
onnanosystemy.upol.cz/upload/76/cpic_boa_final.pdfand devise new forms of the potentials. Fig.1....
45
6 nd European Symposium on Computing π-Conjugated Compounds Olomouc 5 th – 7 th February 2015 Book of Abstracts
Transcript of onnanosystemy.upol.cz/upload/76/cpic_boa_final.pdfand devise new forms of the potentials. Fig.1....

6nd European Symposium
on
Computing π-Conjugated Compounds
Olomouc
5th – 7th February 2015
Book of Abstracts

i
European Symposiums on Computing π-Conjugated Compounds (CπC) is a series of small symposia
on the computations of various properties of π-conjugated systems. This symposium covers from small
π-conjugated molecules up to very large systems as well as computational approaches ranging from
empirical force fields up to high-level quantum chemistry.
Previous CπC symposia were organized in Valencia (2010), Limoges (2011), Mons (2012), Marseille
(2013) and Linköping (2014). These meetings were very successful and stimulated several successful
collaborative activities among participants. We hope that you enjoy CπC meeting in Olomouc and
benefit from the newly established contacts and provoked ideas.
See you in Olomouc!
Michal Otyepka and Karel Berka
Local organizing committee
Michal Otyepka (+420 733 690 624)
Karel Berka (+420 723 953 791)
CπC scientific board
Johannes Gierschner IMDEA Nanoscience, Madrid Spain
Begoña Milián-Medina IMDEA Nanoscience, Madrid Spain
Patrick Trouillas University of Limoges France
Yoann Olivier University of Mons Belgium
Luca Muccioli University of Bologna Italy
Philippe Marsal University of Marseille France
Juan-Carlos Sancho-García University of Alicante Spain
Mathieu Linares University of Linköping Sweden
Michal Otyepka Palacký University Olomouc Czech Rep.
Mari Carmen Ruiz Delgado University of Malaga Spain
Roberto Improta IBB/CNR Naples Italy

ii
Venue
Faculty of Science, Palacký University Olomouc, Czech Republic
tř. 17. listopadu 12, Olomouc
GPS: 49.5924922,17.2632337
3rd
floor, lecture room 3.003 and PC room/refreshment 3.002
Faculty building from Google StreetView
Meeting sponsorship
iii
List of Participants
Karel Berka [email protected] Palacky Uni, Olomouc, CZE
Michal Biler [email protected] Palacky Uni, Olomouc, CZE
Santanu Bhattacharyya [email protected] IMDEA Nanoscience, Madrid, ESP
María del
Carmen Ruiz Delgado
[email protected] Uni Málaga, ESP
Petra Čechová [email protected] Palacky Uni, Olomouc, CZE
Gabriele D'Avino [email protected] Uni Mons, BEL
Florent Di Meo [email protected] IFM, Linköping University, SWE
Matúš Dúbecký [email protected] Palacky Uni, Olomouc, CZE
Fabre Gabin [email protected] Uni Limoges, FRA
Manoj Gali [email protected] Uni Bordeaux, FRA
Hugo Gattuso [email protected] Uni Lorraine, Nancy, FRA
Johannes Gierschner [email protected] IMDEA Nanoscience, Madrid, ESP
Angelo Giussani [email protected] Uni Bologna, ITA
Benjamin Grenier [email protected] CINAM, Marseille, FRA
Roberto Improta [email protected] CNR, Uni degli Studi di Napoli, ITA
Pavel Jelínek [email protected] Inst Physics, AS CR, Prague, CZE
Petr Jurečka [email protected] Palacky Uni, Olomouc, CZE
Stefan Knippenberg [email protected] KTH, Stockholm, SWE
Martin Kubala [email protected] Palacky Uni, Olomouc, CZE
Petra Kührová [email protected] Palacky Uni, Olomouc, CZE
Petr Lazar [email protected] Palacky Uni, Olomouc, CZE
Mathieu Linares [email protected] Linköping Uni, SWE
Philippe Marsal [email protected] CINaM/Uni d'Aix, Marseille, FRA
Micaela Matta [email protected] Uni Bordeaux, FRA
Begoña Milián-Medina [email protected] Uni Valencia, SPA
Antonio Monari [email protected] Uni Lorraine, Nancy, FRA
Monica Moral [email protected] Uni Alicante, ESP
Luca Muccioli [email protected] Uni Bordeaux, FRA
Dana Nachtigallová [email protected] IOCB, AS CR, Prague, CZE
Veronika Navrátilová [email protected] Palacky Uni, Olomouc, CZE
Yoann Olivier [email protected] Uni Mons, BEL
Michal Otyepka [email protected] Palacky Uni, Olomouc, CZE
Martin Pykal [email protected] Palacky Uni, Olomouc, CZE
Olivier Rezazgui [email protected] LCSN, Limoges, FRA
Juan-Carlos
Sancho-García
[email protected] Uni Alicante, ESP
Sunandan Sarkar [email protected] Palacky Uni, Olomouc, CZE
Junqing Shi [email protected] IMDEA Nanoscience, Madrid, ESP
Mária Sudolská [email protected] Palacky Uni, Olomouc, CZE
Jiří Šponer [email protected] Inst Biophysics, AS CR, Brno, CZE
Martin Šrejber [email protected] Palacky Uni, Olomouc, CZE
Patrick Trouillas [email protected] INSERM, Uni Limoges, FRA
Shinto Varghese [email protected] Uni St Andrews, UK
Riccardo Volpi [email protected] Linköping Uni, SWE
Michael Wykes [email protected] IMDEA Nanoscience, Madrid, ESP
Marie Zgarbová [email protected] Palacky Uni, Olomouc, CZE
http://fch.upol.cz/en/cpic

ABSTRACTS
Invited Lectures

2
Electronically Excited States of Nucleic Acid Bases:
Computational Study
Dana Nachtigallová
Institute of Organic Chemistry and Biochemistry AS CR, v.v.i., Czech Republic
Ab initio surface hopping dynamics calculations were performed to study the photophysical
behavior of nucleic acid bases embedded in DNA using a quantum mechanical/molecular
mechanics approach.
The photodynamics of 4-aminopyrimidine used as a model for adenine were performed by
embedding it between two stacking methyl-guanine molecules to determine the effect of spatial
restrictions on the ultrafast photodeactivation mechanism of this nucleobase. During the
dynamics the formation of a significant fraction of intra-strand hydrogen bonding from
4-aminopyrimidine to methyl-guanine above and below is observed. This type of hydrogen bond
may play an important role for the photodynamics within one DNA strand.
The effect of inter-strand hydrogen bonding was investigated for cytosine and guanine and found
to be affected in a completely different way by the hydrogen bonding to the DNA environment.
In case of cytosine, the geometrical restrictions exerted by the hydrogen bonds did not influence
the relaxation time of cytosine significantly due to the generally small cytosine ring puckering
required to access the crossing region between excited and ground state. On the contrary, the
presence of hydrogen bonds significantly altered the photodynamics of guanine.

3
Nature and Magnitude of Aromatic Base Stacking in Nucleic Acids
Jiří Šponer
Institute of Biophysics, Academy of Sciences of the Czech Republic, Královopolská 135, 61265 Brno, Czech
Republic, [email protected]
Vertical (stacking) interactions of nucleic acid bases belong to the most fundamental energy
contributions that are shaping up structure and dynamics of nucleic acids (DNA and RNA). At
the same time, one can hardly find any energy contribution in nucleic acids that is associated
with so many myths, oversimplifications and misunderstandings in the literature.
In my talk, I will briefly explain the physical-chemistry origin of basis stacking as revealed by
modern electronic structure theory. Then I will explain how is it (im)possible to transfer this
knowledge to real structural biology and biochemical problems.
References
[1] J. Sponer, J. Leszczynski, P. Hobza: On the nature of nucleic acid base stacking. Nonempirical ab initio and
empirical potential characterization of 10 stacked base pairs. Comparison of stacked and H-bonded base pairs.
Journal of Physical Chemistry 100, 1996, 5590-5596.
[2] J. Florian, J. Sponer, A. Warshel: Thermodynamic parameters for stacking and hydrogen bonding of nucleic acid
bases in aqueous solution: ab initio/Langevine dipole study. Journal of Physical Chemistry B 103, 1999, 884-892.
[3] P. Hobza, J. Sponer: Structure, energetics, and dynamics of the nucleic acid base pairs: Nonempirical ab initio
calculations. Chemical Reviews 99, 1999, 3247-3276.
[4] J. Sponer, C.A. Morgado, D. Svozil: Comment on "Computational Model for Predicting Experimental RNA and
DNA Nearest-Neighbor Free Energy Rankings. Journal of Physical Chemistry B, 116, 2012, 8331-8332.
[5] J. Sponer, J. E. Sponer, A. Mladek, P. Jurecka, P. Banas, M. Otyepka: Nature and magnitude of aromatic base
stacking in DNA and RNA: Quantum chemistry, molecular mechanics and experiment. Biopolymers, 2013, 99,
978-988.

4
What We Can Learn from High-Resolution AFM/STM Images:
Experiment and Theory
Pavel Jelínek
Institute of Physics ASCR, Cukrovarnická 10, Prague 6, Czech Republic
The recent progress in Atomic Force Microscopy (AFM) and Scanning Tunneling Microscopy
(STM) using functionalized tips provided unprecedented atomic resolution of single organic
molecules [1,2]. However, the origin of the high-resolution AFM/STM imaging contrast is still
not well understood. Here we will present 3D maps of the force, tunneling current and
dissipation over a monolayer of PTCDA molecules deposited on Ag(111) acquired at 1.2 Kelvin
with Xe-functionalized tip (see Fig. 1.). We will compare detailed contrast features of the force
maps at various tip-sample separations to a numerical model [3,4]. The numerical model
describes relaxation of the functionalized tip due to Pauli repulsion and the electrostatic
interaction with surface. Combing the experimental and theoretical evidence, we will explain the
high-resolution imaging mechanism [3], artifacts [3,5] and a possibility to map out
intra-molecular charge distribution in real-space [4]. We will also discuss a possibility to use
experimental measurements to benchmark existing classical interatomic potentials (force fields)
and devise new forms of the potentials.
Fig.1. High resolution AFM/STM images of PTCDA molecules deposited on Ag(111) surface
acquired with Xe-functionalized tip at different tip sample distances.
References
[1] L. Gross et al., Science 325,1110 (2009).
[2] C. Weiss et al., Phys.Rev.Lett. 105, 086103 (2010).
[3] P. Hapala et al., Phys. Rev. B 90, 085421 (2014).
[4] P. Hapala et al., Phys. Rev. Lett. 113, 226101 (2014).
[5] J. Zhang et al., Science 342, 611 (2013).

