
Anemias Professor Nasir Allawi Thalassemias. Definition of Thalassemia A group of inherited...
40
Anemias Professor Nasir Allawi Thalassemias
-
Upload
grant-brooks -
Category
Documents
-
view
225 -
download
0
Transcript of Anemias Professor Nasir Allawi Thalassemias. Definition of Thalassemia A group of inherited...

Anemias
Professor Nasir Allawi
Thalassemias

Definition of Thalassemia
• A group of inherited disorders of Hemoglobin synthesis, characterized by reduced or absent synthesis of one or more of the globin chains of Hemoglobin.
• They are labeled thalassemias, if it is the alpha chain that is affected,
and thalassaemias, if it is the Beta.

β- Thalassaemia
• β-Thalassaemias are inherited defects in the rate of synthesis of β-globin chains of hemoglobin, which are widely distributed throughout the world, with considerable frequencies in certain areas particularly the Mediterranean and Middle Eastern countries, including Kurdistan and Iraq.
• β-Thalassaemia major is an important health problem throughout Iraq, including Kurdistan , where there are more than 800 registered patients in Sulaimani, in a population of about 1500000 .
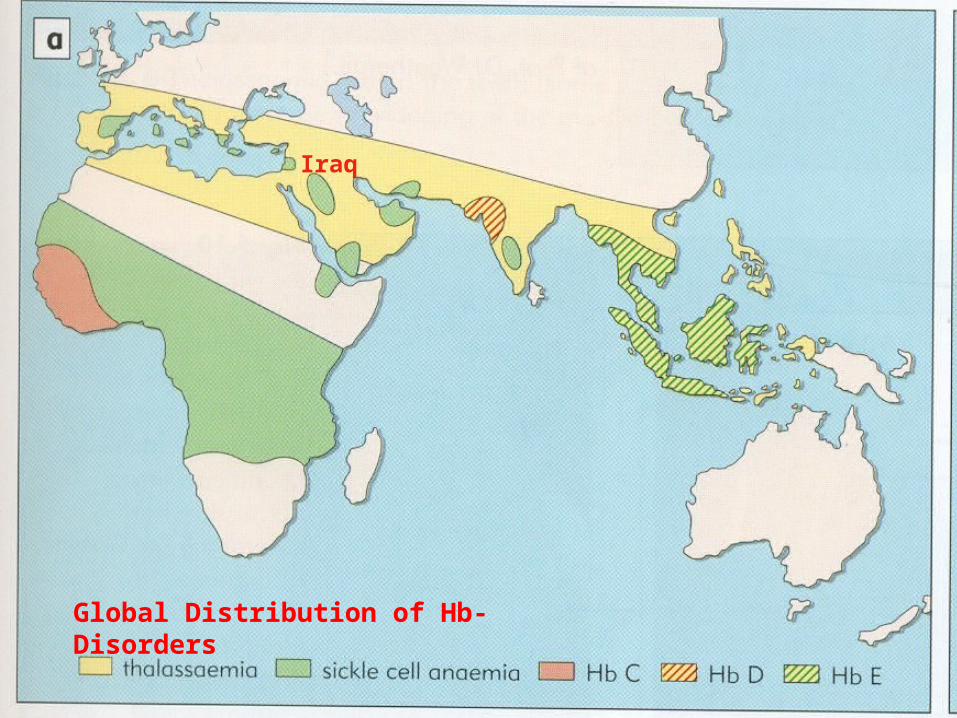
Global Distribution of Hb- Disorders
Iraq

Types of normal Hemoglobins
• All Normal Hemoglobins consists of two pairs of globin chains, at the centre of each is one heme group.
• Hb A ( Adult Hb) : 2 2 (~96%).
• Hb F (Fetal Hb) : 2 2 (<1.0%).
• Hb A2 (minor Adult Hb) : 2 2 (1.8-3.5%).

Types of Normal Hb
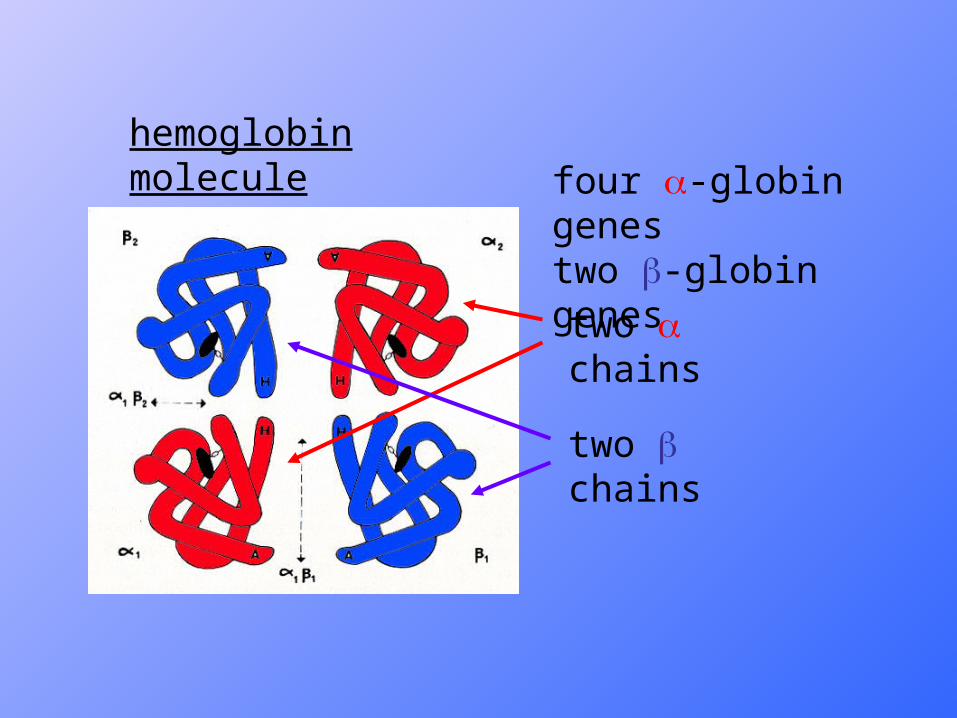
hemoglobin molecule
two chains
two chains
four -globin genestwo -globin genes

