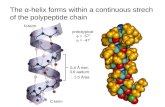
3 Å i i+1 i+2 CαCα CαCα CαCα The (extended) conformation General shape.
34
3 Å i i+ 1 i+ 2 C α C α C α The (extended) conformation • General shape
-
Upload
crystal-wilkerson -
Category
Documents
-
view
219 -
download
1
Transcript of 3 Å i i+1 i+2 CαCα CαCα CαCα The (extended) conformation General shape.

3 Å
i i+1 i+2
Cα
CαCα
The (extended) conformation
• General shape

C’
top view
side view
N’
The (extended) conformation
• Hydrogen bonds

The β conformation
Some facts and statistics:
1. ~30% of globular proteins
2. Strands lie side by side to form a sheet
3. Up to about10 residues per strand
4. 3Ǻ rise per residue
5. Sidechains project upwards and downwards
6. Main-chain amides and carbonyls of different strands H-bond to reduce polarity

7. The sheet has a ~30º right twist
8. The sheet can be parallel, anti-parallel (β-meander) or mixed (only 20% of sheets)
9. Backbone amide-carbonyl H-bonds occur between strands
Main-chain representation
(β-meander)

The (extended) conformation
• Structural motifs
β-α-β motif Twisted β-sheet (thioredoxin)

The (extended) conformation
• Structural motifs
β-barrel

Why are helices and sheets so common?
The energy change (in kcal/mol) of transferring a charged sphere from water into a hydrophobic environment:
ΔE = El – Ew = 166 (q2/r) (1/εl - 1/εw)

c
+
Why are helices and sheets so common?

Why build helices and sheets?
• By pairing polar main-chain amide and
carbonyl groups in H-bonds, helices/sheets
electrostatically mask them from the
hydrophobic core of the protein
Disruption of one H-bond in the protein core:
+5.3 kcal/mol (Ben-Tal et al, 1996)
Disruption of 20 H-bonds (average helix):
+106 kcal/mol
Since the proteins are only marginally stable (5-20 kcal/mol), this
means the disruption of protein structure!

1 2
3
4
Reverse turns and loops
β-turns locations
β-turn structure
Loops

Antigen binding site including 6 loops
Loops• Connect secondary structure elements that create the hydrophobic core of the
protein
• Usually hydrophilic and face the outside of the protein
• Hydrophilic nature results from polar residues and fewer satisfied main-chain H-
bonds
• Often create binding/active sites of receptors and enzymes

Secondary elements are more ordered than loops

Tertiary Structure

FibrousGlobular
Two types of proteins

Unfolded Folded
Folded globular proteins have nonpolar core and overall polar surface

• Globular proteins play different roles in numerous and diverse
cellular activities (enzymes, transporters, immune, and regulatory
proteins)
• This requires some properties that can only be conferred by the
globular shape
Globular proteins

The globular shape allows secondary structures to go in different directions
This allows the protein to achieve:
1. Compactness (an advantage in the extremely dense cytoplasm)
2. Keeping hydrophilic residues outside (confers water solubility) while
maximizing the burial of hydrophobic parts
3. Easy for creating binding sites (cavities)
4. Allows the joining of functional residues that are separated by sequence

Creation of binding site from residues separated by sequence

Some basic characteristics of tertiary structureInteractions that stabilize 3D structure:
1. Covalent interactions (disulfide) – less frequent because they limit protein dynamics
2. Non-covalent – vdW, electrostatic (ionic, H-bond), non-polar (hydrophobic)
[reversible, confer specificity and allow dynamics]
Aromatic ring stacking

The Ca2+-binding ‘EF-hand’ motif (Lewit-Bentely and Rety (2000))
• Ca2+ is involved in many signaling pathways in the cell, as well as in
muscle contraction
• Ca2+ works by binding to signaling proteins (e.g. calmodulin) and inducing
conformational changes that allow further binding to other signaling
proteins
Helical motifs: helix-turn-helix

a
bX, Y: Asp/GlnZ: Asp/Gln/SerY: Main-chain carbonylX: H2OZ: Asp/Glu
Helical motifs: helix-turn-helix
• EF-hand (Ca2+ binding)