5
ABSTRACTS
Oral Communications

6
Excited State Features and Dynamics in Conjugated Organic Materials
Johannes Gierschner, Begoña Milian-Medina, Michael Wykes,
Santanu Bhattacharyya, Shinto Varghese, Junqing Shi,
Rameesha Parambil, Benedikt Dähnekamp, Jorge Moreno
Madrid Institute for Advanced Studies, IMDEA Nanoscience, Madrid, Spain
Having now steady-state and time-resolved absorption and emission spectroscopy fully operative,
our team in Madrid has intensified the studies on excited state dynamics in conjugated organic
materials in solution and in the solid state by an integrative spectroscopic and computational
approach.
Isolated chains: With our expertise in polymer bandgap predictions, we are now investigating
low bandgap homopolymers, as well as co-polymers with strongly localized MOs, and the
impact on charge and exciton transport (Begoña). Following the series of previous fluorination
studies, we discover new symmetry breaking effects in latter-type oligophenylenes leading to
unprecedented optical properties (Benedikt & Begoña). Studying the excited state potential
surfaces of substituted distyrylbenzenes, we are elucidating the selectivity of specific substituent
position on the photophysics (Junqing & Begoña). New branches opened on metal-free organic
phosphors, on reactivity studies for novel fluorine sensors, and on acidochromic
(multi-responsive) materials.
Solid State: Quantum chemical studies finally helped to understand why some organic single
crystals are lasing and others not (Shinto). Careful quantum-chemical studies on molecular
aggregates allow for a detailed understanding of the emission process in aggregates of
-conjugated systems with respect to their AIEE properties (Junqing) and their vibronic structure
(Rameesha & Mike). Surprisingly strong emission properties of donor-acceptor co-crystals were
traced back to the specific nature of the excited singlet & triplet state manifolds (Santanu).

7
MO and Excited State Design of Low Bandgap Copolymers
Begoña Milián-Medina1,2, Johannes Gierschner2
1Department of Physical Chemistry, University of Valencia
c/ Dr. Moliner 50, 46100 Burjassot (Valencia) - (SPAIN) 2Madrid Institute for Advanced Studies, IMDEA Nanoscience
c/ Faraday 9, Ciudad Universitaria de Cantoblanco, 28049 Madrid - (SPAIN)
e-mail: [email protected]
Donor-acceptor low bandgap copolymers are frequently used in organic photovoltaics to
efficiently harvest the sunlight. However, manipulation of the bandgap via the donor-acceptor
approach also produces significant changes in the electronic properties and of the excited state
manifold through frontier MO localization. This might significantly influence optical properties,
intra-chain photophysics, and solid state exciton dynamics, charge transport and (re)combination.
Conceptual quantum-chemistry is used to elucidate the pre-requisites for specifically localized
MOs, and their impact on the copolymers' electronic, optical and photophysical properties.

8
Low-temperature Fine Structure via Atomistic Quantum Chemical
Franck-Condon Herzberg-Teller calculations.
Michael Wykes, Rameesha Mangattu Parambil, Johannes Gierschner
IMDEA Nanoscience, Madrid, Spain
Para-distyrylbenzene (DSB), a model compound for the first polymer (PPV) intensely
investigated for its electroluminescent properties, has been the focus of numerous experimental
and theoretical investigations into the fundamental photophysics of organic semiconductors. The
drastic change in optical properties of DSB upon transfer from the dilute solution phase to the
bulk crystalline phase has been linked to the herring-bone crystal structure of DSB, which leads
to the formation of H-aggregates in which the transition dipole moment of the lowest electronic
transition is greatly reduced. While the vibronic-fine structure of DSB seen in low-temperature
solution spectra has been fully interpreted on the basis of atomistic simulations combining
quantum chemical (QC) calculations and Franck-Condon models of vibronic coupling,1 the
optical spectra of crystal spectra have thus far been simulated using an alternative coarse-grained
phenomenological Holstein-type model in which molecules are treated as sites coupled to a
single effective intramolecular vibrational mode.2 Here we treat single molecules and model
DSB-aggregates on an equal footing, with atomistic models combining QC calculations and
Franck-Condon and Herzberg Teller models of vibronic coupling. We compare our simulated
spectra with coarse-grained Holstein-based simulations and experiments and discuss the
theoretical foundations of the two models as well as their advantages and disadvantages when
applied to molecular crystals.
References
[1] Gierschner, J.; Mack, H.-G.; Lüer, L.; Oelkrug, D. J. Chem. Phys. 2002, 116, 8596.
[2] Spano, F. C. J. Chem. Phys. 2003, 118, 981

9
Simulations of Excited Electronic States and Photochemistry
S. Knippenberg1, R. L. Gieseking2, D. R. Rehn3, M. Wormit3,
S. Mukhopadhyay2, J.-L. Brédas2, M. V. Bohnwagner3, P. H. P. Harbach3,
and A. Dreuw3
1Division of Theoretical Chemistry and Biology, KTH Royal Institute of Technology, Roslagstullsbacken 15, S-106
91 Stockholm, Sweden 2Center for Organic Materials for All-Optical Switching (COMAS), Georgia Institute of Technology, Atlanta,
Georgia 30332-0400 3Interdisciplinary Center for Scientific Computing, Ruprecht-Karls-University, Im Neuenheimer Feld 368, D-69120
Heidelberg, Germany
e-mail: [email protected]
The Algebraic Diagrammatic Construction (ADC) scheme, which is a powerful approach to
investigate excited states and their properties [1], is used for the first time to calculate second
hyperpolarizabilities [2]. A convenient sum-over-states (SOS) expression is applied and the
focus is put upon short and middle-sized streptocyanines. The real part of , denoting the
refraction index, is investigated for all compounds in the band 0.8-0.95 eV (1200-1550 nm),
which is of utmost importance for telecommunication purposes. Comparison has been made with
results obtained using packages which have been often applied in literature, like the powerful
symmetry adapted cluster configuration interaction (SAC-CI) method and the pragmatic
semi-empirical Zindo one. The two-photon absorption cross section has been profoundly focused
on, too. In the case of second order ADC [ADC(2)], it is calculated using a three-state expression
as well as by applying an analytically closed form. The data are found to be very comparable,
enforcing the two-photon SAC-CI, Zindo and higher order ADC results, which are only obtained
by means of the SOS formalism.
Focusing on photochemistry, the BisBODIPY compound and the influences of excitonic
coupling on its absorption spectra are investigated [3]. With the help of model systems, the
electronic coupling is quantized and its influence on energetics and oscillator strengths is
highlighted. For the explanation of the experimental spectrum, orbital interaction effects are
found to be important.
References
[1] J. Schirmer, Phys. Rev. A 26 (1982), 2395; J. Schirmer and A.B. Trofimov, J. Chem. Phys. 120 (2004) 11449;
Wormit, M.; Rehn, D.; Harbach, P. et al., Mol. Phys. 112 (2014), 774.
[2] S. Knippenberg, D. Rehn, M. Wormit et al., J. Chem. Phys. 136 (2012), 064107; S. Knippenberg, R. L.
Gieseking, D. Rehn, et al., to be submitted.
[3] M. Bröring, R. Krüger, S. Link et al., J. Phys. Chem. A 116 (2012), 12321; S. Knippenberg, M. V. Bohnwagner,
P. Harbach, A. Dreuw, submitted.

10
On the Computation of Two-dimensional Electronic Pump-Probe
Spectra
Angelo Giussani, Artur Nenov, Javier Segarra-Martí, Irene Conti,
Ivan Rivalta, Elise Dumont, Vishal K. Jaiswal, Salvatore Altavilla,
Shaul Mukamel, and Marco Garavelli
Dipartimento di Chimica G. Ciamician, Università di Bologna, Via F. Selmi 2, 40126 Bologna, Italy
Two-dimensional electronic spectroscopy is a promising technique expected to become a key
tool for the description of excited state decays. [1] The methodology is based on the detection
into a so-called two-dimensional electronic spectrum of all the intensive electronic transitions
caused by a UV or vis radiation (the pump signal) at time t1 and the subsequent interaction with a
second UV or vis radiation (the probe signal) at time t2 (t2 ≥ t1). Two-dimensional electronic
spectroscopy will then provide both high spectral and temporal resolution allowing the analysis
of ultrafast complex (multi-pathway) reactions.
The theoretical construction of two-dimensional electronic spectra requires at least the
knowledge of all the accessible electronic transitions by a defined pump signal from a particular
geometry and electronic states of the system, and all the accessible electronic transitions by a
defined probe signal from a particular geometry and electronic states. Such information is
obtained through the calculation of a considerable number of excited states energies and the
corresponding transition dipole moments at the pump and probe geometries. In order to obtain
quantitative outcomes for medium size molecules, as the canonical nucleobases, nowadays the
most suitable computational strategies are probably the CASSCF and CASPT2 (or RASSCF and
RASPT2) methods. Having the required data, using the qSpec2DUV program a two-dimensional
spectrum can then be produced.[1]
With the present contribution, the main features and potentialities of two-dimensional electronic
spectroscopy are presented, together with the machinery developed in the Garavelli's group in
order to compute two-dimensional electronic spectra, and the results of its application on the
study of the photophysics and photochemistry of adenine and of the adenine dimer.[2]
References
[1] a) Rivalta I, Nenov A, Cerullo G, Mukamel S, Garavelli M, Int. J. Quantum Chem., 2014, 114, 85–93. b) Nenov
A, Rivalta I, Cerullo G, Mukamel S, Garavelli M, J. Phys. Chem. Lett., 2014, 5, 767-771.
[2] Nenov A, Segarra-Martí J, Giussani A, Conti I, Rivalta I, Dumont E, Jaiswal E, Altavilla S, Mukamel S,
Garavelli M, Accepted for the Faraday discussions: Temporally and spatially resolved molecular sciences, 2014,
Bangalore, India