Thalassaemia
• This is one of the most common inherited hematological disorders in Iraq.
• It was estimated that around 4.4-6.5% of the population of the country carry thalassemia genes (Baghdad : 4.4%, Basra : 4.6%, Mousel : 6.5%, Duhuk 3.7%, Sulaimani 4.1%).

Genetics of thalassemia
• There is one β- globin gene on each chromosome 11 in human genome.
• This form of thalassaemia is mostly caused by point mutations involving various points in and around the beta globin gene.
• The inheritance of this disorder is autosomal recessive, so that heterozygous are usually symptomless, while homozygotes are severely or moderately affected.
• β0 denotes absent β chain synthesis, while β+ means reduced synthesis of β chain .

Clinically β thalassaemia could be classified into :
• β Thalassaemia Major : Severe clinical manifestations presenting before the age of
2 years, usually transfusion dependent. Due usually to homozygosity to β thalassemic gene defect (β0 β0, β+ β+, β0 β+) .
• β Thalassaemia minor : Mild or no clinical manifestations, usually does not require
specific management. Due usually to heterozygosity to β thalassemia gene defect(β0β or β+ β) .
• β Thalassaemia Intermediate : Moderate manifestations, intermediate between major and
minor.
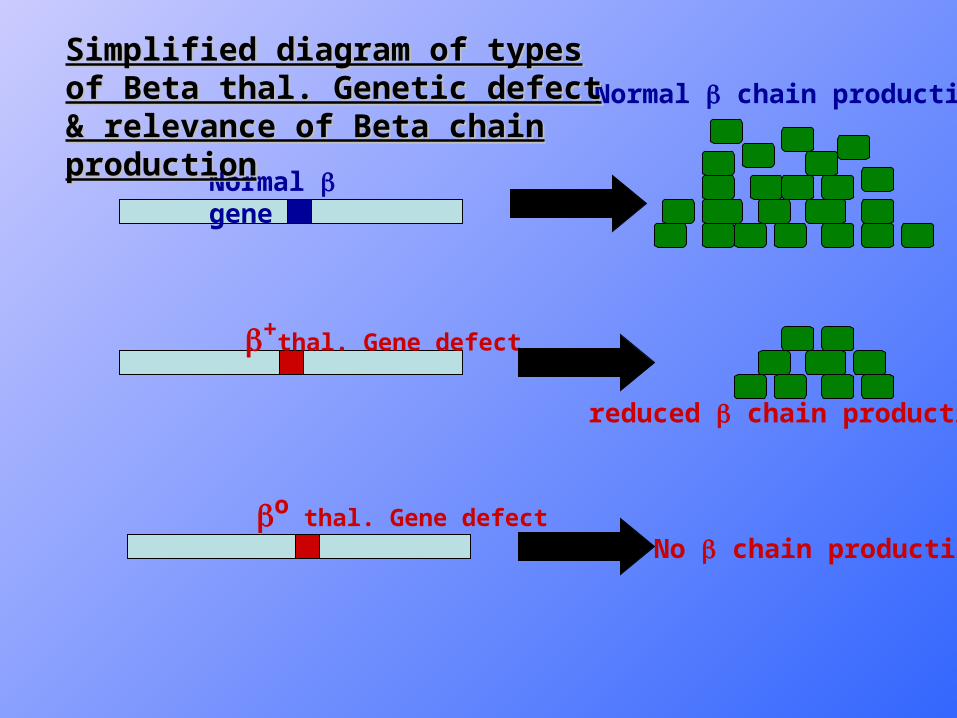
Normal gene
+thal. Gene defect
o thal. Gene defect
Normal chain production
reduced chain production
No chain production
Simplified diagram of types of Beta thal. Simplified diagram of types of Beta thal. Genetic defect & relevance of Beta chain Genetic defect & relevance of Beta chain productionproduction
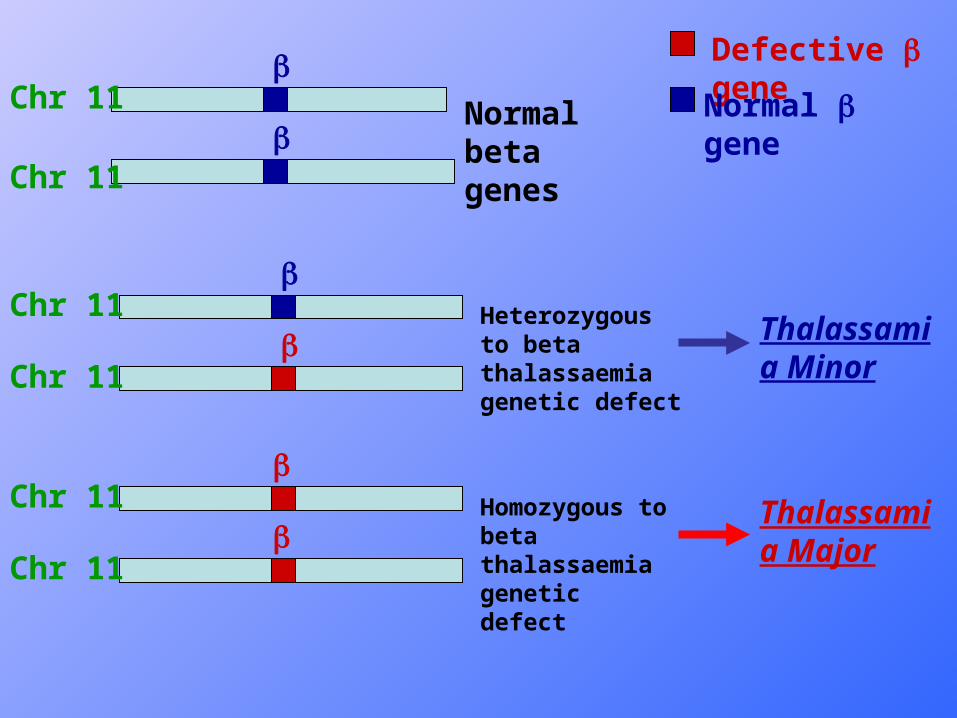
Defective gene
Normal geneNormal beta genes
Heterozygous to beta thalassaemia genetic defect
Homozygous to beta thalassaemia genetic defect
Thalassamia Minor
Thalassamia Major
Chr 11
Chr 11
Chr 11
Chr 11
Chr 11
Chr 11