• The Ca2+ binding motifs in proteins are of
limited configurations
• The most common motif is the ‘EF hand’,
adopted also by bacteria
• This motif was first discovered in the
muscle protein Parvalbumin (Krestinger) (Lewit-Bentley and Rety 2000)
1.1 Helix-loop-helix (HLH) motifs
• The motif is formed by the 5th and 6th helices (termed E, F) in
parvalbumin, hence the name
• Based on this structure and the sequence constraints emerging from
it, Krestinger predicted EF hand motifs in troponin C and
calmodulin, which were later confirmed

(Lewit-Bentley and Rety 2000)
• The ‘hand’ analogy describes both the fold (helix-loop-helix) and the motion
induced by Ca2+ binding (a)
• The Ca2+-binding loop usually includes 12 residues with the pattern X•Y•ZG–Y•–
X••–Z, where X, Y, Z, –X, –Y and –Z are the ligands that participate in metal
coordination (b) and “•” marks any amino acid
• In Parvalbumin, Ca2+is coordinated by the carboxylate sidechains of 5 residues
(Asp/Glu), by main-chain carbonyl groups and by H2O
• The 6th residue of the loop is Gly, preventing disturbance to the structure
X, Y: ~ D/NZ: ~ D/N/S-Y:~peptide carbonyl-X:~ water-Z:~E/D

(Lewit-Bentley and Rety 2000)
• Some EF-hand motifs cannot bind Ca2+ (e.g. p11). In these, the EF-
hand conformation is maintained in the ‘open’ (analogous to Ca2+-
loaded) form by a network of H-bonds (c)
• The motif is detected in small proteins (e.g. calmodulin), or within
the domains of larger proteins (e.g. myosin or calpain)
• EF-hand motifs usually occur in pairs (two, or four in a dimer), with
cooperative binding

cd
Ca2+
Free Bound
myosin light chain
Helical motifs: helix-turn-helix
• The EF-hand motif in calmodulin (CaM)

e f
Helical motifs: helix-turn-helix
• The EF-hand motif in CaM

• DNA-binding proteins (e.g. transcription factors) are able to
recognize nucleotide sequences both specifically and non-
specifically (the difference results from affinity)
Helical motifs: helix-turn-helix

DNA
TF
Helical motifs: helix-turn-helix
• Helix-turn-helix (DNA binding)

• Direct read-out of the DNA usually
requires the protein to penetrate into
the major and/or the minor grooves of
the DNA
• One way to achieve this type of
penetration is by using HTH motifs
(the connection here is a short β-turn)
• The β-turn and first helix position the
second helix in an orientation that
allows it to fit inside the major groove
of the DNA
DNA-binding HTH motifs

• DNA-binding HTH proteins are used
by both bacteria and eukaryotes
• In eukaryotes, they serve in
developmental regulation of gene
expression
• Such are the ‘homeodomain proteins’,
which contain an extended HTH motif
DNA-binding HTH motifs

3 2 1 4
motifs
β hairpin
β meander
Greek key

β-sheetβ-sheet
motifs
• The sandwich motif

VL
CL
VHCH1CH2
CH3
heavy chains
light chains
antigen binding site
• The immunoglobulin motif
motifs

Other motifs
propeller helix


![vulgarisering ϊ ό vulgaritet ϊ i ɔ ç jeg synes vulgaritet er motbydelig … · 2018-01-31 · 2 for, ta en for å være) ώ [p rn ɔ ja] / det overlater jeg til dere/Dem å vurdere](https://static.fdocument.org/doc/165x107/5d162f9188c993152a8e2ae3/vulgarisering-vulgaritet-i-c-jeg-synes-vulgaritet-er-motbydelig-.jpg)