11
The Role of Charge Transfer Excited Electronic States in
Photophysics and Photochemistry of DNA
Roberto Improta
Istituto Biostrutture e BioImmagini-CNR, Via Mezzocannone16, 80134, Napoli, Italy
The possible involvement of Charge Transfer (CT) excited states in the photoactivated dynamics
of DNA is one of the most controversial issues in the photochemistry of nucleic acids. Exploiting
Time Dependent DFT calculations using last generation functions (enabling an accurate
determination of the CT states stability) and taking solvent effect into account by the Polarizable
Continuum Model, we have studied realistic oligonucleotides, including also the
phospho-deoxyribose backbone and counter-ions, by combing stati and quantum dynamical
calculations. We provide clear indications that states with a noticeable, yet different, degree of
CT character are involved in the photophysics and photochemistry of several systems:
oligoAdenine,[1,2] d(TpT)[3,4] and d(TpC)[5] and d(Tp5methylC)[5] dinucleotides, Guanine
Quadruplex helices[6] Guanine nano-ribbons,[7] and Oxoguanine-Ade dinucleotides.[8] Our
predictions are fully consistent with the available experimental results, i.e. Steady State Circular
Dichrois m,[3,6] Time resolved fluorescence [1-7] and InfraRed spectra, which, fo r the first
time, show a clear spectral signature of CT states.[8] For quadruple helices, our studies also
provide a complete assignment of the optical spectra and indications on the fact ors influencing
the excited state dynamics (cation, conformation of the quadruplex...).[6] Finally, it has been
possible to explain the role of 5-methylation in the phot ochemistry of dinucleotides containing
Cytosine.[5].
References
[1] R. Improta, V. Barone, Angew. Chemie. 2011, 50 , 12016.
[2] A. Banyasz, et al. Chem. Eur. J. 2013, 19 , 3762.
[3] A.Banyasz, et al. J. Am. Chem. Soc. 2012, 134, 14834.
[4] R. Improta, J. Phys. Chem. B 2012, 116, 1426.1
[5] L. Esposito et al. J. Am. Chem. Soc. 2014, 136, 10838.
[6] R. Improta, Chem. Eur. J. 2014, 20, 8106.
[7] K. Hunger, et al. Chem. Eur. J. 2013, 19, 5425.
[8] Zhang, Y. et al. Proc. Nat. Acad. Sci. U.S.A. 2014, 111, 11612.
12
Electronic Circular Dichroism Spectra in Stacked Systems
Mathieu Linares
Division of Theoretical Chemistry, Department of Physics, Biology and Chemistry (IFM) Linköping University –
Sweden – [email protected]
During the last two decades Circular Dichroism (CD) has been widely used to understand the
chirality phenomenon. It has been possible to understand the physical, chemical and
conformational properties of chiral molecules, chiral assemblies, peptides and proteins.
The simulation of the ECD response of a single molecule, is a task well suited for standard
time-dependent density functional theory (TD-DFT)1 (or any other standard first-principles
response theory approach) in combination with the use of gauge-origin independent basis sets.2
It is clear that with the development of reduced scaling techniques, DFT can be applied to large
systems such as a cluster model of polymer aggregates. However, even if TD-DFT is suitable to
model small stacks,3 its use is prohibited in large supramolecular assemblies involving a high
density of excited states. A more suitable approach to bypass this problem is the use of a
resonant convergent formulation of response theory known as the complex polarization
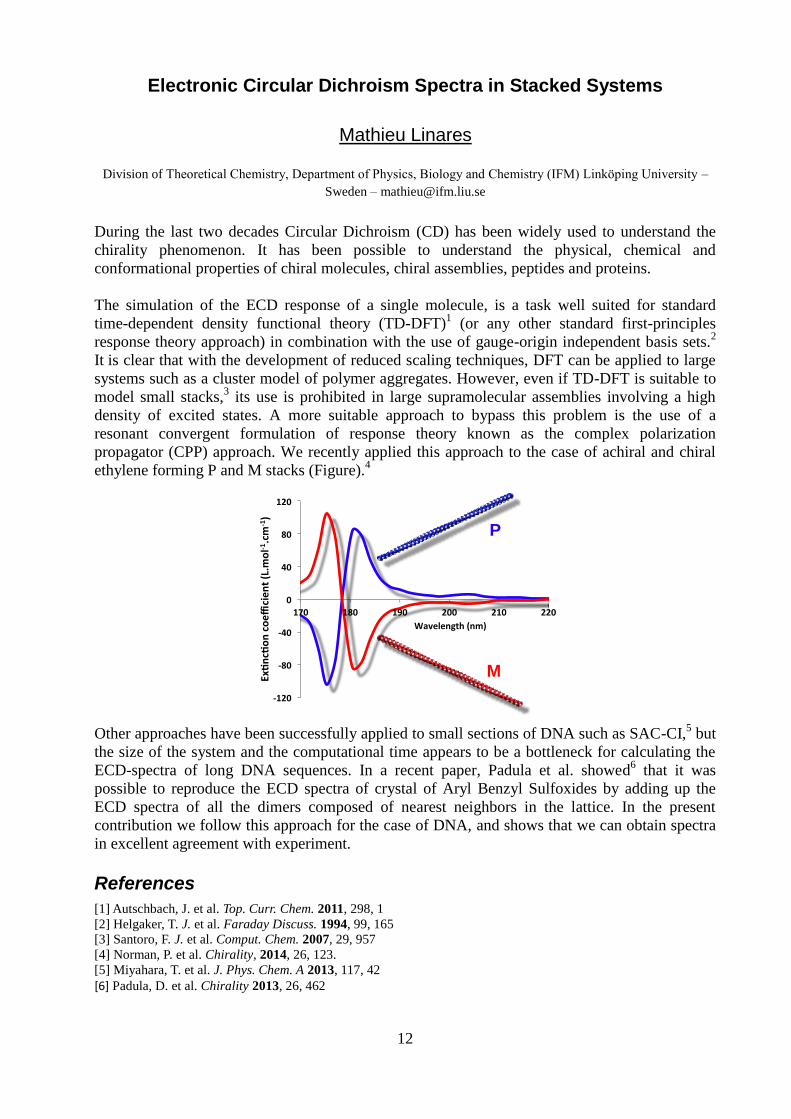
propagator (CPP) approach. We recently applied this approach to the case of achiral and chiral
ethylene forming P and M stacks (Figure).4
Other approaches have been successfully applied to small sections of DNA such as SAC-CI,
5 but
the size of the system and the computational time appears to be a bottleneck for calculating the
ECD-spectra of long DNA sequences. In a recent paper, Padula et al. showed6 that it was
possible to reproduce the ECD spectra of crystal of Aryl Benzyl Sulfoxides by adding up the
ECD spectra of all the dimers composed of nearest neighbors in the lattice. In the present
contribution we follow this approach for the case of DNA, and shows that we can obtain spectra
in excellent agreement with experiment.
References
[1] Autschbach, J. et al. Top. Curr. Chem. 2011, 298, 1
[2] Helgaker, T. J. et al. Faraday Discuss. 1994, 99, 165
[3] Santoro, F. J. et al. Comput. Chem. 2007, 29, 957
[4] Norman, P. et al. Chirality, 2014, 26, 123.
[5] Miyahara, T. et al. J. Phys. Chem. A 2013, 117, 42
[6] Padula, D. et al. Chirality 2013, 26, 462
-120
-80
-40
0
40
80
120
170 180 190 200 210 220
Exnconcoefficient(L.m
ol-1.cm
-1)
Wavelength(nm)
P
M

13
Merging Non-local (van der Waals) Corrections
with Double-Hybrid Density Functionals
J.C. Sancho-García1, J. Calbo2, Y. Olivier3, E. Ortí2, J. Aragó2
1Department of Physical Chemistry, University of Alicante, E-03080, Alicante, Spain
2Institute of Molecular Science, University of Valencia, E-64980 Valencia, Spain
3Laboratory for Chemistry of Novel Materials, University of Mons, B-700 Mons, Belgium
Non-covalent interactions are known to drive the self-assembly and reactivity of molecules at
any length scale, which needs to be successfully and cost-efficiently incorporated into any
intended theoretical treatment of weakly interacting systems [1]. We have coupled a seamless
and truly non-local (NL) correlation functional with the most modern existing expressions for
exchange-correlation effects [2], such as the double-hybrid density functionals (DHDFs) named
as B2-PLYP or revPBE0-DH, providing a family of methods able to deal with any kind of
interactions (covalent and non-covalent) in an accurate way. This is demonstrated through their
excellent performance for a set of recently developed databases (S22, S66, NCCE31, S12L, and
L7) for benchmarking purposes [3,4], as well as for estimating the cohesive or lattice energies of
(bulk) organic molecular semiconductors [5,6].
References
[1] K.E. Riley, M. Pitonak, P. Jurecka, and P. Hobza, Chem. Rev. 110 (2010) 5023.
[2] J.C. Sancho-García, and C. Adamo, Phys. Chem. Chem. Phys. 15 (2013) 14581.
[3] J. Aragó, E. Ortí, and J.C. Sancho-García, J. Chem. Theory Comput. 9 (2013) 3437.
[4] J. Calbo, J.C. Sancho-García, E. Ortí, and J. Aragó, J. Chem. Theory Comput. (submitted).
[5] J.C. Sancho-García, J. Aragó, E. Ortí, and Y. Olivier, J. Chem. Phys. 138 (2013) 204304.
[6] J.C. Sancho-García, A.J. Pérez-Jiménez, and Y. Olivier, J. Chem. Phys. (submitted).

14
Induced Chirality in Porphyrins: Self-Assembly and Systems of
Biological Interest
Florent Di Meo, Mathieu Linares, Patrick Norman
Division of Theoretical Chemistry, Department of Physics, Chemistry and Biology (IFM), Linköping University,
SE-58183 Linköping, Sweden
Email: [email protected]
Porphyrins are an important armamentarium of -conjugated compounds with a broad range of
physico-chemical and biological activities. Even though the porphyrin core is based on a simple
achiral tetrapyrrol core, the possibility to substitute meso positions and/or to include a metal
center leads to a huge variety of original compounds with different conformational features.
Furthermore, the environment (e.g., other porphyrins or biological systems such as DNA) may
deform tetrapyrrol core leading to a specific chiroactive structure. Such induced chirality can be
followed by electronic circular dichroism response in the typical Soret region.
Figure. Examples of (a) chiroactive porphyrin self-assembly and (b) supramolecular
metalloporphyrin G-quadruplex complex.
In the present work, the self-assembly of a chiral porphyrin (Fig. 1a) is first investigated.
Experiments suggest the self-assembly of four porphyrins into a cubic-like shape. Then, each
cube may also undergo a secondary self-assembly process leading to an important increase of the
ECD signal. MD simulations and ZINDO calculations allowed to elucidate (i) the
supramolecular assembly and (ii) the specific ECD response.
Then, the induction of chirality thought adsorption of achiral porphyrins with DNA
G-quadruplexes (e.g., Fig. 1b) is rationalized thanks to MD simulations. Interestingly,
experiments have exhibited an ECD response in the Soret region suggesting an induction of
chirality in the porphyrin core. MD simulations and first-principle calculations rationalized (i)
binding modes and (ii) porphryin deformations leading to an ECD response.
(a) (b)

15
Design and Study of Bio-Inspired Photoherbicide:
From Theory to Experiment
O. Rezazgui, P. Trouillas, S. Leroy-Lhez
1. Laboratoire de Chimie des Substances Naturelles, EA 1069, Université de Limoges,
123 avenue Albert Thomas, 87060 Limoges, France.
2. INSERM UMR-S850, Faculté de Pharmacie, Université de Limoges,
2 rue du Dr.Marcland, 87025 Limoges, France.
It is of utmost importance to prevent contamination of the environment of Earth's surface and of
groundwater. For this purpose, bio-inspired herbicides (e.g., porphyrins) are promising
alternative to the currently used toxic compounds.
To evaluate their biological effects in plants, the new herbicides must be tracked along plant
organs. To do so, the porphyrin herbicide can be associated with a fluorescent tag. This
molecular association needs to be achieved by a linker, which must be carefully chosen.
The linker is critical in the 3D arrangement of the corresponding molecular assembly
porphyrin-linker-fluorophore. DFT (density functional theory) calculations were performed to
evaluate conformational folding. Non-covalent interactions, in particular π-stacking, play a
crucial role in this case, therefore the use of DFT functionals including dispersion (DFT-D)
appeared mandatory. Solvent effects were taken into account implicitly, using PCM (polarizable
continuum model). This preliminary work has led to the selection of some linkers; as well to the
molecular assembly associated synthesis.
From the conformational analysis, the excited states (ES) were characterized by TD (time
dependent)-DFT calculations. UV absorption properties were thus rationalized based on the
molecular orbital scheme, showing the electronic transitions involved in the relevant ES. The
theoretical predictions seem to correlate experimental photo-physical evaluations. The
fluorescence emission analysis has highlighted energy transfer, which is in favor of the folded
conformer.