Clinical features of β thalassaemia Major
• First diagnosis between age of 6 months and 2 years.
• Presentation usually with pallor, poor feeding, failure to thrive , abdominal swelling (due to hepato-splenomegaly) and sometimes Jaundice.
- Deformities in the skull due to bone marrow expansion (Bossing , and mongoloid facies; hair-on-end appearance on skull X-ray).
- Also dental problems and inadequate drainage of sinuses and middle ear, leading to chronic sinusitis and deafness
- Increased frequency of infections.

Clinical features – thal. major

Bossing of the skull- thal. major

Hair-on-end sign on skull X ray in thal. major
Widening of Diploic space – Skull

Blood Picture in Thalassaemia major
• Complete Blood Picture (CBP)• Hemoglobin is usually 3-7 g/dl.• Moderate to sever hypochromic microcytic anemia, with
marked anisopoikylocytosis.• PCV is evidently reduced.• MCV and MCH are both reduced.• Leucocytes : Maybe normal or increased.• Plateletes : may be normal or increased.• Reticulocytes : usually range 2-8%.• Hb electrophoresis:increased Hb F 10-98%. Hb A2 is
variable.
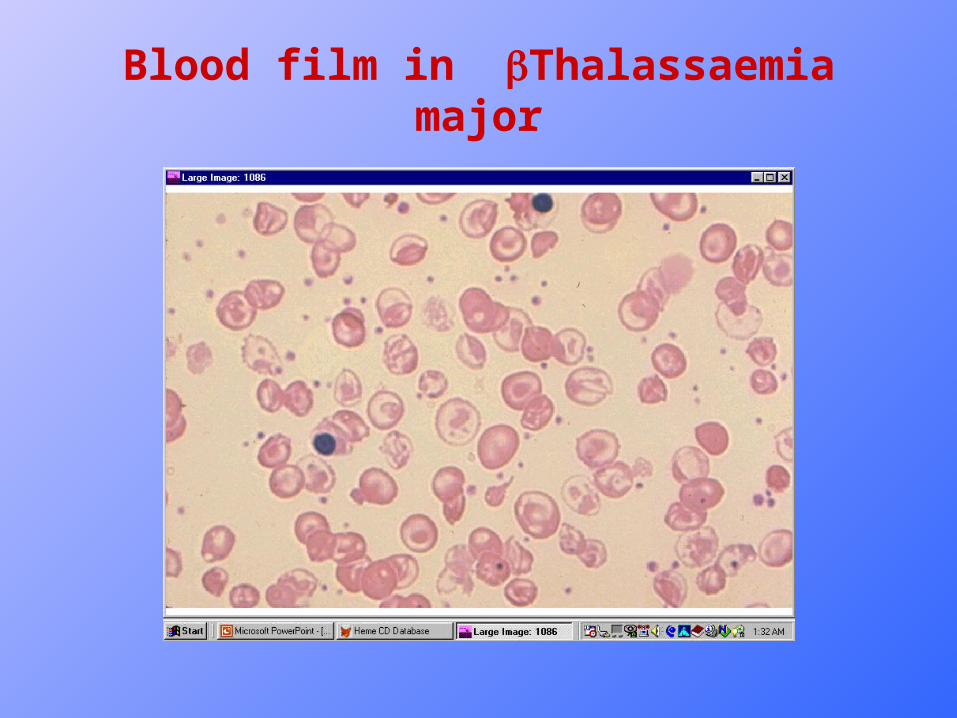
Blood film in Thalassaemia major

Iron Overload

Prognosis in thal major
• If no Transfusions, death usually occurs in the first few years of life.
• If iron overload is allowed to occur then death in 2nd or early third decade, most commonly due to progressive cardiac damage due to iron deposition, with heart failure or arrhythmias, often precipitated by infections.
• However if measures to prevent Iron overload by Iron Chelation are instituted early on, with the transfusion, Iron overload consequences may be limited, although delayed puberty and stunted growth may still be encountered, but otherwise patients may develop normally.

