normalizing for
differences in transfection efficiency by the β-galactosidase plasmid (CMV-β-
Gal). (B) BBS (100 nM) stimulated AP-1 activity in RIE-GRP-R cells by
electrophoretic mobility shift assay. (C) BBS elicited the mRNA abundance of
c-fos and c-jun by Northern blot analyses, and both were blocked by intracellular
Ca2+ cheletos BAPTA. (D) BBS increased promoter activities of c-fos and c-jun
measured by luciferase assays. *=p<0.05 vs. control, n=12.
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

29
Fig. 4 BBS stimulated phosphorylation of ELK-1 (A) and ATF-2 (B) in RIE/GRPR
cells, as measured by immunoblotting of cell lysates. (C) BBS increased
promoter activities of ELK-1 and Sap-1 detected by luciferase assays. *=p<0.05
vs. control, n=12.
Fig. 5 Effect of curcumin on COX-2 mRNA expression and AP-1 activity in response to
BBS. (A) curcumin, an AP-1 binding inhibitor, suppressed the BBS-evoked
COX-2 mRNA abundance in a dose-dependent fashion. (B) Curcumin (10 µM)
completely inhibited BBS-stimulated AP-1 binding activity.
Fig. 6 A summary of intracellular signaling pathways required for BBS-stimulated
COX-2 expression. (A) Time sequence of BBS induction of [Ca2+]i ERKs,
p38MAPK, Elk-1, ATF-2, c-fos, c-jun, COX-2 mRNA and COX-2 protein in
RIE/GRPR cells. (B) Intracellular transduction pathways for bombesin-evoked
COX-2 expression in RIE/GRPR cells. Activation of GRP receptor (GRP-R) by
BBS results in increased phosphorylation of mitogen-activated protein kinases
(ERKs and p38MAPK) and transcriptional factors TCF (ternary complex factor,
includes ELK-1 and Sap-1) and ATF-2; both of the latter then increase the
expression of c-fos and c-jun, respectively, and the binding activity of AP-1. The
activation of AP-1 further stimulates COX-2 promoter and increases the
expression of COX-2 mRNA and protein, as well as the release of PGE2.
Inhibitors of MEK (PD98057), p38MAPK (SB203580) and AP-1 (curcumin) all
suppress BBS-stimulated COX-2 expression. In addition, BBS also increases
[Ca2+]i, which may be required for BBS-evoked COX-2 expression.
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Yan-Shi Guo, Mark R. Hellmich, Xiao Dong Wen and Courtney M. Townsend, Jrin intestinal epithelial cells
AP-1 transcription factor mediates bombesin-stimulated cyclooxygenase-2 expression
published online April 5, 2001J. Biol. Chem.
10.1074/jbc.M101801200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on March 20, 2019
http://ww
w.jbc.org/
Dow
nloaded from