16
Capless HDAC Inhibitors: Molecular Modelling Reveals
the Structural Determinants of Their Selectivity Toward HDAC6
Benjamin Greniera,b, Nathalie Deschampsb, Carolina Dos Santos Passosb,
Alessandra Nurissob, Pierre-Alain Carruptb,
aCentre Interdisciplinaire de Nanoscience de Marseille (CINaM), UPR 3118 CNRS, Aix-Marseille Université,
Campus de Luminy, Case 913, F-13288 Marseille cedex 09, France bSchool of Pharmaceutical Sciences, University of Geneva, University of Lausanne, 30, Quai Ernest-Ansermet,
CH-1211, Geneva, Switzerland
The development of selective histone deacetylase 6 (HDAC6) inhibitors is considered a
challenge not only for the conception of new biological probes but also for the development of
new drug candidates against aging-related and cancer diseases1, 2
. Recently, hydroxamic acid
derivatives without the surface-binding motif, a typical pharmacophoric element present in all
HDAC inhibitors, demonstrated the ability to preferentially inhibit HDAC6 rather than other
isoforms in vitro3. Here, a structural rationalization of such selectivity was carried out.
Comparison between the structural architecture of HDAC2 and HDAC6 catalytic domains
highlighted important areas of diversity. Indeed different pocket shapes are presented by these
two isoforms (Fig.1).Molecular docking and molecular dynamics simulations of three “cap” and
two “capless” compounds showed that HDAC6 selective compounds (an example is represented
in Fig.2) were in interaction by hydrogen bonds and hydrophobic interactions with His 500, Pro
608 and Val 650 HDAC6 residues. These interactions were not observed in HDAC2. So the
pocket size together with the presence of these three residues may be the responsible of the
observed HDAC6 selectivity.
Fig.1 Pocket comparison between
HDAC2 and HDAC6 active site in
gray and green respectively.
a, b, c and d highlight the four
active site areas.
Fig.2 An example of HDAC6
selective ligand, represented in
ball and stick with carbon in gray,
nitrogen in blue and oxygen in
red.
References
[1] Simões-Pires C, Zwick V, Nurisso A, Schenker E, Carrupt PA, Cuendet M (2013) HDAC6 as a target for
neurodegenerative diseases: what makes it different from the other HDACs? Mol Neurodegen 8: 1-16
[2] Yang P, Zhang L, Zhang Y, Zhang J, Xu W (2013) HDAC6: Physiological function and its selective inhibitors for
cancer treatment. JDDT 7: 233-24
[3] Wagner FF, Olson DE, Gale JP, Kaya T, Weïwer M, Aidoud N, Thomas M, Davoine EL, Lemercier BC, Zhang
YL, Holson EB (2013) Potent and selective inhibition of histone deacetylase 6 (HDAC6) does not require a
surface-binding motif. J Med Chem 56: 1772-1776
17
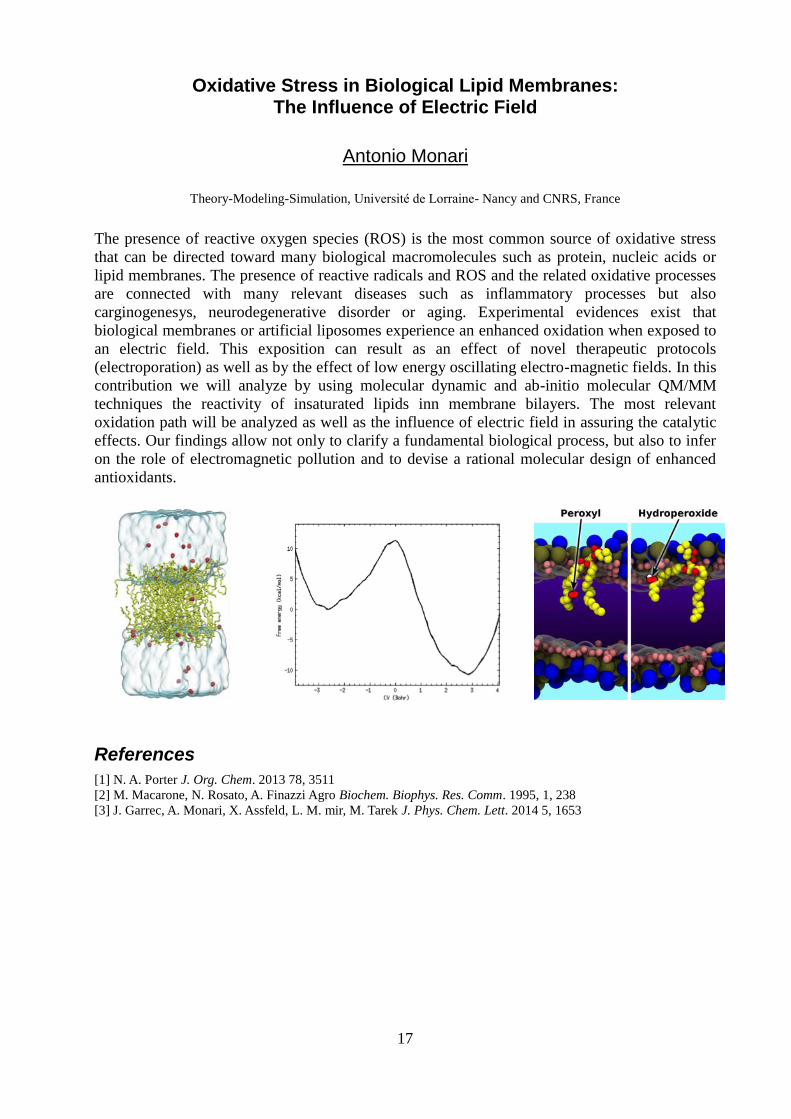
Oxidative Stress in Biological Lipid Membranes: The Influence of Electric Field
Antonio Monari
Theory-Modeling-Simulation, Université de Lorraine- Nancy and CNRS, France
The presence of reactive oxygen species (ROS) is the most common source of oxidative stress
that can be directed toward many biological macromolecules such as protein, nucleic acids or
lipid membranes. The presence of reactive radicals and ROS and the related oxidative processes
are connected with many relevant diseases such as inflammatory processes but also
carginogenesys, neurodegenerative disorder or aging. Experimental evidences exist that
biological membranes or artificial liposomes experience an enhanced oxidation when exposed to
an electric field. This exposition can result as an effect of novel therapeutic protocols
(electroporation) as well as by the effect of low energy oscillating electro-magnetic fields. In this
contribution we will analyze by using molecular dynamic and ab-initio molecular QM/MM
techniques the reactivity of insaturated lipids inn membrane bilayers. The most relevant
oxidation path will be analyzed as well as the influence of electric field in assuring the catalytic
effects. Our findings allow not only to clarify a fundamental biological process, but also to infer
on the role of electromagnetic pollution and to devise a rational molecular design of enhanced
antioxidants.
References
[1] N. A. Porter J. Org. Chem. 2013 78, 3511
[2] M. Macarone, N. Rosato, A. Finazzi Agro Biochem. Biophys. Res. Comm. 1995, 1, 238
[3] J. Garrec, A. Monari, X. Assfeld, L. M. mir, M. Tarek J. Phys. Chem. Lett. 2014 5, 1653

18
Environment Effects on the Spectra of Polycyclic Organic Molecules
Martin Kubala
Dept. of Biophysics, Faculty of Science, Palacký University in Olomouc, Czech Republic
The π -conjugated molecules can be examined in UV/VIS spectroscopy experiments. Although
spectroscopic data usually do not allow direct visualization of the molecular structure, numerous
parameters can be compared to molecular models. The recorded spectrum reflects both the
structure of the examined molecule itself as well as its interaction with other surrounding
molecules, in particular solvent molecules, ligands or macromolecules.
Measurement of absorption and emission spectra in various solvents can yield important
information about the examined molecule. They can concern the molecule itself, such as isomer
identification, characterization of the type of electronic transition (π→π* or n→π*) or
protonation state, or sensitivity to the environmental factors such as polarity or hydrogen
bonding. These observations can be used in further studies examining interaction of small π-conjugated molecules with large biopolymers such as nucleic acids or DNA. Correlation of the
spectroscopic data to some parameters obtained from molecular models can help to
understanding the structural details

19
Investigation of Antiradical and UV/Vis Absorption Properties of
Flavonolignans
Michal Bilera, b, Patrick Trouillasb,c, Martin Kubalaa, Vladimir Krend,
Jan Vaceke
a Department of Biophysics, Centre of the Region Hana for Biotechnological and Agricultural Research, Palacký
University, Šlechtitelů 11, 783 71, Olomouc, Czech Republic b INSERM UMR-S850, Univ. Limoges, School of Pharmacy, University de Limoges, 2 rue du Docteur Marcland,
87 025 Limoges, France c Regional Centre of Advanced Technologies and Materials, Department of Physical Chemistry, Faculty of Science,
Palacký University, tř. 17 listopadu 12, 771 46 Olomouc, Czech Republic d Institute of Microbiology, Laboratory of Biotransformation, Academy of Sciences of the Czech Republic,
Vídeňská 1083, 142 20 Prague, Czech Republic e Department of Medical Chemistry and Biochemistry, Faculty of Medecine and Dentistry, Palacký University,
Hněvotínská 3, 775 15 Olomouc, Czech Republic
Polyphenols are π-conjugated systems containing phenolic OH groups and many of them exhibit
free radical scavenging properties and other protective effects against oxidative stress. Density
functional theory (DFT) has been widely used to elucidate the role of the different OH groups of
polyphenols as radical scavengers. Here we have studied the O-H bond dissociation enthalpies
(BDEs) of a series of flavonolignans (silybin, silychristin, silydianin and their
dehydro-derivatives), which correlate to their capacity to scavenge the DPPH free radical and
cyclic voltammetry results. The spatial spin density distribution is a secondary quantum
descriptor, which accounts for extension of the π-conjugated path and allows rationalization of
the antiradical action.
The theoretical investigation of UV/Vis absorption properties enables a full physical-chemical
characterization of this series of compounds. The experimental measurements were performed in
the Britton-Robinson buffer (pH 2–12). The non-dehydroforms of the flavonolignans exhibit two
main peaks (i) ca. 288 nm with a hypochromic effect vs. pH increase, and (ii) ca. 326 nm with a
hyperchromic and hypsochromic shift vs. pH increase. On the other hand, the absorption spectra
of the dehydro-flavonolignans are bathochromic shifted vs. pH increase. The neutral and
deprotonated forms of the molecules were predicted by TD-DFT, using the B3P86 and
CAM-B3LYP functionals. Overestimation and underestimation of the spectral shifts between the
neutral and deprotonated form are observed with B3P86 and CAM-B3LYP, respectively. Finally,
electronic transitions between molecular orbitals were assigned to each peak of interest.
Acknowledgment
This work was supported by grant no. L01204 National program of Sustainability I, by grant IGA_PrF_2014_029
from Palacky University in Olomouc, and by the French embassy, which financially supports my cotutelle Ph.D.
program.