Blood Picture of thal. minor
• Hemoglobin is usually reduced 1-2 g/dl less than normal for age and sex.
• MCH and MCV are reduced.
• RBC count is > 5 x 1012/L in 85% of cases.
• Reticulocyte count is slightly increased or normal.
• Blood film : slight hypochromia, anisocytosis, poikiocytosis, microcytosis, tear drop cells and target cells.

Diagnostic tests in thal minor:
• Increase in Hb A2 : Normal range of Hb A2 is 1.8-3.5%, in Beta thalassemia minor it is increased to 4-7% .
• Increased Hb A2 is considered diagnostic of Beta thalassemia minor.
• S. Transferrin saturation(S.Iron/TIBC) is usually normal or upper normal.
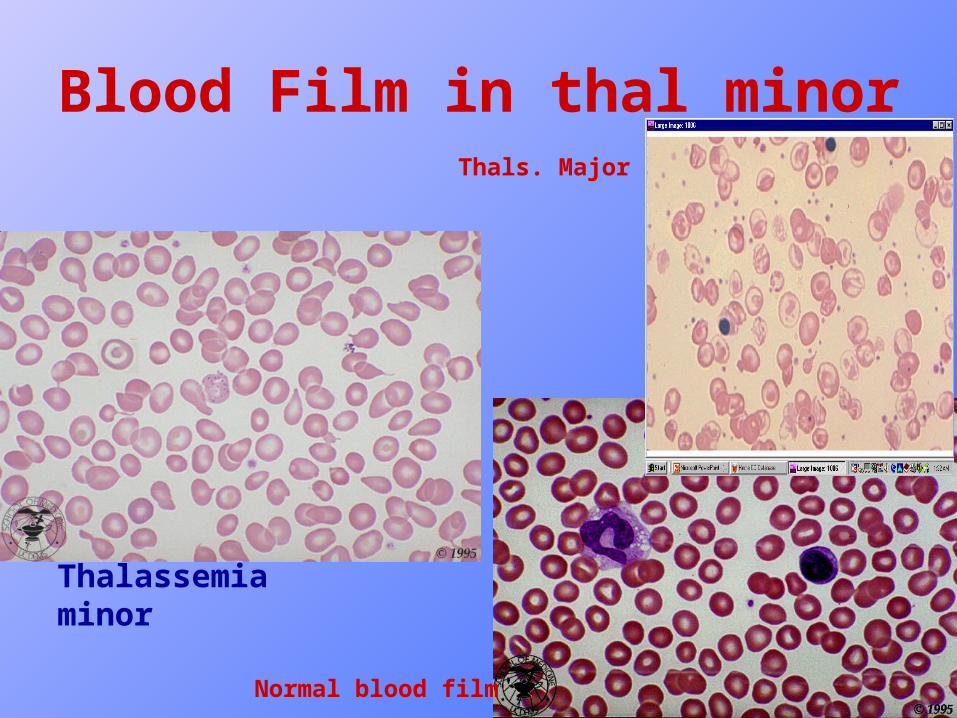
Blood Film in thal minor
Thalassemia minor
Normal blood film
Normal blood film
Thals. Major

Alpha thalassaemias
• Much less common in our country than Beta thalassemias, and of much less clinical significance.
• Due to reduced or absent synthesis of alpha () globin chains of hemoglobin.
• (Alpha () chains are constituents of all three normal Hb A, A2 and F).

Genetics of Alpha thalassemias
• There are two genes on each of chromosome 16, so there is a total of 4 genes in the human genome.
• The defects leading to alpha thalassemias are usually deletions removing one or both alpha genes.
o defects : if both genes are deleted so no alpha chain production by chr. 16.
+ defect : if only one of the genes is deleted, so the production of chains is reduced.
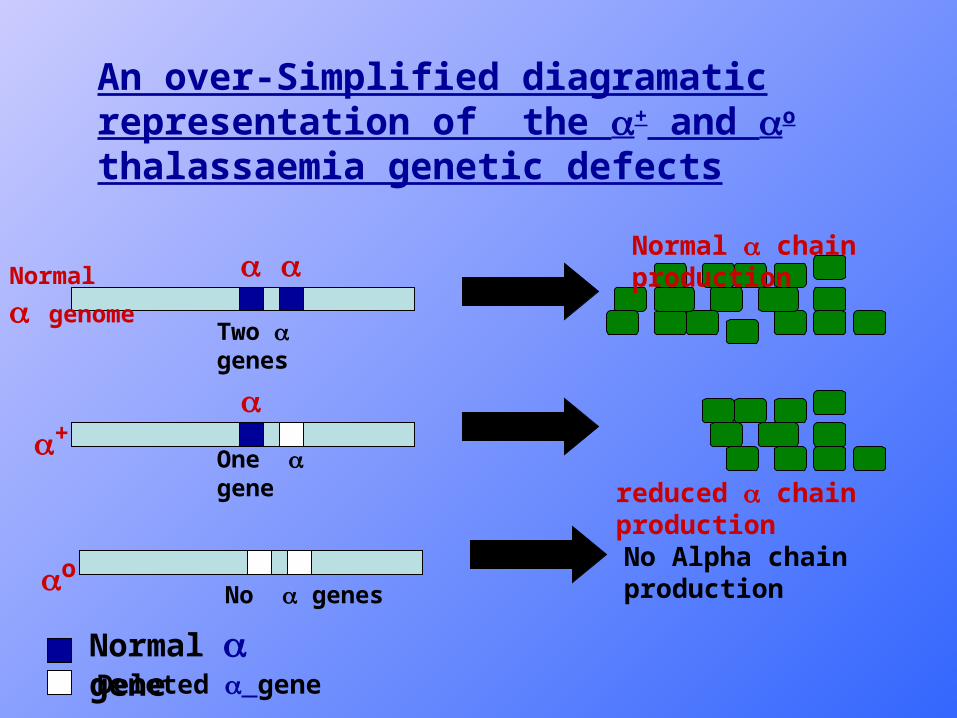
Normal chain production
reduced chain production
+
o
Normal genome
No Alpha chain production
An over-Simplified diagramatic representation of the + and o thalassaemia genetic defects
Normal geneDeleted gene
Two genes
One gene
No genes
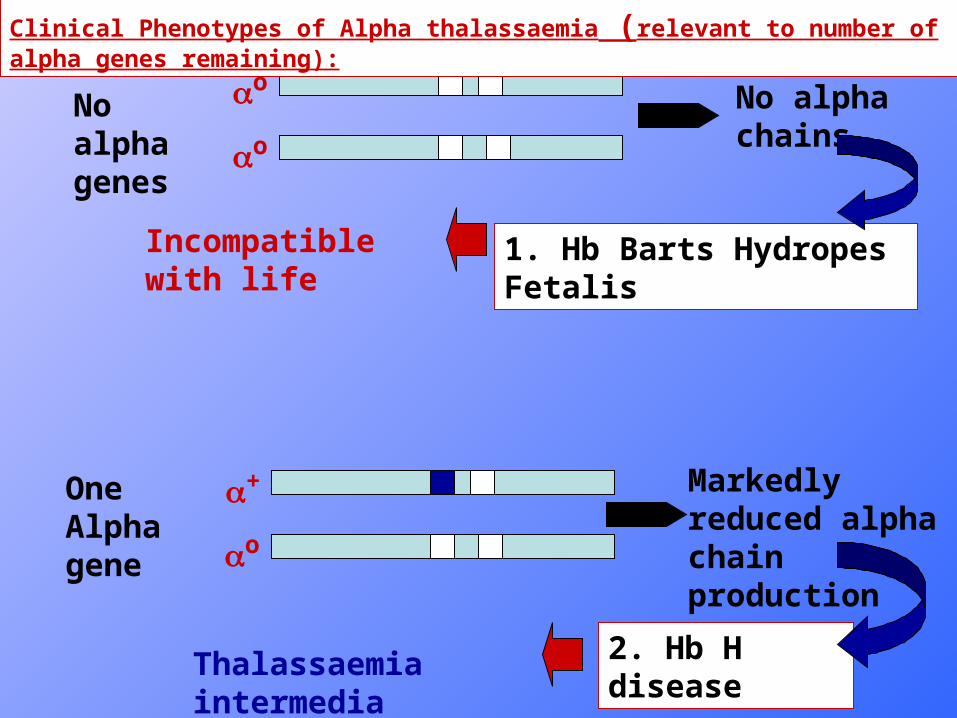
o
oNo alpha genes
No alpha chains
1. Hb Barts Hydropes Fetalis
+
o
One Alpha gene
Markedly reduced alpha chain production
2. Hb H disease
Incompatible with life
Clinical Phenotypes of Alpha thalassaemia (relevant to number of alpha genes remaining):
Thalassaemia intermedia phenotype
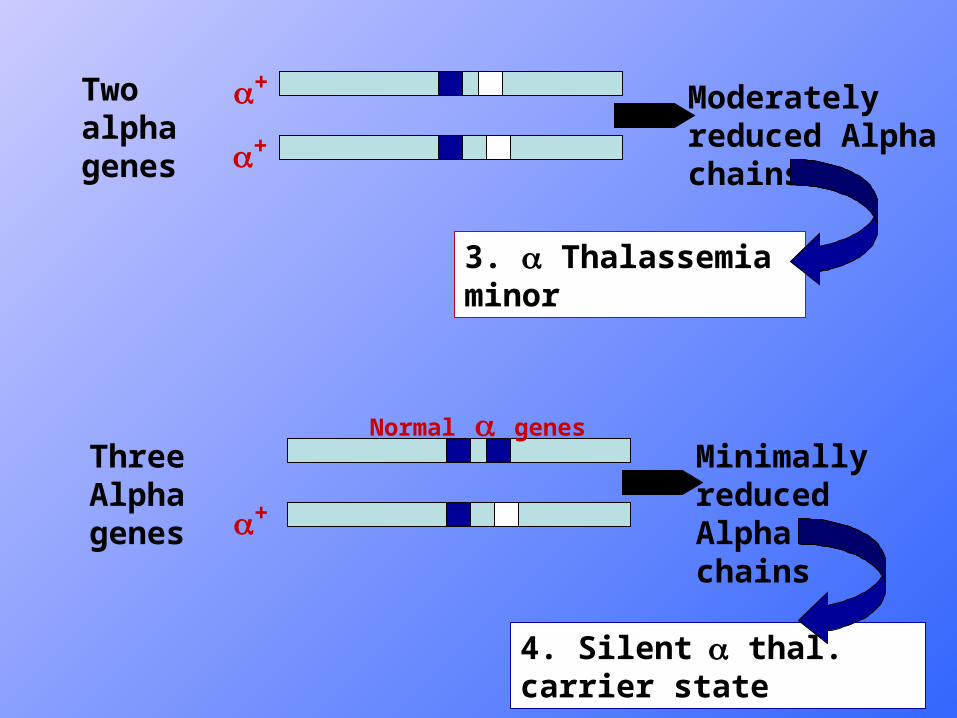
Two alpha genes
Three Alpha genes
Moderately reduced Alpha chains
Minimally reduced Alpha chains
3. Thalassemia minor
4. Silent thal. carrier state
+
+
+
Normal genes