20
Excited State Dynamics
in Mixed Stack Donor-Acceptor Charge Transfer Co-Crystals
Santanu Bhattacharyyaa, Sang Kyu Parkb, SooYoung Parkb, Larry Luera,
Johannes Gierschnera
a) Madrid Institute for Advanced Studies, IMDEA Nanoscience, Calle Faraday 9, Ciudad Universitaria de
Cantoblanco, 28049 Madrid, Spain
b) Center for Suprameolcular Optoelectronic Materials and WCU Hybrid Materials Program, Department of
Materials Science and Engineering, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 151-744, Korea
Solid state organic semiconductors have gained much attention in modern material research due
to applications in optoelectronics, such as organic light emitting diode (OLED), field effect
transistors (OFET’s), photovoltaics (OPV) etc. For many applications, ambipolar transport is
desired [1]
for which charge transfer (CT) complexes consisting of donor (D) and acceptor (A)
molecules were identified as potential materials. [2]
The field was recently pushed by a novel
approach using mixed stack DA CT co-crystals consisting of isosteric D = distrylbenzene and A
= dicyanodistrylbenzene molecules, providing balanced charge transport and surprisingly
efficient deep red CT luminescence,[3]
which further caught our attention. Keeping in mind that
CT states strongly impact the state ordering of the singlet & triplet manifolds, [4]
we here further
investigate the excited state dynamics in both single- and poly-crystalline samples by ultrafast
time resolved spectroscopy and quantum chemical calculations.
The strong frontier MO offset and close -stacking of the alternatively packed D, A molecules
affect significantly fundamental optical and electronic properties. Strongly red-shifted absorption
and emission are assigned to a strong, low lying CT band, while the main absorption originates
from the D-localized S0S3 transition Pumping in S3, the relaxation to the CT states (S1, S2) is
relatively slow due to the large S3-S2 energy difference. Emission from S1 is non-exponential,
exhibiting a long component of 200 ns. The latter is ascribed to thermally activated delayed
fluorescence (TADF), originating from T1 due the small S-T gap as generated by the large CT
character of the lowest excited state. At high laser flux, emission at em is effectively quenched
by singlet-singlet annihilation due to the high excited state density at em as generated by the
strong DA -overlap, preventing lasing. Our studies suggest this DA system as the first
example of highly efficient, intermolecular TADF emitter, which might be an interesting new
route for triplet harvesting in OLEDs.
References
[1] V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey, J.-L. Brédas, Chem. Rev. 2007, 107,
926-952
[2] Zhu, L.; Yi, Y.; Li, Y.; Kim, E.-G.; Coropceanu, V.; Brédas, J.-L. J. Am. Chem. Soc. 2012, 134, 2340
[3] S. K. Park, S. Varghese, J. H. Kim, S.-J. Yoon, O. K. Kwon, B.-K. An, J. Gierschner, S. Y. Park, J. Am. Chem.
Soc. 2013, 135, 4757.
[4] B. Milián-Medina, J. Gierschner, Org. El. 13 (2012) 895
[5] S. Bhattacharyya, S. Kyu Park, S. Varghese, S. Y. Park, Larry Luer, J. Gierschner, in preparation

21
Electronic Ferroelectricity and Magnetoelectric Phenomena
in Mixed-stack Charge Transfer Crystals
Gabriele D’Avino1,2, Matthieu J. Verstraete1
1Physics Department, University of Liège and ETSF, Belgium
2Laboratory for Chemistry of Novel Materials, University of Mons, Belgium
Mixed-stack charge transfer (CT) crystals (e.g. TTF-CA, TTF-BA) are a spectacular example of
multifunctionality among organics materials. In these quasi-1D systems, featuring alternating
π-stacked electron donor (D) and acceptor (A) molecules (…DADA…), the entanglement of
charge, spin and lattice degrees of freedom leads to ferroelectric phases with an exceptionally
strong electronic polarization, magnetoelectric effects, and photoinduced phase transitions
triggered by multiexcitonic phenomena.
We present a theoretical investigation of the ferroelectricity of mixed-stack CT crystals, based on
the Peierls-Hubbard model [1] and first-principles calculations for its parame- trization. This
approach is first validated by reproducing the temperature-induced transition of TTF-CA and
TTF-BA, and then applied to a novel series of hydrogen- bonded CT crystals, for which room
temperature ferroelectricity has recently been claimed [2]. Our analysis shows that the
hydrogen-bonded systems present a very low degree of CT and hence support a negligible
polarization [3]. A critical reexamination of experimental data supports our findings, shedding
doubts on the ferroelectricity of these systems. The qualitative differences between the transition
of TTF-CA and TTF-BA, the coexistence of electrical polarization and antiferromagnetic order,
and the magnetic-field control over ferroelectricity will be also discussed.
References
[1] G. D’Avino et al., Phys. Rev. Lett. 99, 156407 (2007); G. D’Avino et al., Phys. Rev. B 83, 161105R (2011)
[2] A. Tayi et al., Nature 488, 485 (2012)
[3] G. D’Avino and M. Verstrate, Phys. Rev. Lett., accepted (2014)

22
Charge Transport in π-conjugated Polymers:
A Combined Classical-Quantum Approach
to Establish Structure-Property Relationships
Y. Olivier, V. Lemaur, R. Lazzaroni, D. Beljonne, J. Cornil
University of Mons, Belgium
Organic conjugated polymers have attracted an increasing interest over the years for use in
organic opto-electronic devices such as light-emitting diodes, solar cells, or field-effect
transistors as a result of their low cost, light weight and ease of processing from solution. One of
the main challenges to improve the efficiencies of opto-electronic devices is to get a deep
understanding, at the molecular scale, of the charge transport properties of the organic layer.
Indeed, charge transport is known to be very sensitive to the relative position of the molecules in
the organic layer [1]. Based on quantum-chemically parameterized force fields, the
supramolecular organization of p- and n-type conjugated polymers has been investigated [2-5].
The efficiency of charge transfer has then been estimated through the use of the Marcus theory
by evaluating, at the quantum-chemical level, the main parameters governing charge transport,
i.e., the reorganization energies and intermolecular hopping transfer integrals. This strategy
provides a deep insight into structure-property relationships of organic conjugated compounds in
the solid state and paves the way towards the design of a new generation of materials with
enhanced charge transport properties.
References
[1] J.L. Brédas, J.P. Calbert, D.A. da Silva Filho, J. Cornil, PNAS 2002, 99, 5804.
[2] P. Brocorens, A. Van Vooren, M.L. Chabinyc, M.F. Toney, M. Shkunov, M. Heeney, I. McCulloch, J. Cornil, R.
Lazzaroni, Adv. Mat. 2009, 21, 1193.
[3] D. Niedzialek, V. Lemaur, D. Dudenko, J. Shu, M.R. Hansen, J.W. Andreasen, W. Pisula, K. Müllen, J. Cornil, D.
Beljonne, Adv. Mat. 2013, 25, 1939.
[4] G.A.H. Wetzelaer, M. Kuik, Y. Olivier, V. Lemaur, J. Cornil, S. Fabiano, M.A. Loi, P.W.M. Blom, Phys. Rev. B
2012, 86, 165203.
[5] V. Lemaur, L. Muccioli, C. Zannoni, D. Beljonne, R. Lazzaroni, J. Cornil, Y. Olivier, Macromolecules 2013, 46,
8171.

23
Study of Blue Emitters Compounds for OLEDs
Using a New (TADF) Mechanism
M. Moral1, L. Muccioli2, Y. Olivier3, J. C. Sancho-García1
1Dpto. Química Física, Universidad de Alicante, 03080 Alicante, Spain
2Laboratoire de Chimie des Polymères Organiques, University of Bordeaux, 33607 Pessac, France
3Laboratory for Chemistry of Novel Materials, University of Mons, 7000 Mons, Belgium
Since the discovery of OLEDs (Organic Light Emitting Diode) almost two decades ago,
1 their
use have been extensively reported for the display fabrication of smartphones, screens and in
other solid-light emitting applications. In the last years, their efficiency have largely been
improved using metal (phosphorescence) emitters, with an external quantum efficiency reaching
up to 20%.2
Recently, a new mechanism called Thermally Activated Delayed Fluorescence (TADF) has been
promisingly investigated (see Figure 1) to increase the efficiency of OLEDs without resorting to
rare-earth metals. For the most efficient triplet-to-singlet conversion, and always dealing with
conjugated molecules, a small energy gap (ΔEST) between singlet (S1 state) and triplet
(T1 state) excitons is required, which is strongly connected with a large spatial separation
between the highest occupied (HO) and lowest unoccupied (LU) molecular orbital (MO).3,4
N
N N
NO
NO
O
N N
NO
NO
O
N N
NO
N
N N
NO
A
B
2-A
2-B
Figure 1: Energy diagram showing the main light
emitting process for OLEDs aplications.
Figure 2: Chemical structure of the molecule
studied.
In this work, aimed at rationalizing the underlying physical principle behind the TADF
mechanism and motivated by their experimental use as suitable candidates [5], we have
theoretically studied the use of a set of molecules containing electron-withdrawing (oxadiazole
and triazole) together with electron-donating (phenyl-phenoxazine) moieties (see Figure 2). Our
results help to understand the different behaviour of these candidate molecules in real device.5
References
[1] C.W. Tang and S.A. Vanslyke, Appl. Phys. Lett., 1987, 51, 913.
[2] C. Adachi, M.A. Baldo, M.E. Thompsono, S.R. Forrest, J. Appl. Phys., 2001, 90, 5048.
[3] M.Moral, L. Muccioli, W-J. Son, Y. Olivier, J.C. Sancho-García, JCTC, 2014, in press.
[4] J. Gierschner and B. Milian-Medina, Org. Electr. 13 (2012) 985.
[5] J. Lee, K. Shizu. H. Tanaka, H. Nomura, T. Yasuda, C. Adachi, J. Phys. Chem. C., 2013, 1, 4599.
∆EST
Singlet Excited
State (S1)
Triplet excited
state (T1)
Singlet Ground
State (S0)
FL
UO
RE
SC
EN
CE
PH
OS
PH
OR
ES
CE
NC
E
e- : h+
25 %
75 %
TA
DF

24
Charge Transport
in an Amorphous Triphenylamine-fluorene Copolymer
S. M. Gali1, G. D'Avino2,3, L. Muccioli1,3, C. Zannoni3,
T. Papadopoulos 4,5, Y. Yi4, V. Coropceanu4, J.-L. Brédas4,6
1 Laboratoire de Chimie des Polymères Organiques, Université de Bordeaux, France
2 Department of Physics, University of Liège, Belgium
3 Dipartimento di Chimica Industriale “Toso Montanari”, University of Bologna, Italy
4 School of Chemistry & Biochemistry, Georgia Institute of Technology, United States
5 Department of Engineering, University of Bolton, United Kingdom
6 Solar & Photovoltaic Engineering Research Center, KAUST, Saudi Arabia
The alternate copolymer poly[(9,9-dioctylfluorenyl-2,7-diyl)-co-(4,4′-(N-(4-sec- butylphenyl))
diphenylamine, also known as TFB, is widely used as hole transporting and electron blocking
polymer layer in OLEDs, and also constitutes a model system for more fundamental studies [2],
because of its rather good semiconducting properties and its ease of processing. Actually,
discovering the microscopic origin of its hole mobility [3], surprisingly high for an amorphous
system, and possibly learning some lessons applicable to other amorphous system, is the main
objective of this computational study. We first customized a united-atom force field adapt to
model TFB, and then used it to produce the morphology of bulk TFB pentamers at room
temperature with molecular dynamics simulations. Then we evaluated the charge transport
parameters (electronic coupling and reorganization energies) with semiempirical calculations
along the simulation trajectory, and injected them in a kinetic Monte Carlo scheme to evaluate
the charge carrier mobility [4]. In this talk, we will compare the results produced by
Miller-Abrahams and Marcus kinetic models, and discuss the physical and chemical origins of
TFB hole mobility.
References
[1] A. W. Hains, T. J. Marks, Appl. Phys. Lett. 92, 023504 (2008); L.-P. Lu, D. Kabra, K. Johnson, R. H. Friend, Adv.
Funct. Mater 22, 144. (2012)
[2] A. Bruno, L. X. Reynolds, C. Dyer-Smith, J. Nelson, S. A. Haque, J. Phys. Chem. C 117, 19832 (2013)
[3] H. H. Fong, A. Papadimitratos, G. G. Malliaras, Appl. Phys. Lett. 89, 172116 (2006); R. U. A. Khan, D.
Poplavskyy, T. Kreouzis, D. D. C. Bradley, Phys. Rev. B 75, 035215 (2007)
[4] Y. Olivier, L. Muccioli, V. Lemaur, Y. H. Geerts, C. Zannoni, J. Cornil, J. Phys. Chem. B, 113, 14102 (2009)

25
The Origin of Photoluminescence from
Oxygen Containing Graphene Nanoflakes:
A TDDFT Approach
Sunandan Sarkar, Michal Otyepka
Regional Centre of Advanced Technologies and Materials, Department of Physical Chemistry,Faculty of Science,
Palacky University, tr. 17. listopadu 12, 771 46 Olomouc, Czech Republic
Over the recent years, the intensive research of graphene,1 chemically derived graphene
nanoflakes (GNFs) have attracted great interest due to the candidate status instead of graphene in
the aspects of facile synthesis and potential applications.2,3
The large optical absorptivity and
widely tunable band gap of GNFs makes them promising nanomaterials for optoelectronic
devices.4 To improve the performance in optical devices, functionalization of GNFs is an
important way which controls the interaction between the GNFs and the other active components
in the devices. One of the functionalized form is graphene oxide nanoflakes (GONFs), which
consists of various types of oxygen-containing functional groups on the basal plane and at the
edge sites allow to serve as building blocks of the complex carbon based nanomaterials such as
graphene oxides (GOs) and carbon dots (CDs).5,6
Due to the irregular and inhomogeneous
structure, the origin of the photoluminescence (PL) properties of both GOs and CDs materials is
now an important topic of discussion.5-8
Despite the plenty endeavour on the PL properties of the
oxygen containing complex carbon nanomaterials, theoretical studies addressing the optical
properties are scarce. Here we present a systematic investigation on the optical properties from
oxygen containing GNFs using time dependent density functional theory (TDDFT) which could
be a possible description of the origin of PL behaviour from the complex carbon nanomaterials.
References
[1] Novoselov, K. S.; Geim, A. K.; Morozov, S. V.; Jiang, D.; Katsnelson, M. I.; Grigorieva, I. V.; Dubonos, S. V.;
Firsov, A. A. Nature 2005, 438, 197-200.
[2] Pan, D. Y.; Zhang, J. C.; Li, Z.; Wu, M. H. Adv. Mater. 2010, 22, 734-738.
[3] Yan, X.; Cui, X.; Li, L. S. J. Am. Chem. Soc. 2010, 132, 5944-5945.
[4] Zhu, S. J.; Tang, S. J.; Zhang, J. H.; Yang, B. Chem. Commun. 2012, 48, 4527-4539.
[5] Eda, G.; Chhowalla, M. Adv. Mater. 2010, 22, 2392-2415.
[6] Baker, S. N.; Baker, G. A. Angew. Chem., Int. Ed. 2010, 49, 6726-6744.
[7] Loh, K. P.; Bao, Q.; Eda, G.; Chhowalla, M. Nature Chemistry 2010, 2, 1015-1024.
[8] Bourlinos, A. B.; Stassinopoulos, A.; Anglos, D.; Zboril, R.; Georgakilas, V.; Giannelis, E. P. Chem. Mater. 2008,
20, 4539-4541.

26
Exciton Simulations:
From Liquid Crystals to Porphyrin-CNT Aggregates
Micaela Matta1,2, Francesco Zerbetto1
1 Dipartimento di Chimica ”G. Ciamician”, Università di Bologna, Italy
2 Laboratoire de Chimie des Polymères Organiques, UMR 5629, Université de Bordeaux, France
The photophysical behaviour of π-interacting molecular aggregates depends on several factors,
such as: i) the aggregate size, ii) the degree of order, iii) the environment. In order to fully
characterize these systems it is often necessary to employ a joint theoretical and experimental
approach.
In this regard, an algorithm[1]
for the simulation of exciton dynamics has been implemented and
used to reproduce the spectroscopical features of different self-organized systems with
applications in organic photovoltaics. It has been employed successfully to reproduce the
emission spectrum of substituted oligo-phenyleneethynylene (OPE) liquid crystals[2]
, which
presents a strong red shift due to both excimer formation and aggregation in the solid state. The
model has been also adapted to describe the absorption spectrum of protonated porphyrins
(H4P2+
) and carbon nanotubes (CNTs) aggregates[3]
, that is characterized by the simultaneous
presence of H- and J-aggregate bands.
References
[1] L. K. Gallos, A. V. Pimenov, I. G. Scheblykin, M. Van der Auweraer, G. Hungerford, O. P. Varnavsky,
A. G. Vitukhnovsky and P. Argyrakis, J. Phys. Chem. B, 104 (2000), 3918-3923.
[2] K. Toth, J. K. Molloy, M. Matta, B. Heinrich, D. Guillon, G. Bergamini, F. Zerbetto, B. Donnio, P. Ceroni and
D. Felder-Flesch, Angew. Chem. Int. Ed. 52 (2013), 12303-12307.
[4] T. Hasobe, S. Fukuzumi and P. V. Kamat, J. Am. Chem. Soc. 127 (2005), 11884-11885.
[5] T. Hasobe, S. Fukuzumi and P. V. Kamat, J. Phys. Chem. B, 110 (2006), 25477-25484.

27
Tuning the Electronic Properties in Benzochalcogenodiazole-Based
D−A Type Copolymers by Heteroatom Substitution
M. Carmen Ruiz Delgado,1 D. Herrero-Carvajal,1 G.L. Gibson,2
D.S. Seferos,2 J.T. López Navarrete1
1Department of Physical Chemistry, University of Málaga, Spain
2Lash Miller Chemical Lab, Department of Chemistry, University of Toronto, Canada
Donor−acceptor (D−A) approach to conjugated polymer design has become a widely used
method for preparing conjugated polymers with narrow band gaps.1 One outstanding D−A
polymer is poly(cyclopentadithiophene)benzothiadiazole, PCPDTBT (P1 in Figure 1), for which
power conversion efficiencies in solar cells of 4.5-5.5% are reported.2 In this work, we use
resonance Raman (RR) and density functional theory (DFT) calculations to investigate the tuning
of the electronic and structural properties of cyclopentadithiophene-benzochalcogenodiazole
D−A polymers, wherein a single atom in the benzochalcogenodiazole unit is varied from sulfur
to selenium to tellurium (Fig. 1).3 By analyzing the enhancement pattern in the RR spectra using
Raman excitation wavelengths coincident with the various transitions in the copolymers, it was
possible to describe excited state geometric distortion, which in turn is used to define the nature
of the electronic excitation. These experimental findings are corroborated by DFT calculations
using long-range corrected functionals, considering both tuned and default range-separation
parameters.
Figure 1. D-A copolymers under study. Their synthesis are reported In Ref. 4
References
[1] (a) P. M. Beaujuge, C. M. Amb, J. R. Reynolds, Acc. Chem. Res. 2010, 43, 1396−1407; (b) X. Guo, M.D. Watson,
Macromolecules 2011, 44, 6711−6716.
[2] (a) S. Albrecht, S. Janietz, W. Schindler, J. Frisch, J. Kurpiers, J. Kniepert, S. Inal, P. Pingel, K. Fostiropoulos, N.
Koch, D.J. Neher, J. Am. Chem. Soc. 2012, 134, 14932−14944; (b) F. S. U. Fischer, K. Tremel, A. K. Saur, S. Link,
N. Kayunkid, M. Brinkmann, D. Herrero-Carvajal, J.T. López Navarrete, M.C. Ruiz Delgado, S. Ludwigs,
Macromolecules 2013, 46, 4924-4931.
[3] D. Herrero-Carvajal, G. L. Gibson, D. S. Seferos, S. Salman, V. Hernández, J.T. López Navarrete, M. Carmen
Ruiz Delgado, in preparation
[4] G. L. Gibson, T. M. McCormick, D. S. Seferos, J. Am. Chem.Soc. 2012, 134, 539−547.

28
ABSTRACTS
Posters

29
Optical Properties of
π-conjugated Azo Derivatives and Related Conformational Features
Petra Čechováa, Patrick Trouillasb,c, Martin Kubala,a Bruno Therriend
a Department of Biophysics, Centre of the Region Hana for Biotechnological and Agricultural Research, Palacký
University, Šlechtitelů 11, 783 71, Olomouc, Czech Republic b INSERM UMR-S850, Univ. Limoges, School of Pharmacy, University de Limoges, 2 rue du Docteur Marcland,
87 025 Limoges, France c Regional Centre of Advanced Technologies and Materials, Department of Physical Chemistry, Faculty of Science,
Palacký University, tř. 17 listopadu 12, 771 46 Olomouc, Czech Republic d Institute of Chemistry, University of Neuchatel, Ave de Bellevaux 51, CH-2000 Neuchatel, Switzerland
Bipyridines may be used to synthetize Ru-cationic metallarectangles. The specific molecular
structure of these rectangles provides various biological activities including antiproliferative
effects (on A2780 ovarian cancer cells).1 Bipyridines derivatives can exist in their E- or Z-
forms. The E→Z transformation is induced by UV light. Various other linkers can be envisaged
(azo derivatives) to synthetized metallarectangles, for which this transformation must be
perfectly rationalized. Time-dependent density functional theory (TD-DFT) has been widely
used to study the opto-electronic properties of π-conjugated compounds. A series of symmetrical
pyridyl and benzyl derivatives was studied for their conformational features in the E- and Z-
forms, with respect to UV-light absorption. The comprehensive evaluation of potential energy
surfaces in ground state (GS) and excited states (ES) has been initiated for two relevant
prototypes, namely azobenzene and 4,4'-azopyridine. The E→Z transformation is likely and
unlikely for the former and the latter compound, respectively. Significant differences in UV/Vis
absorption spectra were observed, which is discussed in terms of electronic transitions between
the molecular orbitals involved in the GS→ES transition.
Figure: Chemical structures of 4,4'-azopyridine (right) and azobenzene (left)
References
[1] J. Mattsson, P. Govindaswamy, A. K. Renfrew, P. J. Dyson, P. Štĕpnička, G. Süss-Fink, B. Therrien,
Organometallics 28 (2009), 4350
Acknowledgments:
This work was supported by grant no. L01204 National program of Sustainability I, and by grant
IGA_PrF_2014_029 from Palacky University in Olomouc