Hb Bart’s Hydrops Fetalis
• Common only in SEA.• Genetics : Due to inheritance of o defect from both parents, so (--/--).
So no Hb F but 4 (Hb Barts) which is ineffective as an Oxygen carrier.
• Clinical : Death in utero, or within hours of birth.CBPSever hypochromic anemia with marked
anisopoikylocytosis.Hb electrophoresis;• Electrophoresis : ~ 80% Hb Barts (4),
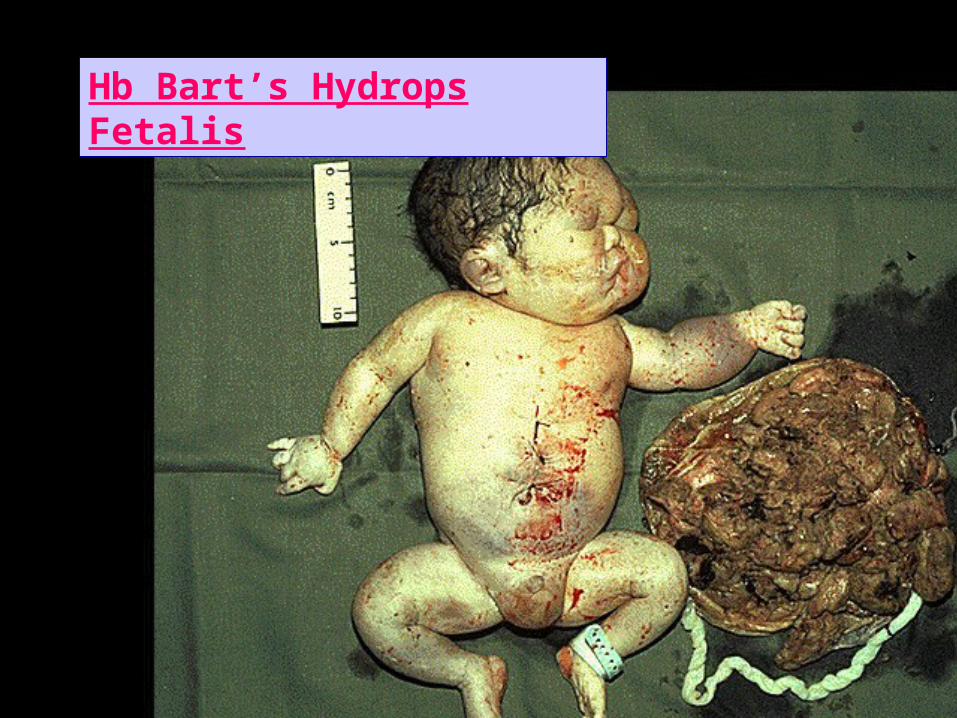
Hb Bart’s Hydrops Fetalis

Hemoglobin H disease
• Common in South East Asia, less so in Mediterranean countries. Sporadic in Iraq.
• The only clinical phenotype of alpha thalassaemia of clinical significance.
• Due to deletion of three of the four normal alpha genes… so only one functional alpha gene is left with associated marked reduction in alpha chain production.
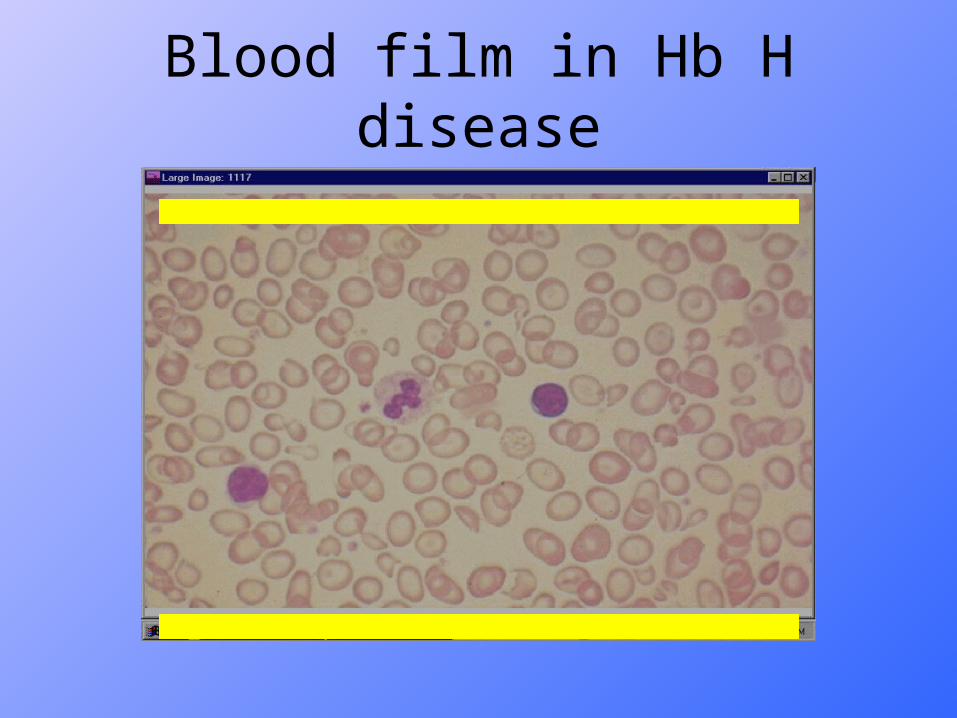
Blood film in Hb H disease