30
Modeling the Electronic Circular Dichroism of DNA and
Photosensitized DNA
Hugo Gattuso, Xavier Assfeld, Antonio Monari
Université de Lorraine, Théorie-Modélisation-Simulation, SRSMC UMR 7565, Vandoeuvre-Lès-Nancy, France
CNRS, Théorie-Modélisation-Simulation, SRSMC UMR 7565, Vandoeuvre-Lès-Nancy, France
The modeling of DNA photochemistry and its interactions with photosensitizers have recently
gained interest because it allows the understanding of DNA photodegradation processes.
Moreover photosensitization can be exploited for cancer treatment or simply DNA probing. One
of the key aspects that need to be better characterized are the specific modes with whom
sensitizers interact with DNA as well as the structural modifications induced in its global
structure.
To do so, we studied the electronic circular dichroism (ECD) of two types of B-DNA, the
poly(d[AT]) and poly(d[CG]) double strands, with and without photosensitizers and we
compared the evolution occurring in the spectra. ECDs were simulated using the Frenkel exciton
theory and the conformational space was explored using classical molecular dynamics. Excited
states were obtained using QM/MM methods at the TD-DFT level of theory.

31
Are Waters Around RNA More Than Just a Solvent? – An Insight from
Molecular Dynamics Simulations
Petra Kührová,1 Michal Otyepka,1,2 Jiří Šponer,2,3 and Pavel Banáš*1,2
1 Regional Centre of Advanced Technologies and Materials, Department of Physical Chemistry, Faculty of Science,
Palacky University, tr. 17. Listopadu 12, 771 46, Olomouc, Czech Republic
2 Institute of Biophysics, Academy of Sciences of the Czech Republic, Kralovopolska 135, 612 65 Brno, Czech
Republic
3 CEITEC – Central European Institute of Technology, Campus Bohunice, Kamenice 5, 625 00 Brno, Czech
Republic
Hydrating water molecules are believed to be an inherent part of the RNA structure and have a
considerable impact on RNA conformation. However, the magnitude and mechanism of the
interplay between water molecules and the RNA structure are still poorly understood. In
principle, such hydration effects can be studied by molecular dynamics (MD) simulations. In our
recent MD studies,(1, 2) we observed that the choice of water model has a visible impact on
the predicted structure and structural dynamics of RNA, and in particular, has a larger effect than
type, parameterization and concentration of the ions. Furthermore, the water model effect is
sequence dependent and modulates the sequence dependence of A-RNA helical parameters.
Clearly, the sensitivity of A-RNA structural dynamics to the water model parametrization is a
rather spurious effect that complicates MD studies of RNA molecules. These results nevertheless
suggest that the sequence dependence of the A-RNA structure, usually attributed to base stacking,
might be driven by the structural dynamics of specific hydration. Here, we present a systematic
MD study that aimed to (i) clarify the atomistic mechanism of the water model sensitivity, and
(ii) discover whether and to what extent specific hydration modulates the A-RNA structural
variability. We carried out an extended set of MD simulations of canonical A-RNA duplexes
with TIP3P,(3) TIP4P/2005,(4) TIP5P(5)and SPC/E(6) explicit water models and found that
different water models provided a different extent of water bridging between 2’-OH groups
across the minor groove, which in turn influences their distance and consequently also
inclination, roll and slide parameters. Minor groove hydration is also responsible for the
sequence dependence of these helical parameters.
References
[1] I. Besseova et al., Phys Chem Chem Phys 11, 10701 (2009).
[2] I. Besseova et al., J. Phys.Chem. B 116, 9899 (Aug 23, 2012).
[3] W. L. Jorgensen et al., Journal of Chemical Physics 79, 926 (1983).
[4] J. L. F. Abascal et al., J Chem Phys 123, (Dec 15, 2005).
[5] M. W. Mahoney et al., J Chem Phys 112, 8910 (May 22, 2000).
[6] H. J. C. Berendsen et al., J Phys Chem 91, 6269 (Nov 19, 1987).

32
The Strength of Noble Metal-Graphene Interaction
Petr Lazar
Regional Centre of Advanced Technologies and Materials, Department of Physical Chemistry, Faculty of Science,
Palacky University, tr. 17. Listopadu 12, 771 46, Olomouc, Czech Republic
We present a combined theoretical and experimental study of the noble metal-graphene
interactions. We studied the adsorption of Au, Ag, Cu, and Pd atoms and small clusters on
graphene using the density functional theory including the exact exchange (EE) and
non-empirical van der Waals (vdW-DF) corrections. We measured the strength of
metal-graphene interaction using dynamic atomic force microscopy with probes coated by
various metals. The combination of advanced AFM technique measuring forces at ambient
conditions with the density functional theory calculations enabled us to i) quantify the interaction
force between metalized AFM tips and graphene, and ii) test to the possibility of AFM-based
identification of derivatives of graphene (fluorographene, graphene oxide).

33
Lipid Enhanced Exfoliation for Production of Graphene Nanosheets
M. Pykal,† K. Šafářová,† K. Machalová Šišková,† P. Jurecka,†
A. B. Bourlinos,‡ R. Zbořil,† M. Otyepka†
†Regional Centre of Advanced Technologies and Materials, Department of Physical Chemistry, Faculty of Science,
Palacký University Olomouc, 17. Listopadu 12, 771 46 Olomouc, Czech Republic ‡Physics Department, University of Ioannina, 45110 Ioannina, Greece
Liquid-phase exfoliation of graphite is a widely used method to obtain graphene nanosheets, and
therefore the development of a simple and efficient exfoliation procedure remains challenging.
Here, we present a one-step method of graphene exfoliation in lecithin/chloroform solution. The
graphene nanosheets produced by the lecithin assisted exfoliation method were analyzed by
microscopy techniques, including statistical analysis of atomic force microscopy images and
Raman spectroscopy, which both indicate the presence of few-layer graphene nanosheets,
including substantial content of three-layer sheets. Molecular dynamics simulations on the time
scale of 0.5+ μs suggested that stability of the obtained colloid might originate from formation
of lecithin reverse hemimicelles and micelles, which prevent the aggregation of exfoliated
graphene flakes by entropic repulsion of the lipid hydrophobic chains.

34
Solid State Luminescence Enhancement in Dicyano-Distyrylbenzenes:
Intra- and Intermolecular Contributions
Junqing Shi,1 Seong-Jun Yoon,2 Sang Kyu Park,2 Soo Young Park,2
Shinto Varghese,1 Begoña Milián-Medina,1 Johannes Gierschner1
1 Madrid Institute for Advanced Studies - IMDEA Nanoscience, Madrid, Spain
Tel: +34 912998765 E-mail: [email protected] 2 Center for Supramolecular Optoelectronic Materials, Department of Materials Science and Engineering, Seoul
National University, Seoul, South Korea
Within the last few decades, aggregation-induced enhanced emission (AIEE) of conjugated
organic compounds has drawn much attention, in particular due to applications in
optoelectronics.[1]
However, the mechanism is not yet fully elucidated, being a complex
synergetic process determined by both intra- and intermolecular factors.[2,3]
Hence, gaining
deeper insights into the AIEE mechanism is highly beneficial towards targeted design strategies
for optoelectronic applications.
We investigate here structure–property relationships of functionalized dicyano-distyrylbenzenes
(DCS, Fig. 1), being a AIEE prototype material. Intra- and intermolecular factors are
elucidated independently both in solution and in the solid state through an integrative approach
combining steady-state and time-resolved fluorescence and pump-probe techniques with
computational methods.[4]
Our library of molecules with systematic variation of the
cyano-substituent position (α/β), the presence of additional alkoxy substituents in the central
and/or terminal phenyl rings, allows for systematic tuning of solution and solid state
luminescence properties, providing an in-depth understanding of the AIEE mechanism and
suggesting design strategies for highly effective functionalized DSB-based materials.[4]
Fig. 1: Functionalization of DSB
References
[1] (a) S.-J. Yoon et al, J. Am. Chem. Soc. 132 (2010) 13675. (b) X. Luo et al, J. Phys. Chem. C 116 (2012) 21967. [2] J. Gierschner et al, J. Phys. Chem. Lett. 4 (2013) 2686. [3] J. Gierschner, S. Y. Park, J. Mater. Chem. C 1 (2013) 5818. [4] J. Shi et al, in preparation.

35
Cytochrome P450 Oxidoreductase Simulations:
Cofactors Movement and Structural Changes
Martin Šrejber, Veronika Navrátilová, Karel Berka
Department of Physical Chemistry, Faculty of Science, Palacky University Olomouc, Czech Republic
The NADPH-dependent Cytochrome P450 oxidoreductase (CYPOR) is large 677 amino-acid
long microsomal multidomain enzyme responsible for electron donation to its redox partner
cytochrome P450 (CYP). Electron transfer chain is mediated by two riboflavin - based cofactors
flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) within their respective
domains and nicotinamide adenine dinucleotide phosphate (NADPH). During this electron
transfer CYPOR undergoes several structural changes in open and closed state of both domains
in different degree of contact. In spite of the fact that CYP-CYPOR complexes play a key role in
drug metabolism, no complete atomistic mechanism of structural rearrangements during
complex electron transfers has been described yet. Here we present the results of our preliminary
study on structural changes during CYPOR multidomain complex movement between individual
electron transfers using molecular dynamics (MD) simulations with cofactors of NADPH, FAD
and FMN in resting state.
Homology model of human CYPOR was embedded into pure dioleoylphosphatidylcholine
(DOPC) membrane composed of 250 lipids in each layer in several orientations. After system
equilibration, structural changes of protein, anchor and cofactor movement were studied. Anchor
did not move upper or deeper into the membrane, but we were able to suggest possible
CYPOR-membrane orientation which would allow interaction with cytochrome P450. As for the
internal motions - FMN and FAD cofactor remained in close van der Waals contact during the
100-ns long simulation stabilized by π stack interaction of FAD with Trp676, whereas continual
movement of NADPH weakens its π stack interaction with FAD. The results so far pointed on
the necessity of reparametrization of cofactors in various oxidation states in order to fully
comprehend the domain movements during whole electron transfer cycle.

36
Exciton Diffusion Measurements in DTS Derivatives
Shinto Varghese and Ifor D. W. Samuel
Organic Semiconductor Optoelectronics, School of Physics and Astronomy
University of St-Andrews, UK.
E-mail : [email protected]
Exciton diffusion is one of the basic process in the operation of organic optoelectronic devices
which needs to be understood and tuned to utilise the full potential of the device. In a bulk hetero
junction solar cell, the exciton formed in one component (donor) of the blend must diffuse to an
interface with the acceptor, at which the offset of the energy levels lead to charge separation.
Understanding organic solar cells therefore requires the understanding of exciton diffusion.1
Materials with long exciton diffusion lengths are appreciable as solar cell materials, as this will
ensure the excitons generated in the active layer reaches the interface and generate charges.
However the scenario is just the opposite in light emitting devices such as organic light emitting
diodes and laser. Long Exciton diffusion length is detrimental for the high efficiency as the
exciton can met another exciton in its lifetime and decay to ground state non-radiatively. This
process is called Exciton annihilation. This will reduce the efficiency of the device and is quiet
critical in the optoelectronic devices that works at very high exciton densities such as LASERs.
Exciton diffusion can be investigated by different methods however all the methods have its own
advantages and weakness. The methods we utilised for the determination of the exciton diffusion
lengths are volume quenching, surface quenching and exciton-exciton Annihilation.1,2
The
quenching efficiency will be monitored by the time resolved fluorescence measurements and
modelled to appropriate quenching model. Our investigation to a class of small molecule donor
which exhibits high efficiency bulk hetero junction solar cell will be presented in the poster.
References
[1] Ruseckas A., Shaw P. E., Samuel I. D.,W. Dalton Trans, 2009, 10040-10043.
[2] Ward A. J., Ruseckas, A., Samuel I. D. W.J. Phys. Chem C. 2012,116, 23931-23937.

37
Transition Fields in Organic Materials:
From Percolation to Inverted Marcus Regime.
A Consistent Monte-Carlo Method
Riccardo Volpi, Sven Stafström, Mathieu Linares
Department of Physics, Chemistry and Biology (IFM), Linköping University
In the last decades organic semiconductors, and in particular polymeric materials, have attracted
a huge interest among scientists. They are low cost to produce and exhibit peculiar
characteristics that can lead to new promising applications such as: organic light-emitting diodes
(OLED) and organic solar cells (OPV). Theoretical studies are very useful for the improvement
of such devices in terms of the understanding of their supramolecular organization and to help
the polymers design process. The intrinsic probabilistic nature of the conduction, together with
the structural disorder exhibited by these materials, render difficult an analytical expression of
the conductivity; however these aspects are instead suitable to be modeled in a Monte Carlo
simulation [1]. In this study we focus on the mobility dependence in organic materials for a
broad range of electric field strenghts. It is well known in literature that to analyze correctly this
field dependence, the spatial correlation effects in the energetic landscape has to be taken into
account [2-3]. We propose here a model suitable for an extended electric field range, [0,1.6e7]
V/cm. The mobility field dependence is not reducible to a single functional expression, such as
Poole-Frenkel [4], instead we have to take into account the existence of different operating
regions. In particular we identify the transition fields between these regions and explain their
physical origin. In principle, this allows us to characterize the mobility field dependence for any
organic material, changing the parameters in the transition fields equation. Monte-Carlo
simulations of holes hopping in disordered PPV are presented and analyzed to support our thesis.
References
[1] M. Jakobsson, M. Linares, S. Stafström. Monte Carlo simulations of charge transport in organic systems with
true off-diagonal disorder. J. Chem. Phys. 137, 114901 (2012)
[2] D. Dunlap, V. Kenkre, P. Parris. What is behind the √E J. Imag. Sci. and Tech. 43, 437 (1999)
[3] S. Novikov, A. Vannikov. Cluster Structure in the Distribution of the Electrostatic Potential in a Lattice of
Randomly Oriented Dipoles. J. Phys. Chem. 99, 14573 (1995)
[4] J. Frenkel. On Pre-Breakdown Phenomena in Insulators and Electronic Semi-Conductors. Phys. Rev. 54, 647
(1938).

38
Toward Improved Description of DNA Backbone
Marie Zgarbová,1 Michal Otyepka,1 Pavel Banáš,1 F. Javier Luque,2
Thomas E. Cheatham, III,3 Jiří Šponer,4 Petr Jurecka1
1Regional Centre of Advanced Technologies and Materials, Department of Physical Chemistry, Faculty of Science,
Palacky University, 17. listopadu 12, 77146 Olomouc, Czech Republic 2Department de Fisicoquímica and Institut de Biomedicina (IBUB), Facultat de Farmàcia, Universitat de Barcelona,
Avgda Diagonal 643, Barcelona 08028, Spain
3Department of Medicinal Chemistry, College of Pharmacy, University of Utah, Salt Lake City, Utah, United States
4Institute of Biophysics, Academy of Sciences of the Czech Republic, Královopolská 135, 612 65 Brno, Czech
Republic
e-mail: [email protected], [email protected]
Torsion parameters are known to strongly influence description of nucleic acids backbone in
molecular dynamics (MD) simulations. Recently, we have introduced a reparameterization of the
epsilon and zeta torsion parameters of the Cornell et al. force field, which relies on high-level
quantum-mechanical calculations and accounts for the conformation-dependent solvation effects
that are not covered by the standard explicit solvent models in MD simulations. Here we present
an exhaustive analysis of the performance of the refined parameters based on extended
molecular dynamics simulations for several representative systems. The results showed that the
epsilon/zeta refinement improves the backbone description of B-DNA double helices. The
resulting twist and sequence dependence is in better agreement with solution NMR data.
Furthermore, the parameters improved the description of groove widths and substantially
reduced the RMSD of the sugar-phosphate backbone as compared to the ff99bsc0 MD
simulations. The balance between populations of BI and BII backbone sub-states was shifted
towards the BII state, in better agreement with ensemble-refined solution experimental results.
Tests were also performed on other DNA structures, including DNA A-tracts, G-DNA and
Z-DNA. The refinement is suggested as a possible alternative to the current epsilon/zeta torsion
potential, which may enable more accurate modeling of nucleic acids.
39
Map for
Dinners and Lunches
Map of Olomouc with directions

Program
Thursday 5th
February 2015
19:00 Meeting at Holy Trinity Column (UNESCO), Horní náměstí, Olomouc
19:30 Dinner at Michalský Výpad restaurant, Blažejské náměstí 10, Olomouc
Friday 6th
February 2015 17. listopadu 12, Olomouc, room 3.003
Excited States of Conjugated Systems chair: Patrick Trouillas
8:00-8:15 Welcome
8:15-8:30 Gierschner Excited State Features and Dynamics in Conjugated Organic Materials
8:30-8:55 Milián-Medina MO and Excited State Design of Low Bandgap Copolymers
8:55-9:20 Wykes
Fluorescence in Distyrylbenzene Single-crystals: Interpreting the
Low-temperature Fine Structure via Atomistic Quantum Chemical
Franck-Condon Herzberg-Teller calculations
9:20-9:45 Knippenberg Simulations of Excited Electronic States and Photochemistry
9:45-10:00 Giussani On the Computation of Two-dimensional Electronic Pump-Probe Spectra
10:00-10:30 Coffee Break (3.002) + Posters (atrium 3rd
floor)
10:30-11:15 Nachtigallová Electronically Excited States of Nucleic Acid Bases: Computational
Study
11:15-11:40 Improta The Role of Charge Transfer Excited Electronic States in The
Photophysics and Photochemistry of DNA
11:40-12:05 Linares Electronic Circular Dichroism Spectra in Stacked Systems
12:05-12:30 Sancho-García Merging Non-local (van der Waals) Corrections with Double-Hybrid
Density Functionals
12:30-14:00 Lunch (3.002)
13:15-14:00 Committee Meeting (3.031)
Conjugated Systems in Biology chair: Michal Otyepka
14:00-14:45 Šponer Nature and Magnitude of Aromatic Base Stacking in Nucleic Acids
14:45-15:10 Di Meo Induced Chirality in Porphyrins: Self-Assembly and Systems of Biological
Interest
15:10-15:35 Rezazgui Design and Study of Bio-Inspired Photoherbicide: From Theory to
Experiment
15:35-16:00 Grenier Capless HDAC Inhibitors: Molecular Modelling Reveals the Structural
Determinants of Their Selectivity Toward HDAC6
16:00-17:00 Coffee Break (3.002) + Posters (atrium 3rd
floor)
17:00-17:25 Monari Oxidative Stress in Biological Lipid Membranes: The Influence of Electric
Field
17:25-17:50 Kubala Environment Effects on the Spectra of Polycyclic Organic Molecules
17:50-18:15 Biler Investigation of Antiradical and UV/Vis Absorption Properties of
Flavonolignans
18:15-18:30 Concluding Remarks of the Day
19:00 Dinner at Moravská restaurant, Horní náměstí 23, Olomouc

Saturday 7th
February 2015 17. listopadu 12, Olomouc, room 3.003
Conjugation in Material Science chair: Mathieu Linares
8:00-8:45 Jelínek What We Can Learn from High-Resolution AFM/STM Images:
Experiment and Theory
8:45-9:10 Bjattacharrya Excited State Dynamics in Mixed Stack Donor-Acceptor Charge Transfer
Co-Crystals
9:10-9:35 D'Avino Electronic Ferroelectricity and Magnetoelectric Phenomena in Mixed-stack
Charge Transfer Crystals
9:35-10:00 Olivier Charge Transport in π-conjugated Polymers: A Combined
Classical-quantum Approach to Establish Structure-Property Relationships
10:00-10:25 Moral Study of Blue Emitters Compounds for OLEDs Using a New (TADF)
Mechanism
10:30-11:00 Coffee Break (3.002) + Posters (atrium 3rd
floor)
11:00-11:25 Gali Charge Transport in an Amorphous Triphenylamine-fluorene Copolymer
11:25-11:50 Sarkar The Origin of Photoluminescence from Oxygen Containing Graphene
Nanoflakes : A TDDFT Approach
11:50-12:15 Matta Exciton Simulations: From Liquid Crystals to Porphyrin-CNT Aggregates
12:15-12:40 Carmen Ruiz
Delgado
Tuning the Electronic Properties in Benzochalcogenodiazole-Based D−A
Type Copolymers by Heteroatom Substitution
12:40-12:50 Concluding Remarks
13:30-16:00 Lunch at M3 restaurant, Masarykova třída 889/3, Olomouc (200 CZK)
List of Posters
Čechová Optical Properties of π-conjugated Azo Derivatives and Related Conformational
Features
Gattuso Modeling the Electronic Circular Dichroism of DNA and Photosensitized DNA
Kührová Are Waters Around RNA More Than Just a Solvent? – An Insight from Molecular
Dynamics Simulations
Lazar The Strength of Noble Metal-Graphene Interaction
Pykal Lipid Enhanced Exfoliation for Production of Graphene Nanosheets
Shi Solid State Luminescence Enhancement in Dicyano-Distyrylbenzenes: Intra- and
Intermolecular Contributions
Šrejber Cytochrome P450 Oxidoreductase Simulations: Cofactors Movement and Structural
Changes
Varghese Exciton Diffusion Measurements in DTS Derivatives
Volpi Transition Fields in Organic Materials: From Percolation to Inverted Marcus Regime.
A Consistent Monte-Carlo Method.
Zgarbová Toward Improved Description of DNA Backbone

