Clinical Features• Very variable, variable pallor.• Variable degrees of splenomegaly.• Sometimes Jaundice.• Most unusual to see severe thalassaemic skeletal changes or growth
retardation.• Usually survive to adult life.• Anemia aggravated by infections, oxidant drugs.CBPSever hypochromic anemia with marked anisopoikylocytosis.Hb electrophoresis;• Electrophoresis : ~ Shows Hb A with 5-40% Hb H.• On modification of the retics stain : characteristic Hb H
inclusions could be seen in RBCs( Golf ball appearance).
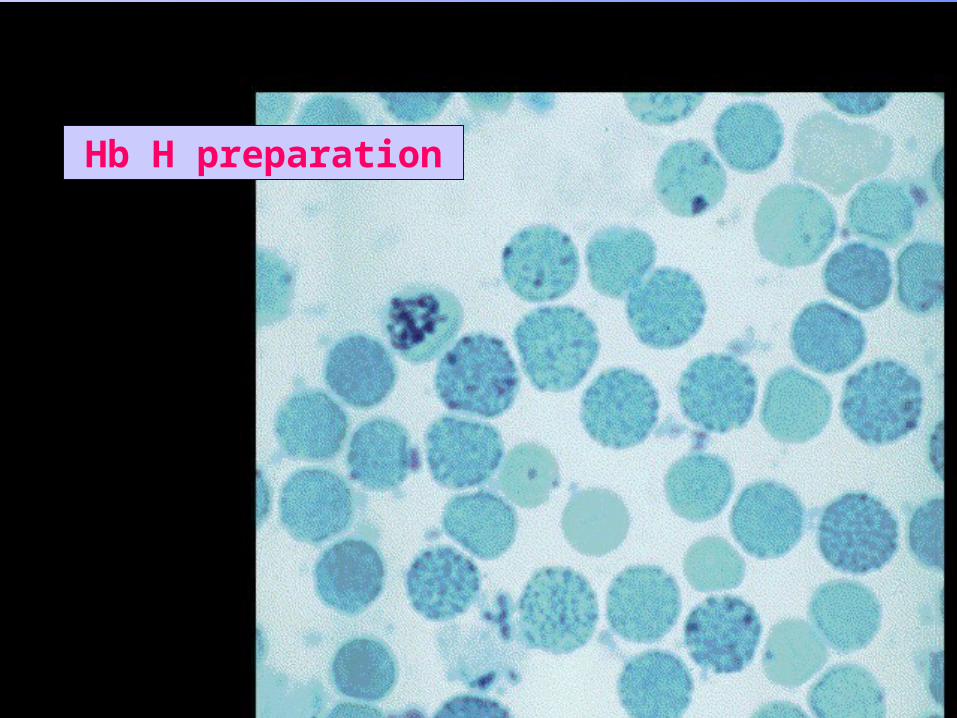
Hb H preparation
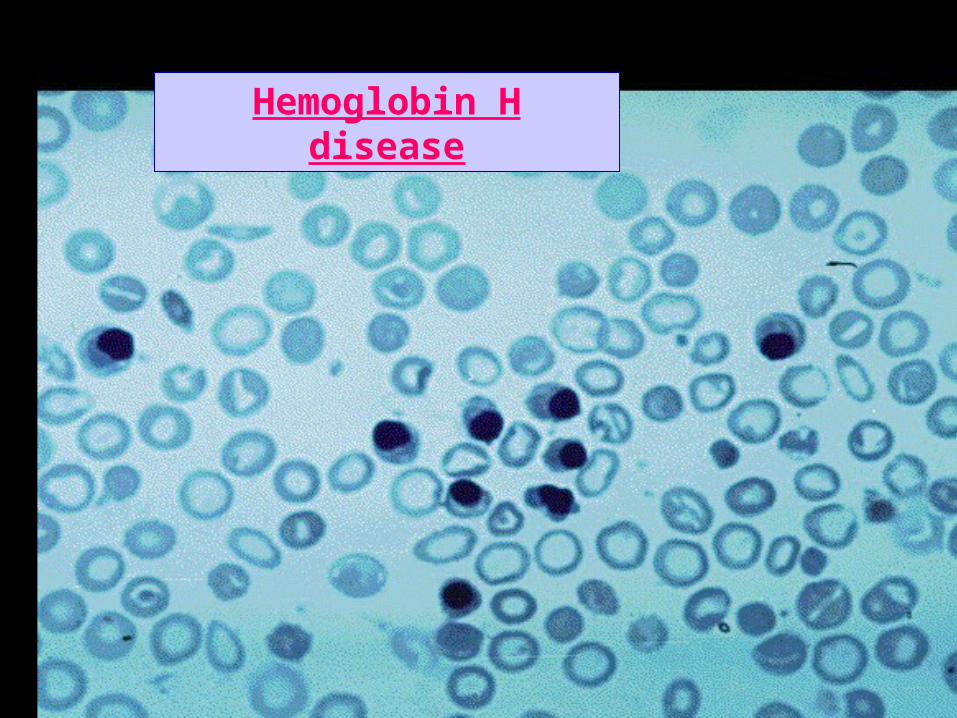
Hemoglobin H disease

Alpha thalassemia minor
• Due to deletion of two alpha genes, leaving only two alpha genes, so only moderate reduction of alpha chain production.
• Clinical and blood picture, exactly the same as beta thalassaemia minor.
• Hb electrophoresis shows Hb A, with normal or reduced Hb A2 and normal Hb F.
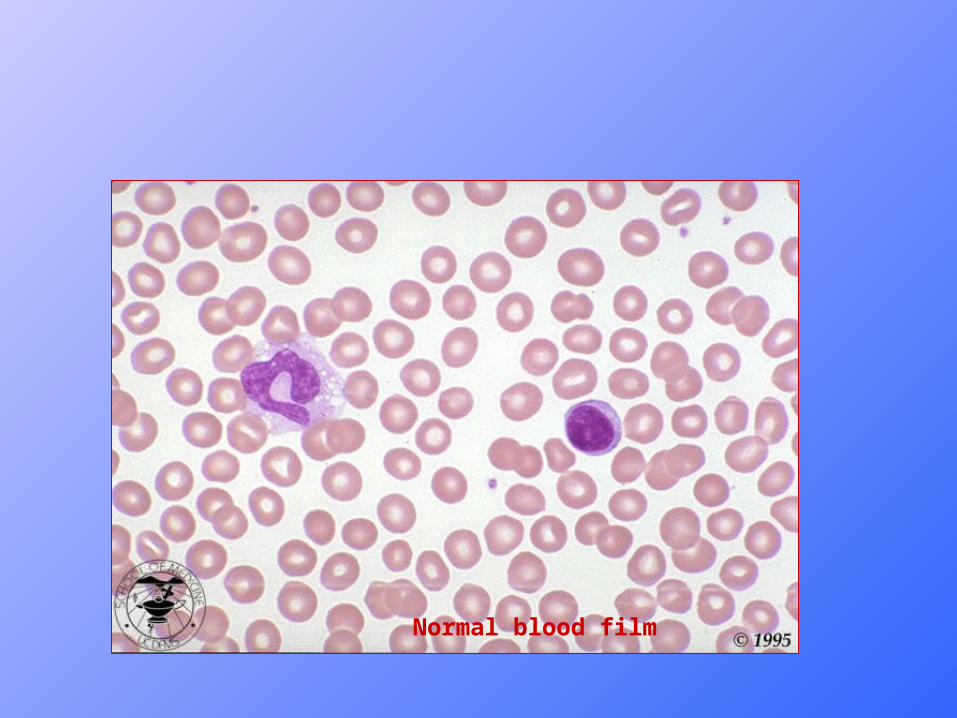
Normal blood film

Iron Deficiency Anemia
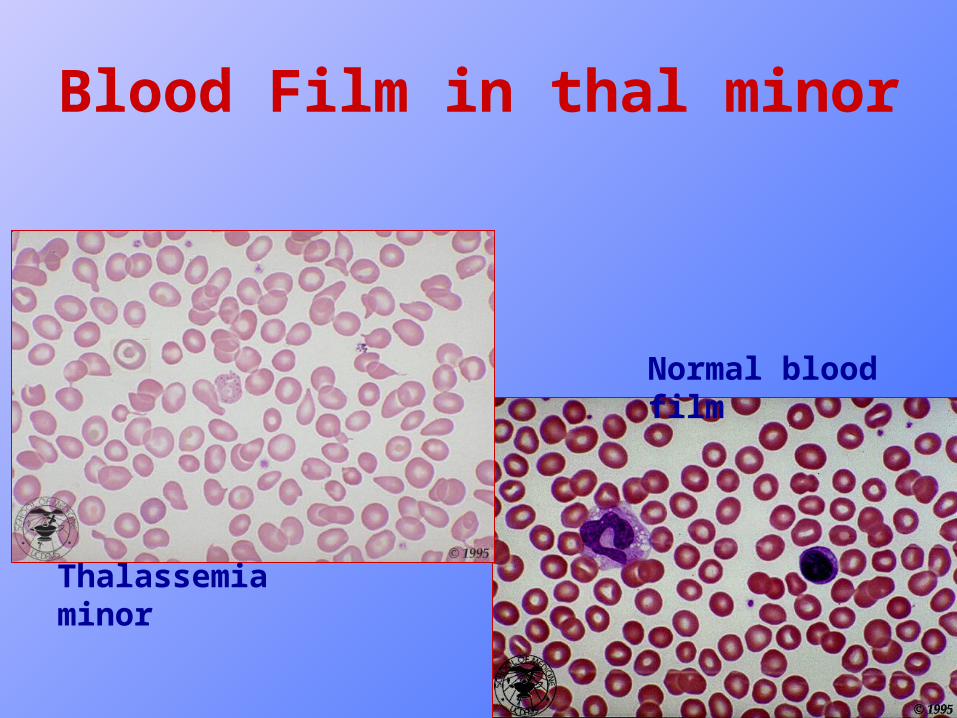
Blood Film in thal minor
Thalassemia minor
Normal blood film











![TheOngoingChallengeofHematopoieticStemCell-Based ...downloads.hindawi.com/journals/sci/2011/987980.pdf · Thalassemias are caused by more than 200 mutations ... [27], while in primates](https://static.fdocument.org/doc/165x107/602afc26aeb6bc151050ebdc/theongoingchallengeofhematopoieticstemcell-based-thalassemias-are-caused-by.jpg)




